
Anion Recognition by Neutral and Cationic Iodotriazole Halogen Bonding Scaffolds




Abstract
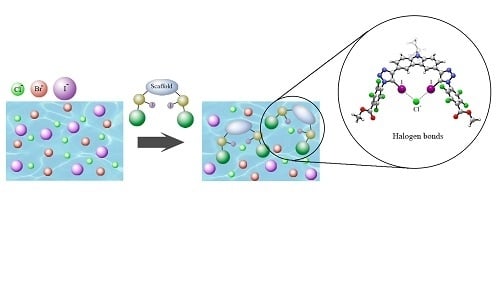
1. Introduction
2. Results
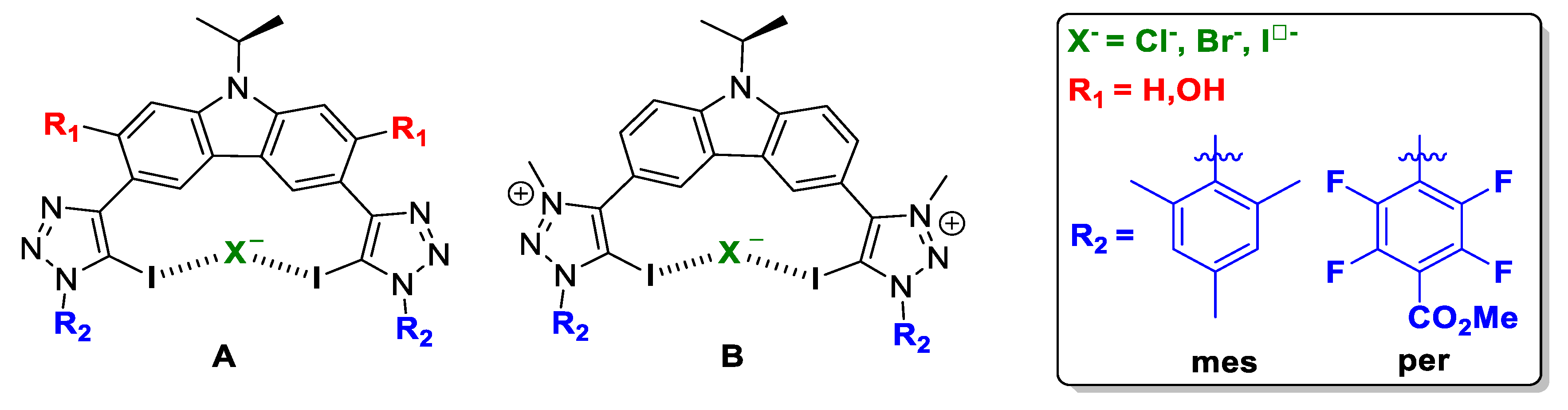
2.1. Bis-Iodotriazole Carbazole-Based XB Donor Motif
2.1.1. MEPs Monomers
2.1.2. Structural Analysis
2.1.3. Energetic Analysis
2.2. Bis-Iodotriazole Pyridinium-Based XB Donor Motif
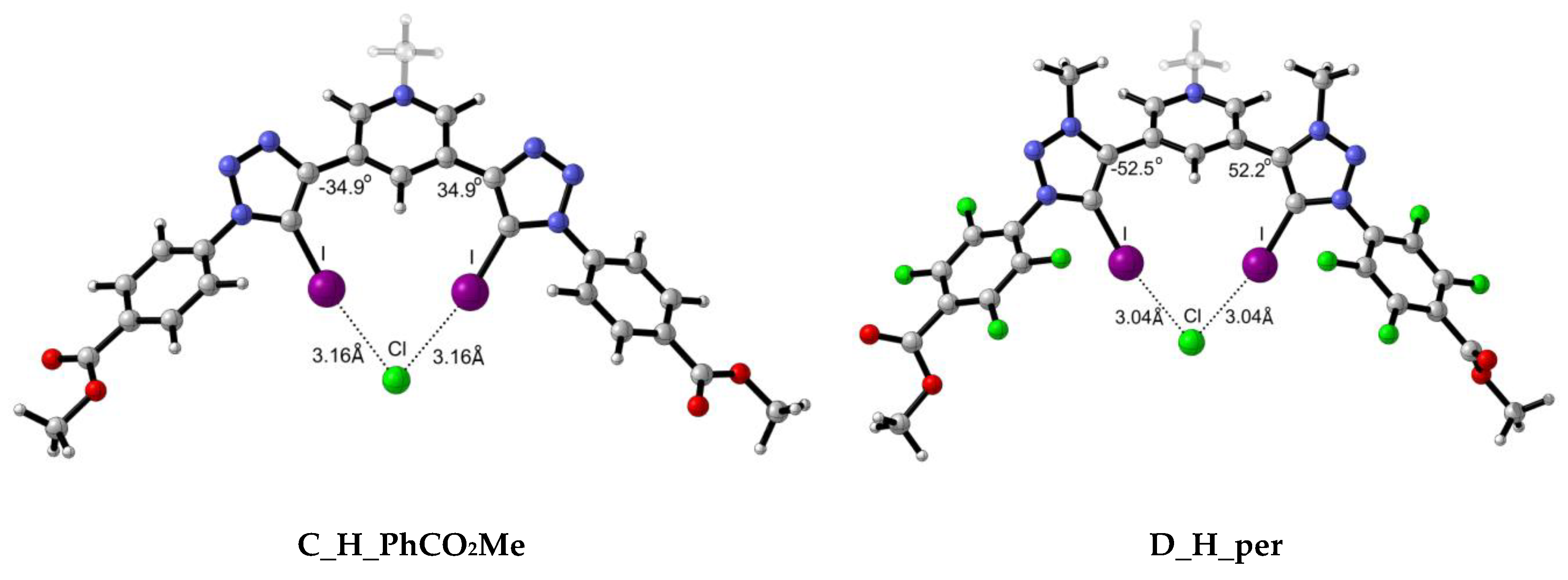
2.2.1. Structural Analysis
2.2.2. Energetic Analysis
3. Discussion
4. Computational Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lim, J.Y.C.; Beer, P.D. Sigma-Hole Interactions in Anion Recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Scheiner, S. Highly Selective Halide Receptors Based on Chalcogen, Pnicogen, and Tetrel Bonds. Chem. Eur. J. 2016, 22, 18850–18858. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Assembly of Effective Halide Receptors from Components. Comparing Hydrogen, Halogen, and Tetrel Bonds. J. Phys. Chem. A 2017, 121, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Comparison of halide receptors based on H, halogen, chalcogen, pnicogen, and tetrel bonds. Faraday Discuss. 2017, 203, 213–226. [Google Scholar] [CrossRef]
- Decato, D.A.; Riel, A.M.S.; Berryman, O.B. Anion Influence on the Packing of 1,3-Bis(4-Ethynyl-3-Iodopyridinium)-Benzene Halogen Bond Receptors. Crystals 2019, 9, 522. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C. Cyclohexane-Based Scaffold Molecules Acting as Anion Transport, Anionophores, via Noncovalent Interactions. J. Chem. Inf. Model. 2019, 59, 2212–2217. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C. Improvement of Anion Transport Systems by Modulation of Chalcogen Interactions: The influence of solvent. J. Phys. Chem. A 2018, 122, 1369–1377. [Google Scholar] [CrossRef]
- Benz, S.; Macchione, M.; Verolet, Q.; Mareda, J.; Sakai, N.; Matile, S. Anion Transport with Chalcogen Bonds. J. Am. Chem. Soc. 2016, 138, 9093–9096. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen bonding: A general route in anion recognition and coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef]
- Tepper, R.; Schulze, B.; Jäger, M.; Friebe, C.; Scharf, D.H.; Görls, H.; Schubert, U.S. Anion Receptors Based on Halogen Bonding with Halo-1,2,3-triazoliums. J. Org. Chem. 2015, 80, 3139–3150. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for Halogen Bond Covalency in Acyclic and Interlocked Halogen-Bonding Receptor Anion Recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.; Schulze, B.; Görls, H.; Bellstedt, P.; Jäger, M.; Schubert, U.S. Preorganization in a Cleft-Type Anion Receptor Featuring Iodo-1,2,3-Triazoles As Halogen Bond Donors. Org. Lett. 2015, 17, 5740–5743. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.; Concha, M. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Bunchuay, T.; Docker, A.; Martinez-Martinez, A.J.; Beer, P.D. A Potent Halogen-Bonding Donor Motif for Anion Recognition and Anion Template Mechanical Bond Synthesis. Angew. Chem. Int. Ed. 2019, 58, 13823–13827. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, version 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Engelage, E.; Schulz, N.; Heinen, F.; Huber, S.M.; Truhlar, D.G.; Cramer, C.J. Refined SMD Parameters for Bromine and Iodine Accurately Model Halogen-Bonding Interactions in Solution. Chem. Eur. J. 2018, 24, 15983–15987. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Jmol: An Open-source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 10 December 2019).
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A.; Aicken, F.M.; O’Brien, S.E. Atoms In Molecules: An introduction, Prentice Hall, Harlow, England. Chem. Model. Appl. Theory 2000, 1, 143–198. [Google Scholar]
- Keith, T.A. TK Gristmill Software. Available online: Aim.tkgristmill.com (accessed on 10 December 2019).
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
Sample Availability: Not available |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomers | 1 | 2 |
|---|---|---|
| A_H_mes | 0.0512 | 0.0512 |
| A_H_per | 0.0614 | 0.0618 |
| A_OH_mes | 0.0605 | 0.0605 |
| A_OH_per | 0.0705 | 0.0711 |
| ρ | ∇2ρ | d(I⋯X) | Dihedral | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complexes | Cl | Br | I | Cl | Br | I | Cl | Br | I | Cl | Br | I |
| A_H_mes | 0.0178 | 0.0156 | 0.0136 | 0.0521 | 0.0409 | 0.0329 | 3.17 | 3.35 | 3.56 | −29.36 | −31.10 | −32.17 |
| A_H_per | 0.0195 | 0.0170 | 0.0152 | 0.0564 | 0.0441 | 0.0358 | 3.13 | 3.31 | 3.51 | −31.26 | −32.40 | −32.81 |
| A_OH_mes | 0.0189 | 0.0168 | 0.0146 | 0.0548 | 0.0437 | 0.0347 | 3.14 | 3.30 | 3.52 | −14.07 | −14.92 | −16.27 |
| A_OH_per | 0.0206 | 0.0184 | 0.0158 | 0.0589 | 0.0470 | 0.0369 | 3.10 | 3.26 | 3.48 | −20.59 | −20.02 | −24.22 |
| B_H_mes | 0.0232 | 0.0204 | 0.0178 | 0.0643 | 0.0501 | 0.0394 | 3.03 | 3.21 | 3.43 | −40.36 | −43.14 | −42.88 |
| B_H_per | 0.0254 | 0.0222 | 0.0195 | 0.0683 | 0.0529 | 0.0414 | 2.99 | 3.17 | 3.38 | −41.56 | −44.34 | 43.68 |
| Eb (kJ/mol) | ΔGb (kJ/mol) | K | ||||||
|---|---|---|---|---|---|---|---|---|
| Complexes | Cl | Br | I | Cl | Br | I | Cl−/Br-− | Cl−/I-− |
| A_H_mes | −49.28 | −46.90 | −44.92 | −15.91 | −13.30 | −11.00 | 2.86 | 7.24 |
| A_H_per | −58.73 | −56.40 | −52.83 | −24.82 | −24.07 | −19.54 | 1.36 | 8.45 |
| A_OH_mes | −57.06 | −53.84 | −50.78 | −19.47 | −19.37 | −17.57 | 1.04 | 2.16 |
| A_OH_per | −66.39 | −62.59 | −58.69 | −35.14 | −30.53 | −29.95 | 6.42 | 8.13 |
| B_H_mes | −113.91 | −109.10 | −104.30 | −77.92 | −79.48 | −81.98 | 0.53 | 0.19 |
| B_H_per | −125.23 | −120.85 | −114.57 | −95.14 | −92.42 | −86.63 | 2.99 | 3.09 |
| E(2) (kJ/mol) | |||
|---|---|---|---|
| Complexes | Cl | Br | I |
| A_H_mes | 122.67 | 125.56 | 116.94 |
| A_H_per | 109.04 | 97.61 | 90.96 |
| A_OH_mes | 79.04 | 73.55 | 70.37 |
| A_OH_per | 119.24 | 111.34 | 98.24 |
| B_H_mes | 167.03 | 141.63 | 72.01 |
| B_H_per | 170.37 | 154.56 | 144.81 |
| ρ | ∇2ρ | d(X⋯I) | Dihedral | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complexes | Cl | Br | I | Cl | Br | I | Cl | Br | I | Cl | Br | I |
| C_H_PhCO2Me | 0.0184 | 0.0169 | 0.0147 | 0.0537 | 0.0438 | 0.0351 | 3.16 | 3.32 | 3.53 | −34.88 | −36.34 | −37.14 |
| C_H_per | 0.0192 | 0.0177 | 0.0155 | 0.0554 | 0.0453 | 0.0363 | 3.14 | 3.29 | 3.50 | −34.45 | −35.81 | −37.24 |
| C_OH_PhCO2Me | 0.0194 | 0.0177 | 0.0155 | 0.0560 | 0.0454 | 0.0364 | 3.13 | 3.29 | 3.50 | −8.21 | −8.76 | −7.30 |
| C_OH_per | 0.0204 | 0.0187 | 0.0164 | 0.0582 | 0.0471 | 0.0377 | 3.11 | 3.27 | 3.47 | −1.60 | −1.49 | −1.17 |
| D_H_PhCO2Me | 0.0221 | 0.0201 | 0.0178 | 0.0618 | 0.0497 | 0.0396 | 3.06 | 3.22 | 3.43 | −52.24 | −52.25 | −52.57 |
| D_H_per | 0.0234 | 0.0212 | 0.0190 | 0.0642 | 0.0512 | 0.0408 | 3.04 | 3.20 | 3.40 | −52.46 | −52.81 | −53.01 |
| ΔEb (kJ/mol) | ΔGb (kJ/mol) | K | ||||||
|---|---|---|---|---|---|---|---|---|
| Complexes | Cl | Br | I | Cl | Br | I | Cl−/Br− | Cl−/I− |
| C_H_PhCO2Me | −59.42 | −58.21 | −56.10 | −28.80 | −27.51 | −28.16 | 1.68 | 1.29 |
| C_H_per | −64.34 | −62.79 | −60.67 | −33.75 | −34.47 | −30.58 | 0.75 | 3.60 |
| C_OH_PhCO2Me | −62.67 | −60.91 | −58.18 | −28.31 | −29.06 | −33.90 | 0.74 | 2.70 |
| C_OH_per | −69.11 | −67.24 | −64.13 | −35.48 | −34.06 | −33.02 | 1.78 | 2.70 |
| D_H_PhCO2Me | −83.64 | −81.56 | −78.05 | −49.20 | −54.26 | −49.65 | 0.13 | 0.83 |
| D_H_per | −90.66 | −88.32 | −84.36 | −53.14 | −55.26 | −51.07 | 0.42 | 2.30 |
| E(2) (kJ/mol) | |||
|---|---|---|---|
| Complexes | Cl | Br | I |
| C_H_PhCO2Me | 94.81 | 91.34 | 82.93 |
| C_H_per | 101.42 | 98.58 | 89.79 |
| C_OH_PhCO2Me | 103.01 | 99.16 | 90.50 |
| C_OH_per | 111.92 | 108.24 | 99.50 |
| D_H_PhCO2Me | 128.74 | 124.35 | 115.06 |
| D_H_per | 140.71 | 135.31 | 127.24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iribarren, I.; Sánchez-Sanz, G.; Trujillo, C. Anion Recognition by Neutral and Cationic Iodotriazole Halogen Bonding Scaffolds. Molecules 2020, 25, 798. https://doi.org/10.3390/molecules25040798
Iribarren I, Sánchez-Sanz G, Trujillo C. Anion Recognition by Neutral and Cationic Iodotriazole Halogen Bonding Scaffolds. Molecules. 2020; 25(4):798. https://doi.org/10.3390/molecules25040798
Chicago/Turabian StyleIribarren, Iñigo, Goar Sánchez-Sanz, and Cristina Trujillo. 2020. "Anion Recognition by Neutral and Cationic Iodotriazole Halogen Bonding Scaffolds" Molecules 25, no. 4: 798. https://doi.org/10.3390/molecules25040798
APA StyleIribarren, I., Sánchez-Sanz, G., & Trujillo, C. (2020). Anion Recognition by Neutral and Cationic Iodotriazole Halogen Bonding Scaffolds. Molecules, 25(4), 798. https://doi.org/10.3390/molecules25040798