Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective



Abstract
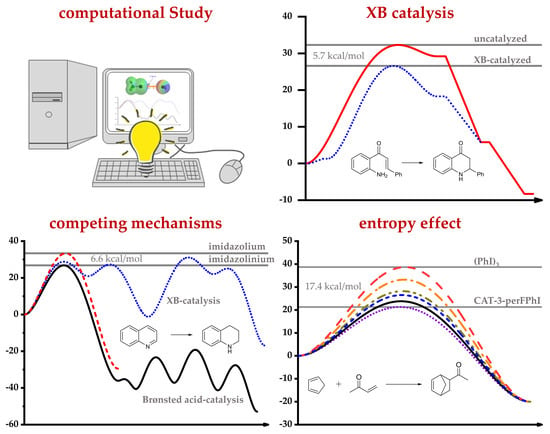
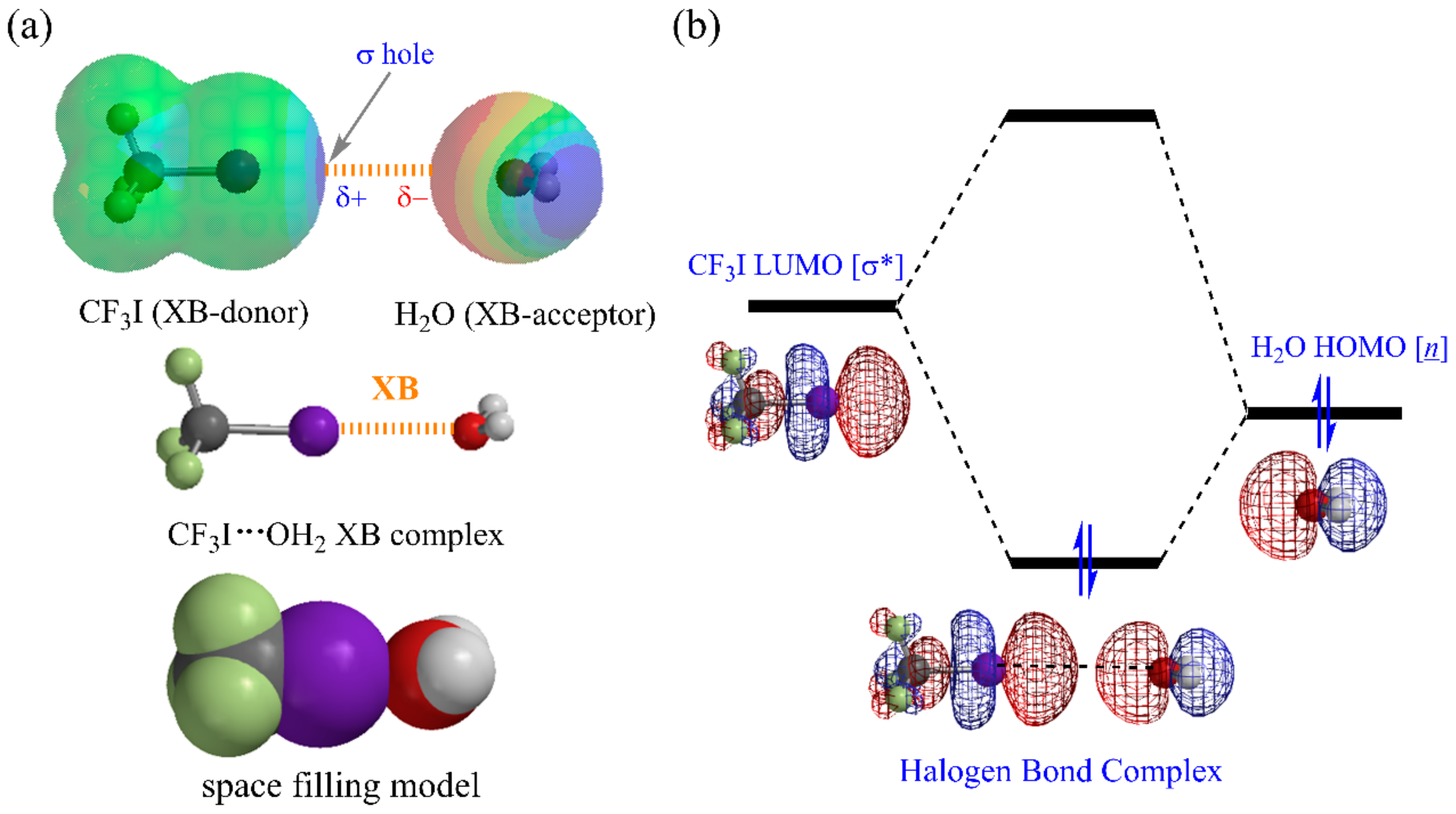
1. Introduction
2. Computational Methods for XB Organocatalysis
3. Computational XB Organocatalysis Studies
3.1. Transition State and Binding XB Complex Studies
3.2. Neutral Halogen Bond Catalysts
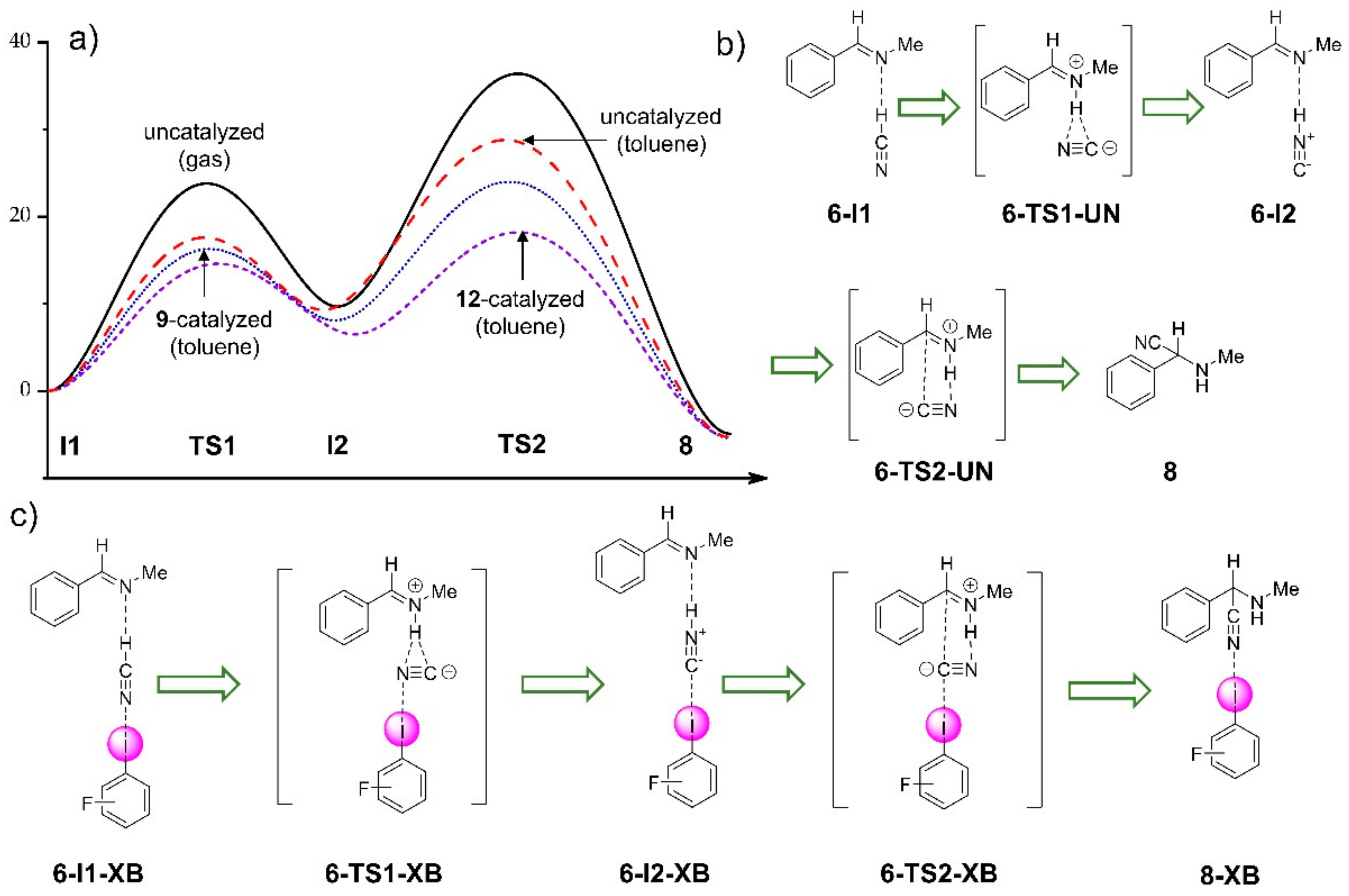
3.2.1. Computational Study of XB-Catalyzed Hydrocyanation of Imines
3.2.2. Computational Design of Neutral XB Catalysts
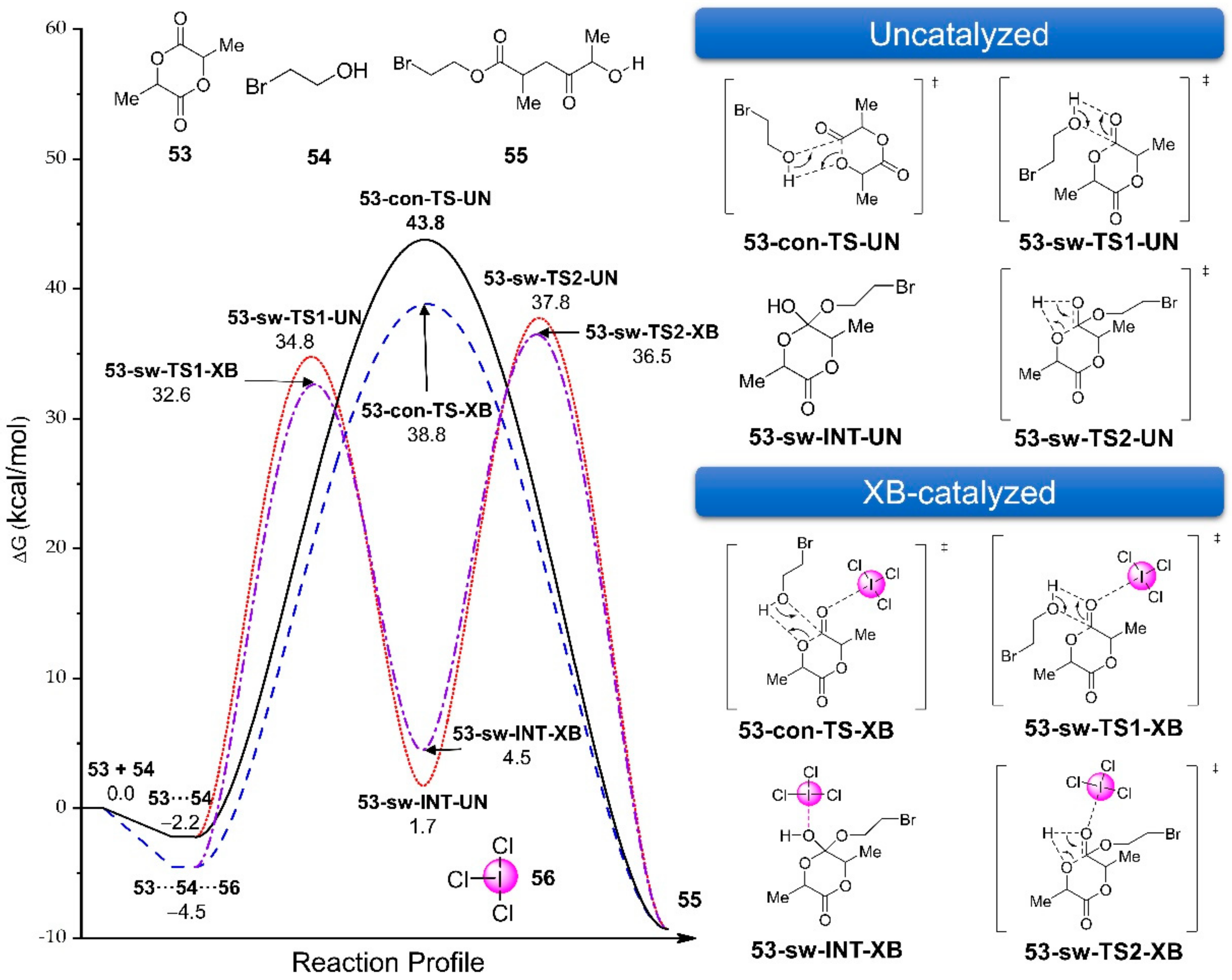
3.2.3. I2 as an XB Catalyst in Organic Reactions
3.2.4. I2-Catalyzed iso-Nazarov Cyclization of Conjugated Dienals
3.2.5. Dihalogen-Catalyzed Michael Addition Reactions
3.2.6. XB in Metal Acetate-Catalyzed Halolactonization
3.3. Cationic XB Catalysts
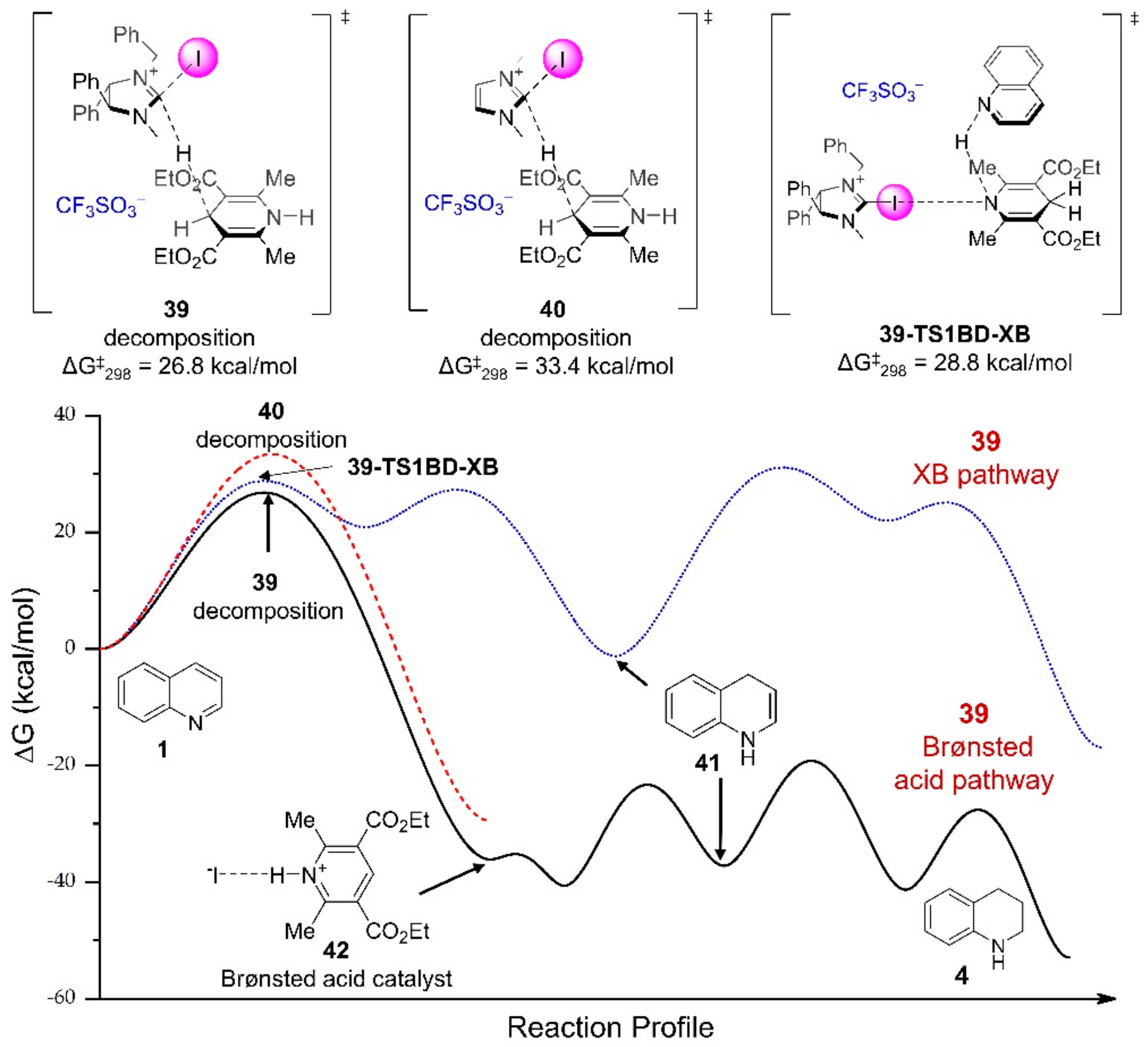
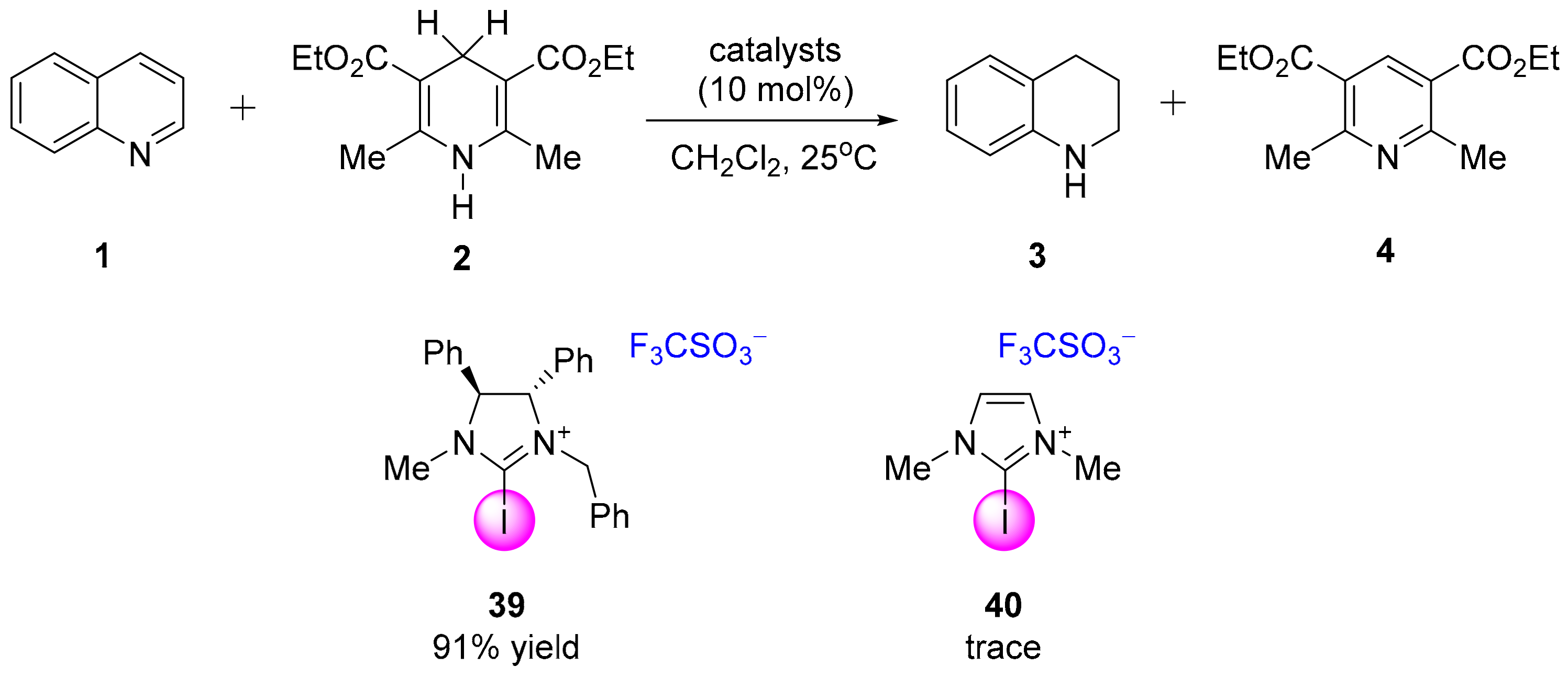
3.3.1. XB-Catalyzed Reduction of Quinolines by Hantzsch Ester
3.3.2. XB-Catalyzed Conjugate Addition of Thiophenes to Enones and Enals
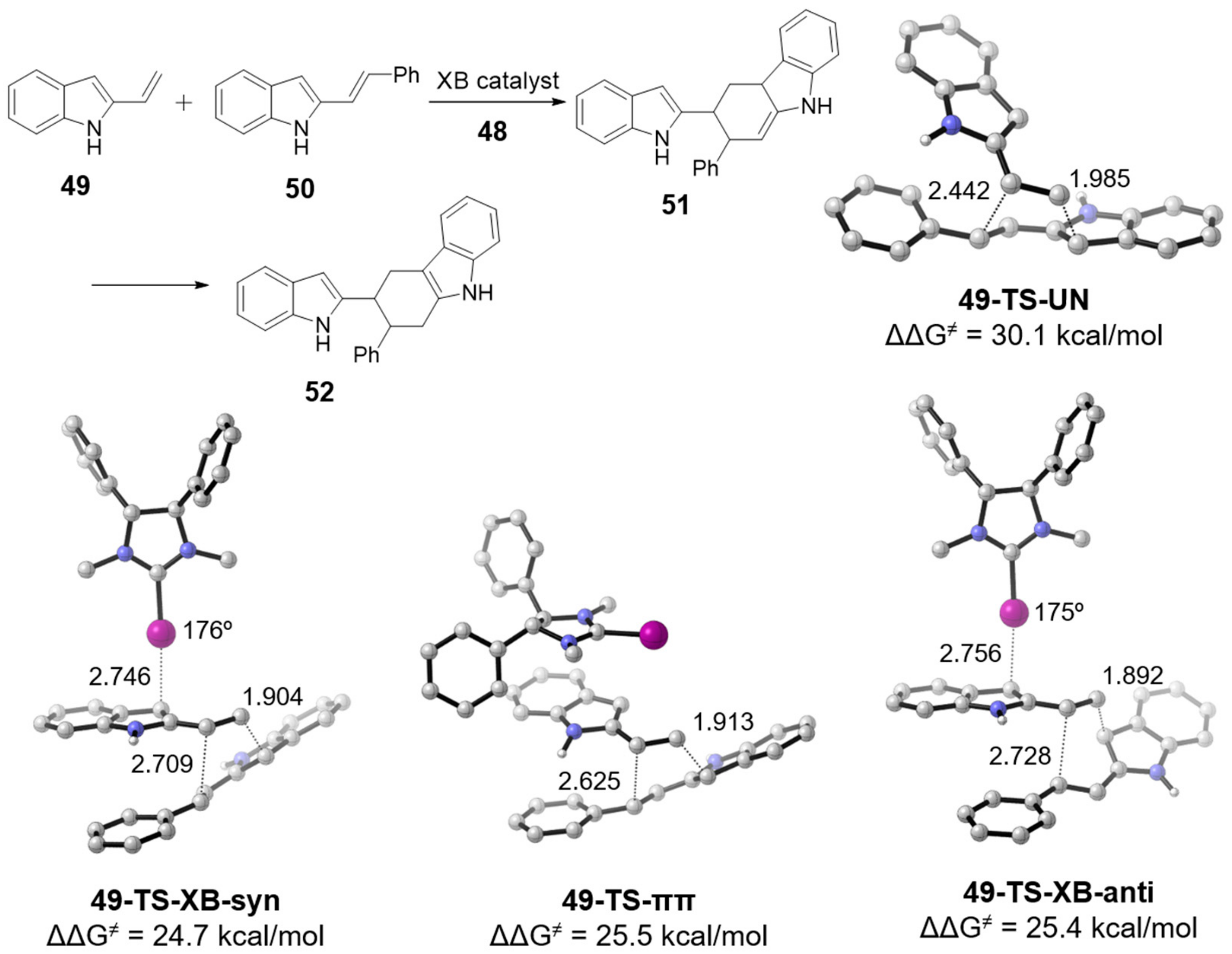
3.3.3. Catalysis by C–I···π Type of XB Interaction
3.4. Hypervalent Iodine(III)-Catalyzed Reactions
ICl3-Catalyzed Ring-Opening Polymerization of l-Lactide
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, September 5th-9th, 2005. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.J.; Mak, A.M.; Sullivan, M.B.; Wong, M.W. Site specificity of halogen bonding involving aromatic acceptors. Phys. Chem. Chem. Phys. 2018, 20, 8685–8694. [Google Scholar] [CrossRef] [PubMed]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Ang, S.J.; Mak, A.M.; Wong, M.W. Nature of halogen bonding involving π-systems, nitroxide radicals and carbenes: A highlight of the importance of charge transfer. Phys. Chem. Chem. Phys. 2018, 20, 26463–26478. [Google Scholar] [CrossRef]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On the nature of the halogen bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for halogen bond covalency in acyclic and interlocked halogen-bonding receptor anion recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef]
- Mustoe, C.L.; Gunabalasingam, M.; Yu, D.; Patrick, B.O.; Kennepohl, P. Probing covalency in halogen bonds through donor K-edge X-ray absorption spectroscopy: Polyhalides as coordination complexes. Faraday Discuss. 2017, 203, 79–91. [Google Scholar] [CrossRef]
- Erakovic, M.; Cincic, D.; Molcanov, K.; Stilinovic, V. A crystallographic charge density study of the partial covalent nature of strong NBr halogen bonds. Angew. Chem. Int. Ed. 2019, 58, 15702–15706. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Lane, P. σ-Hole bonding and hydrogen bonding: Competitive interactions. Int. J. Quantum Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Bolm, C.; Bruckmann, A.; Pena, M. Organocatalysis through halogen-bond activation. Synlett 2008, 2008, 900–902. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen bonding in organic synthesis and organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Sutar, R.L.; Huber, S.M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Walter, S.M.; Kniep, F.; Herdtweck, E.; Huber, S.M. Halogen-bond-induced activation of a carbon–heteroatom bond. Angew. Chem. Int. Ed. 2011, 50, 7187–7191. [Google Scholar] [CrossRef]
- Kniep, F.; Jungbauer, S.H.; Zhang, Q.; Walter, S.M.; Schindler, S.; Schnapperelle, I.; Herdtweck, E.; Huber, S.M. Organocatalysis by neutral multidentate halogen-bond donors. Angew. Chem. Int. Ed. 2013, 52, 7028–7032. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.C.; Tan, C.H. Halogen-bonding-induced hydrogen transfer to C=N bond with Hantzsch ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef]
- Jungbauer, S.H.; Bulfield, D.; Kniep, F.; Lehmann, C.W.; Herdtweck, E.; Huber, S.M. Toward molecular recognition: Three-point halogen bonding in the solid state and in solution. J. Am. Chem. Soc. 2014, 136, 16740–16743. [Google Scholar] [CrossRef]
- Jungbauer, S.H.; Huber, S.M. Cationic multidentate halogen-bond donors in halide abstraction organocatalysis: Catalyst optimization by preorganization. J. Am. Chem. Soc. 2015, 137, 12110–12120. [Google Scholar] [CrossRef]
- Bergamaschi, G.; Lascialfari, L.; Pizzi, A.; Martinez Espinoza, M.I.; Demitri, N.; Milani, A.; Gori, A.; Metrangolo, P. A halogen bond-donor amino acid for organocatalysis in water. Chem. Commun. 2018, 54, 10718–10721. [Google Scholar] [CrossRef]
- Dreger, A.; Engelage, E.; Mallick, B.; Beer, P.D.; Huber, S.M. The role of charge in 1,2,3-triazol(ium)-based halogen bonding activators. Chem. Commun. 2018, 54, 4013–4016. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl iodides: An efficient scaffold for halogen bonding catalysts with an sp3-hybridized carbon–iodine moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Schulz, N.; Sokkar, P.; Engelage, E.; Schindler, S.; Erdelyi, M.; Sanchez-Garcia, E.; Huber, S.M. The interaction modes of haloimidazolium salts in solution. Chem. Eur. J. 2018, 24, 3464–3473. [Google Scholar] [CrossRef]
- Squitieri, R.A.; Fitzpatrick, K.P.; Jaworski, A.A.; Scheidt, K.A. Synthesis and evaluation of azolium-based halogen-bond donors. Chem. Eur. J. 2019, 25, 10069–10073. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Zablotny, S.; Bryce, D.L. Halogen bonding as a supramolecular dynamics catalyst. Nat. Commun. 2019, 10, 916. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef]
- Walter, S.M.; Kniep, F.; Rout, L.; Schmidtchen, F.P.; Herdtweck, E.; Huber, S.M. Isothermal calorimetric titrations on charge-assisted halogen bonds: Role of entropy, counterions, solvent, and temperature. J. Am. Chem. Soc. 2012, 134, 8507–8512. [Google Scholar] [CrossRef]
- Von der Heiden, D.; Bozkus, S.; Klussmann, M.; Breugst, M. Reaction mechanism of iodine-catalyzed Michael additions. J. Org. Chem. 2017, 82, 4037–4043. [Google Scholar] [CrossRef]
- Chan, Y.C.; Yeung, Y.Y. Halogen-bond-catalyzed addition of carbon-based nucleophiles to N-acylimminium ions. Org. Lett. 2019, 21, 5665–5669. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. Halogen bonds: Benchmarks and theoretical analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Manna, D.; Sylvetsky, N.; Martin, J.M.L. The X40 × 10 halogen bonding benchmark revisited: Surprising importance of (n-1)d subvalence correlation. J. Phys. Chem. A 2018, 122, 2184–2197. [Google Scholar] [CrossRef]
- Anderson, L.N.; Aquino, F.W.; Raeber, A.E.; Chen, X.; Wong, B.M. Halogen bonding interactions: Revised benchmarks and a new assessment of exchange vs. dispersion. J. Chem. Theory Comput. 2018, 14, 180–190. [Google Scholar] [CrossRef]
- Ang, S.J.; Ser, C.T.; Wong, M.W. Modeling halogen bonding with planewave density functional theory: Accuracy and challenges. J. Comput. Chem. 2019, 40, 1829–1835. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Ser, C.T.; Yang, H.; Wong, M.W. Iodoimidazolinium-catalyzed reduction of quinoline by Hantzsch ester: Halogen bond or Brønsted acid catalysis. J. Org. Chem. 2019, 84, 10338–10348. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Engelage, E.; Schulz, N.; Heinen, F.; Huber, S.M.; Truhlar, D.G.; Cramer, C.J. Refined SMD parameters for bromine and iodine accurately model halogen-bonding interactions in solution. Chem. Eur. J. 2018, 24, 15983–15987. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Yepes, D.; Neese, F.; List, B.; Bistoni, G. Unveiling the delicate balance of steric and dispersion interactions in organocatalysis using high-level computational methods. J. Am. Chem. Soc. 2020, 142, 3613–3625. [Google Scholar] [CrossRef]
- Liakos, D.G.; Neese, F. Is it possible to obtain coupled cluster quality energies at near density functional theory cost? Domain-based local pair natural orbital coupled cluster vs. modern density functional theory. J. Chem. Theory Comput. 2015, 11, 4054–4063. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Fernandez, I.; Bickelhaupt, F.M. How dihalogens catalyze Michael addition reactions. Angew. Chem. Int. Ed. 2019, 58, 8922–8926. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of halogen bonding in solution: Substituent, structural, and solvent effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, S.H.; Walter, S.M.; Schindler, S.; Rout, L.; Kniep, F.; Huber, S.M. Activation of a carbonyl compound by halogen bonding. Chem. Commun. 2014, 50, 6281–6284. [Google Scholar] [CrossRef] [PubMed]
- Gliese, J.P.; Jungbauer, S.H.; Huber, S.M. A halogen-bonding-catalyzed Michael addition reaction. Chem. Commun. 2017, 53, 12052–12055. [Google Scholar] [CrossRef] [PubMed]
- Dreger, A.; Wonner, P.; Engelage, E.; Walter, S.M.; Stoll, R.; Huber, S.M. A halogen-bonding-catalysed Nazarov cyclisation reaction. Chem. Commun. 2019, 55, 8262–8265. [Google Scholar] [CrossRef] [PubMed]
- Stoesser, J.; Rojas, G.; Bulfield, D.; Hidalgo, P.I.; Pasán, J.; Ruiz-Pérez, C.; Jiménez, C.A.; Huber, S.M. Halogen bonding two-point recognition with terphenyl derivatives. New J. Chem. 2018, 42, 10476–10480. [Google Scholar] [CrossRef]
- Linke, A.; Jungbauer, S.H.; Huber, S.M.; Waldvogel, S.R. Potent affinity material for tracing acetone and related analytes based on molecular recognition by halogen bonds. Chem. Commun. 2015, 51, 2040–2043. [Google Scholar] [CrossRef]
- Coulembier, O.; Meyer, F.; Dubois, P. Controlled room temperature ROP of L-lactide by ICl3: A simple halogen-bonding catalyst. Polym. Chem. 2010, 1, 434–437. [Google Scholar] [CrossRef]
- Chan, Y.C.; Yeung, Y.Y. Halogen bond catalyzed bromocarbocyclization. Angew. Chem. Int. Ed. 2018, 57, 3483–3487. [Google Scholar] [CrossRef]
- Togo, H.; Iida, S. Synthetic use of molecular iodine for organic synthesis. Synlett 2006, 2006, 2159–2175. [Google Scholar] [CrossRef]
- Thakur, A.; Das, S.; Borah, R.; Devi, R. Molecular iodine in protection and deprotection chemistry. Synlett 2008, 2008, 2741–2762. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Iodine-catalyzed transformation of molecules containing oxygen functional groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S.; Tilve, S.G. Recent developments in the synthesis of five- and six-membered heterocycles using molecular iodine. Chem. Eur. J. 2012, 18, 5460–5489. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.-M.; Cai, C.; Yang, R.-C. Molecular iodine-catalyzed multicomponent reactions: An efficient catalyst for organic synthesis. RSC Adv. 2013, 3, 7182–7204. [Google Scholar] [CrossRef]
- Breugst, M.; von der Heiden, D. Mechanisms in iodine catalysis. Chem. Eur. J. 2018, 24, 9187–9199. [Google Scholar] [CrossRef]
- Hibbert, H. Use of iodine as a dehydrating and condensing agent. J. Am. Chem. Soc. 1915, 37, 1748–1763. [Google Scholar] [CrossRef]
- Heinen, F.; Engelage, E.; Dreger, A.; Weiss, R.; Huber, S.M. Iodine(III) derivatives as halogen bonding organocatalysts. Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [Google Scholar] [CrossRef]
- Corey, E.J.; Grogan, M.J. Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Heinz, N.; Dolg, M.; Berkessel, A. A theoretical study of imine hydrocyanation catalyzed by halogen-bonding. J. Comput. Chem. 2015, 36, 1812–1817. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Takeuchi, S.; Sugita, K. Iodoalkyne-based catalyst-mediated activation of thioamides through halogen bonding. Chem. Asian J. 2016, 11, 2863–2866. [Google Scholar] [CrossRef]
- Kee, C.W.; Wong, M.W. In silico design of halogen-bonding-based organocatalyst for Diels–Alder reaction, Claisen rearrangement, and Cope-type hydroamination. J. Org. Chem. 2016, 81, 7459–7470. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. A combined kinetic−quantum mechanical model for assessment of catalytic cycles: Application to cross-coupling and Heck reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Uhe, A.; Kozuch, S.; Shaik, S. Automatic analysis of computed catalytic cycles. J. Comput. Chem. 2011, 32, 978–985. [Google Scholar] [CrossRef]
- Mitsumori, S.; Zhang, H.; Ha-Yeon Cheong, P.; Houk, K.N.; Tanaka, F.; Barbas, C.F. Direct asymmetric anti-Mannich-type reactions catalyzed by a designed amino acid. J. Am. Chem. Soc. 2006, 128, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Shinisha, C.B.; Sunoj, R.B. Bicyclic proline analogues as organocatalysts for stereoselective aldol reactions: An in silico DFT study. Org. Biomol. Chem. 2007, 5, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Neel, A.J.; Hehn, J.P.; Tripet, P.F.; Toste, F.D. Asymmetric cross-dehydrogenative coupling enabled by the design and application of chiral triazole-containing phosphoric acids. J. Am. Chem. Soc. 2013, 135, 14044–14047. [Google Scholar] [CrossRef]
- Markad, D.; Mandal, S.K. Design of a primary-amide-functionalized highly efficient and recyclable hydrogen-bond-donating heterogeneous catalyst for the Friedel–Crafts alkylation of indoles with β-nitrostyrenes. ACS Catal. 2019, 9, 3165–3173. [Google Scholar] [CrossRef]
- Yang, H.; Wong, M.W. b-amino acid catalyzed asymmetric Michael additions: Design of organocatalysts with catalytic acid/base dyad inspired by serine proteases. J. Org. Chem. 2011, 76, 7399–7405. [Google Scholar] [CrossRef]
- Breugst, M.; Detmar, E.; von der Heiden, D. Origin of the catalytic effects of molecular iodine: A computational analysis. ACS Catal. 2016, 6, 3203–3212. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Marsili, L.A.; Pergomet, J.L.; Gandon, V.; Riveira, M.J. Iodine-catalyzed iso-Nazarov cyclization of conjugated dienals for the synthesis of 2-cyclopentenones. Org. Lett. 2018, 20, 7298–7303. [Google Scholar] [CrossRef]
- Koenig, J.J.; Arndt, T.; Gildemeister, N.; Neudorfl, J.M.; Breugst, M. Iodine-catalyzed Nazarov cyclizations. J. Org. Chem. 2019, 84, 7587–7605. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Zeist, W.J.; Bickelhaupt, F.M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kuester, W.E.; Burk, M.T. Catalytic, asymmetric halofunctionalization of alkenes—A critical perspective. Angew. Chem. Int. Ed. 2012, 51, 10938–10953. [Google Scholar] [CrossRef]
- Cheng, Y.A.; Yu, W.Z.; Yeung, Y.-Y. Recent advances in asymmetric intra- and intermolecular halofunctionalizations of alkenes. Org. Biomol. Chem. 2014, 12, 2333–2343. [Google Scholar] [CrossRef]
- Guha, S.; Kazi, I.; Nandy, A.; Sekar, G. Role of Lewis-base-coordinated halogen(I) intermediates in organic synthesis: The journey from unstable intermediates to versatile reagents. Eur. J. Org. Chem. 2017, 2017, 5497–5518. [Google Scholar] [CrossRef]
- Arai, T.; Horigane, K.; Watanabe, O.; Kakino, J.; Sugiyama, N.; Makino, H.; Kamei, Y.; Yabe, S.; Yamanaka, M. Association of halogen bonding and hydrogen bonding in metal acetate-catalyzed asymmetric halolactonization. iScience 2019, 12, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.-C.; Yang, H.; Heusler, A.; Chua, Z.; Wong, M.W.; Tan, C.-H. Halogen-bonding-induced conjugate addition of thiophenes to enones and enals. Chem. Asian J. 2019, 14, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis based on C−I⋅⋅⋅π halogen bonds: Electrophilic activation of 2-alkenylindoles by cationic halogen-bond donors for [4+2] cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Sun, C.; Meng, L.; Zeng, Y. The mechanism of ring-opening polymerization of L-lactide by ICl3 catalysts: Halogen bond catalysis or participating in reactions? J. Comput. Chem. 2019, 40, 2827–2833. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate chiral bis(imidazolium)-based halogen bond donors: Synthesis and first applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFT a | ΔH298 (kcal/mol) | ΔG298 b (kcal/mol) | DXB (Å) |
|---|---|---|---|
| MN15 | −10.1 | −1.0 | 2.75 |
| B3LYP | −1.6 | 7.2 | 2.87 |
| B3LYP-D3 | −8.1 | 1.4 | 2.79 |
| M06-2X | −8.2 | 1.2 | 2.80 |
| ω-B97XD | −6.9 | 2.4 | 2.85 |
| PBE0 | −4.1 | 5.0 | 2.78 |
| PBE0-D3 | −8.2 | 1.1 | 2.74 |

| Uncatalyzed | 18-Catalyzed | |||
|---|---|---|---|---|
| T(k) | ΔG≠ (kcal/mol) | kKeq (M h−1) | ΔG≠n-hexane (kcal/mol) | TOF (h−1) |
| 318.15 | 26.5 | 5.3×10−4 | 28.3 | 8.6×10−4 |
| 298.15 | 26.5 | 3.3×10−5 | 27.3 | 2.3×10−4 |
| 273.15 | 26.4 | 5.3×10−7 | 26.0 | 2.5×10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Wong, M.W. Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules 2020, 25, 1045. https://doi.org/10.3390/molecules25051045
Yang H, Wong MW. Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules. 2020; 25(5):1045. https://doi.org/10.3390/molecules25051045
Chicago/Turabian StyleYang, Hui, and Ming Wah Wong. 2020. "Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective" Molecules 25, no. 5: 1045. https://doi.org/10.3390/molecules25051045
APA StyleYang, H., & Wong, M. W. (2020). Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules, 25(5), 1045. https://doi.org/10.3390/molecules25051045