MicroRNA-155 Participates in Smoke-Inhalation-Induced Acute Lung Injury through Inhibition of SOCS-1

Abstract
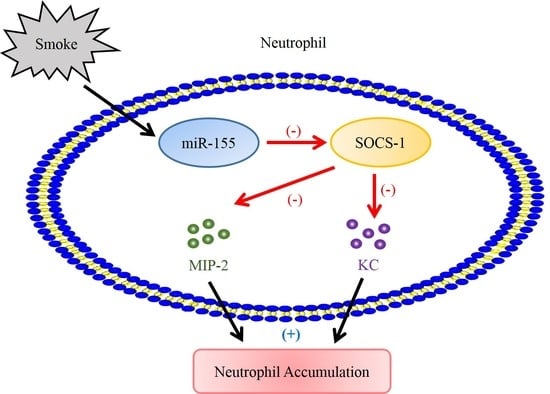
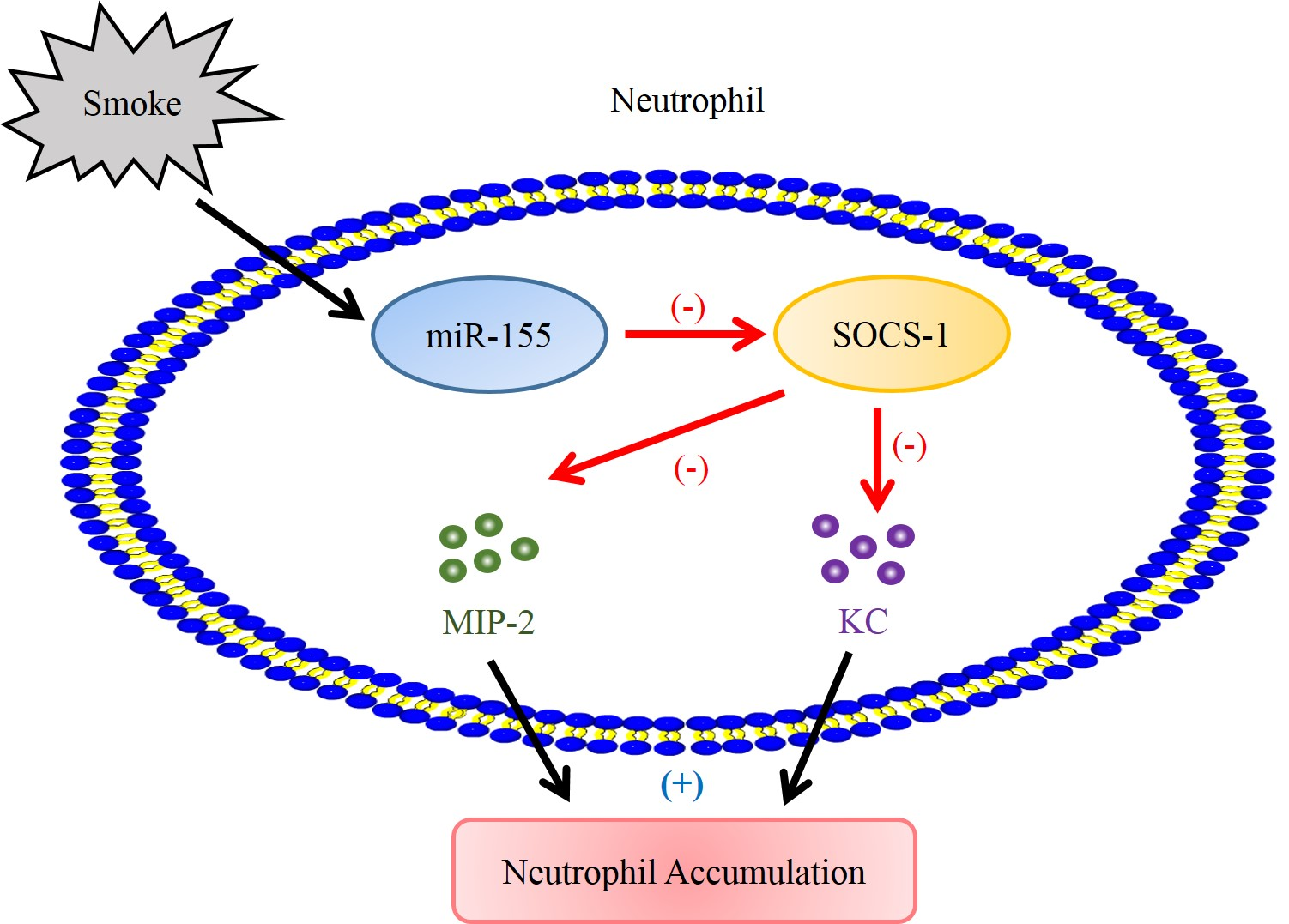{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
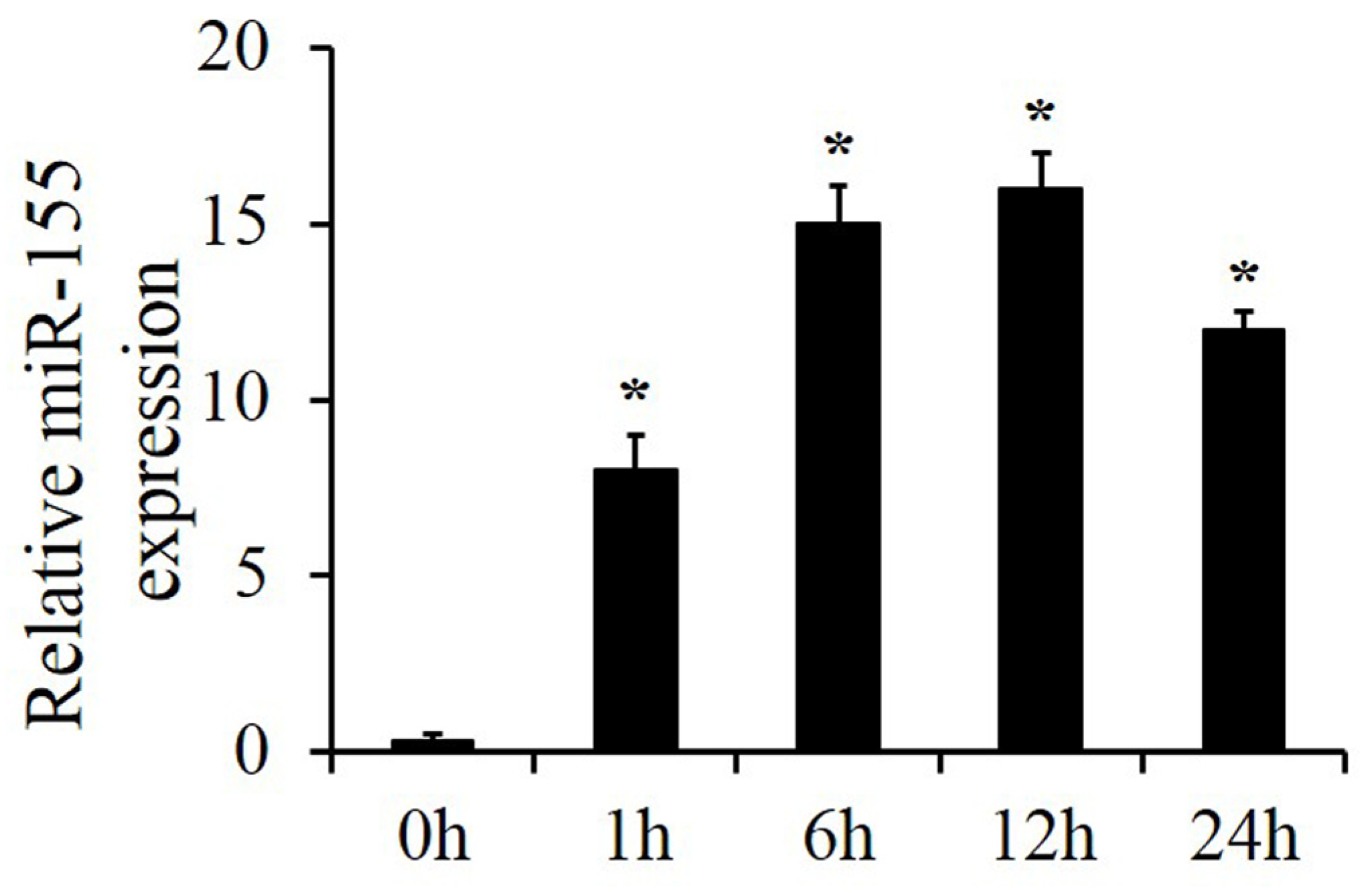
2.1. MiR-155 Expression Increased after Smoke Inhalation
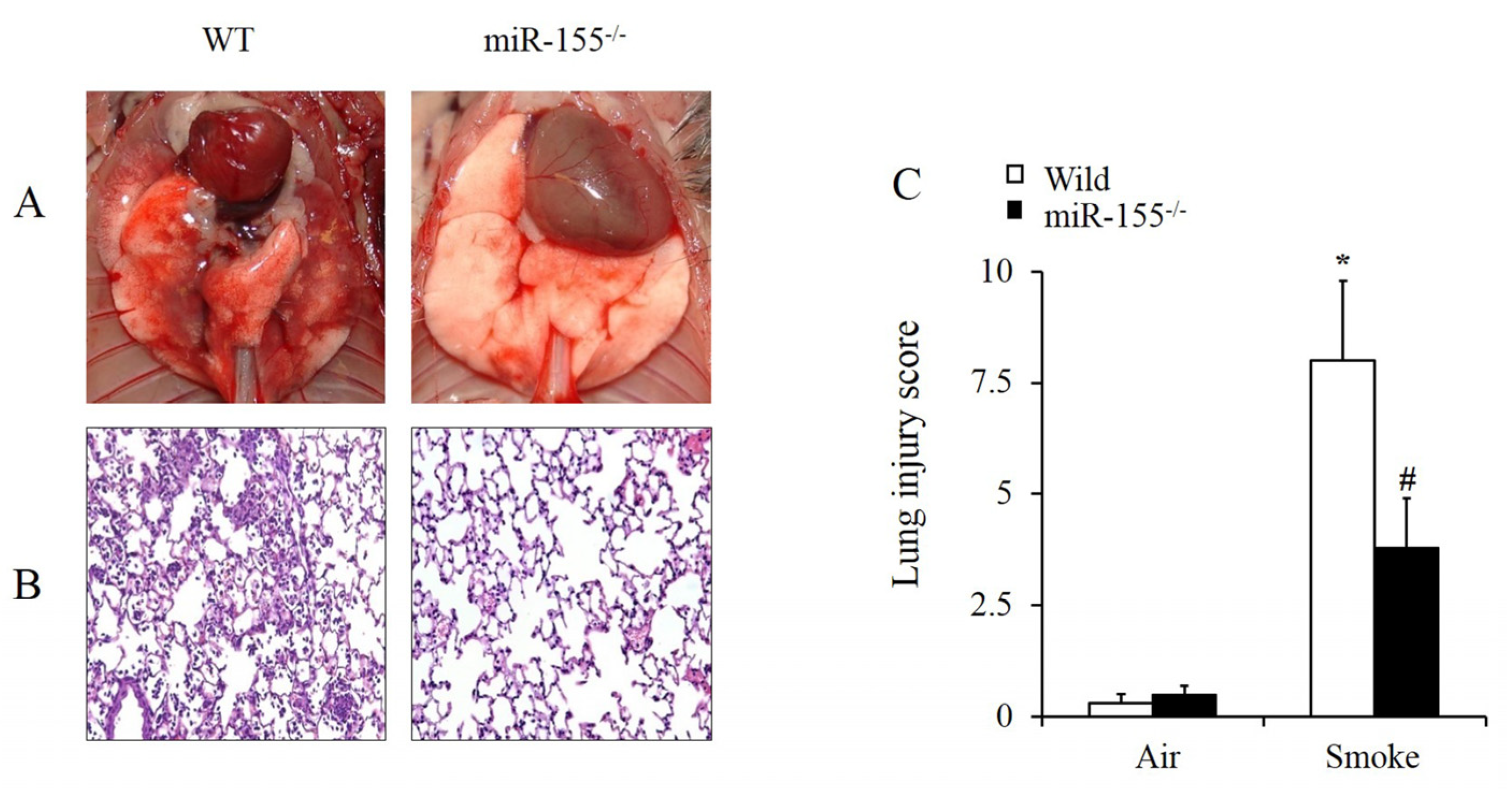
2.2. Absence of Mir-155 Relieved Smoke-Inhalation-Induced ALI
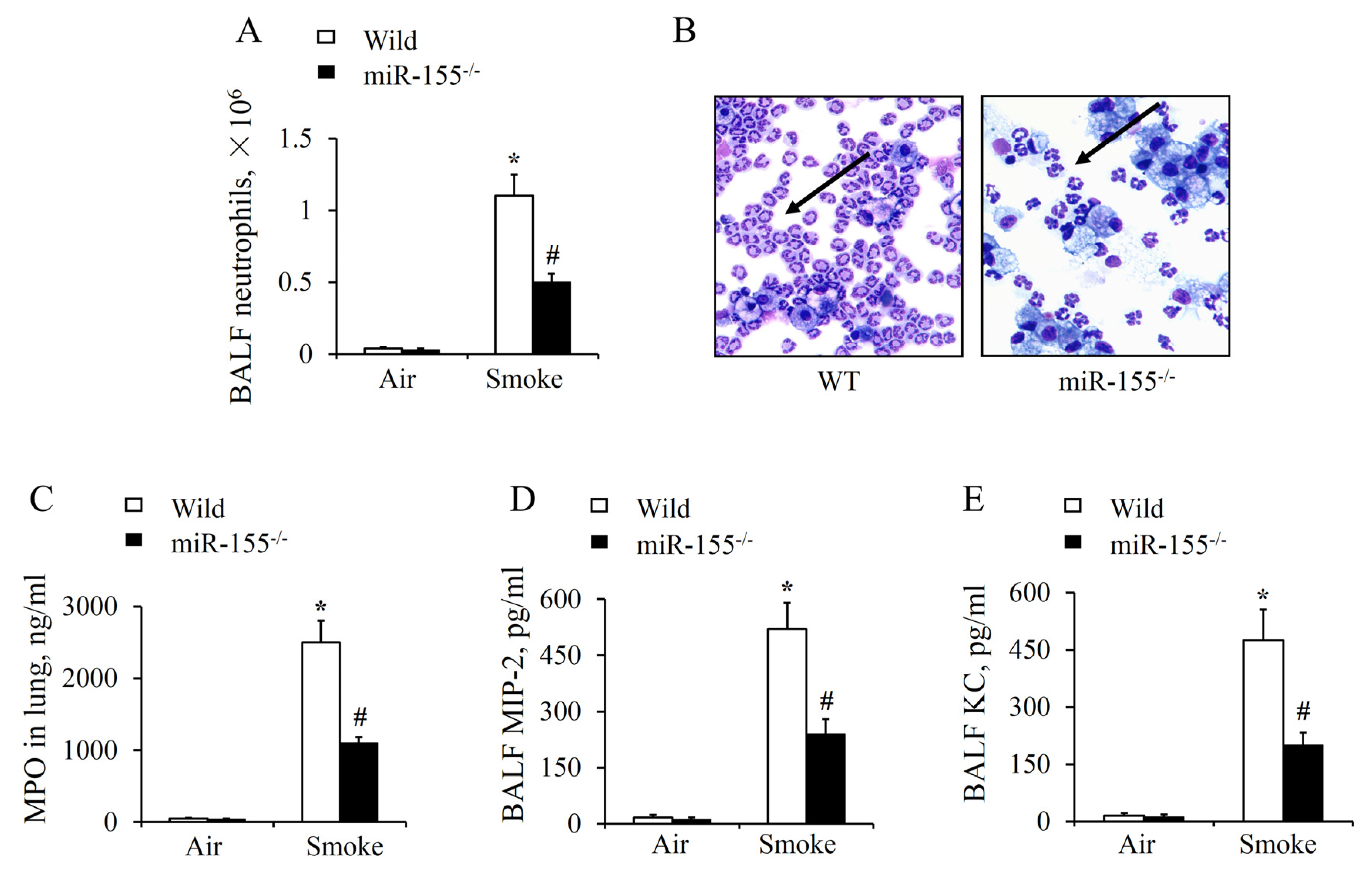
2.3. MiR-155 KO Reduced Neutrophil Aggregation and Inflammatory Cytokines Release in the Lung
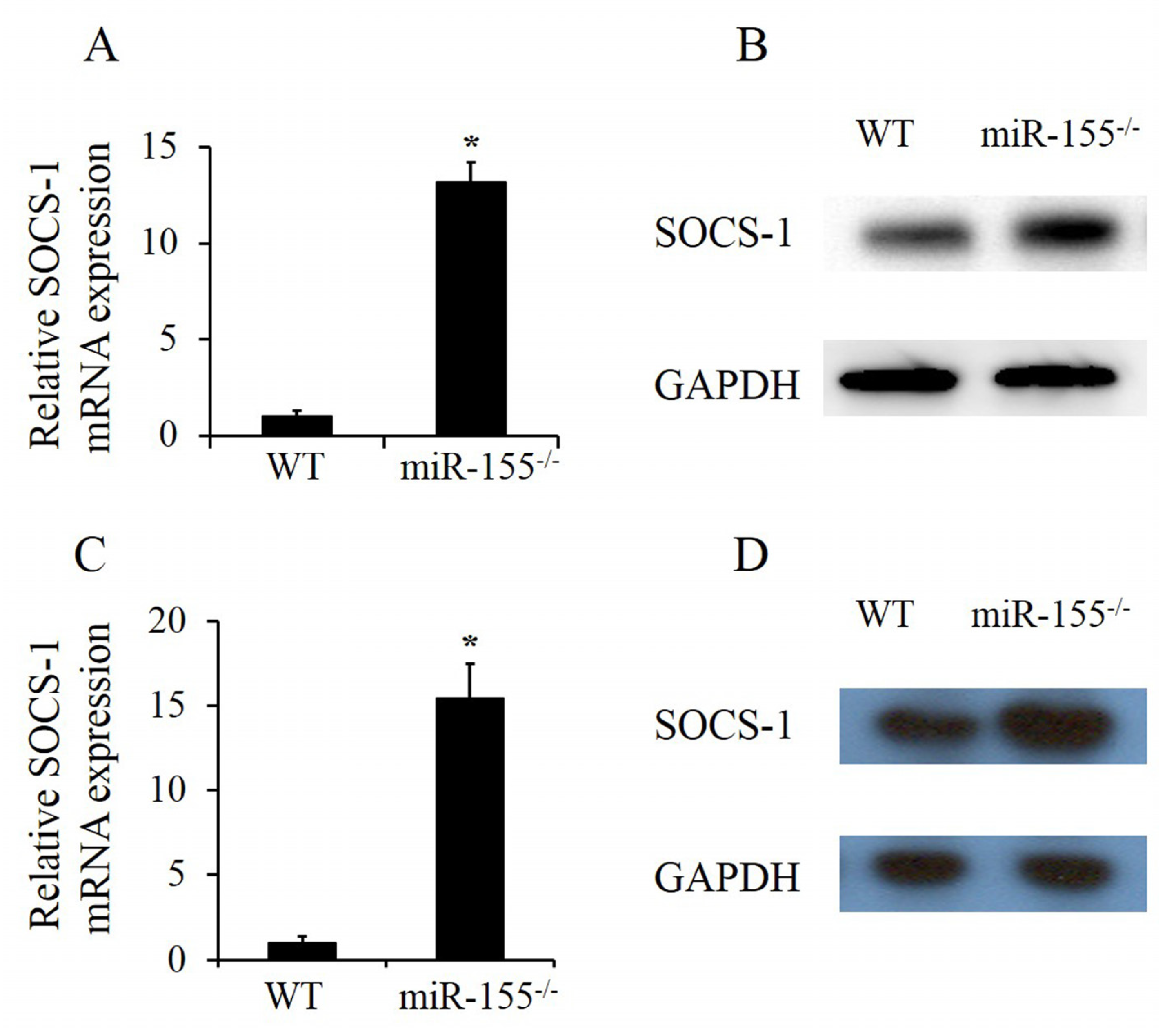
2.4. MiR-155 Deficiency Made SOCS-1 Expression Increase in Both Lung Tissues and Neutrophils after Smoke Exposure
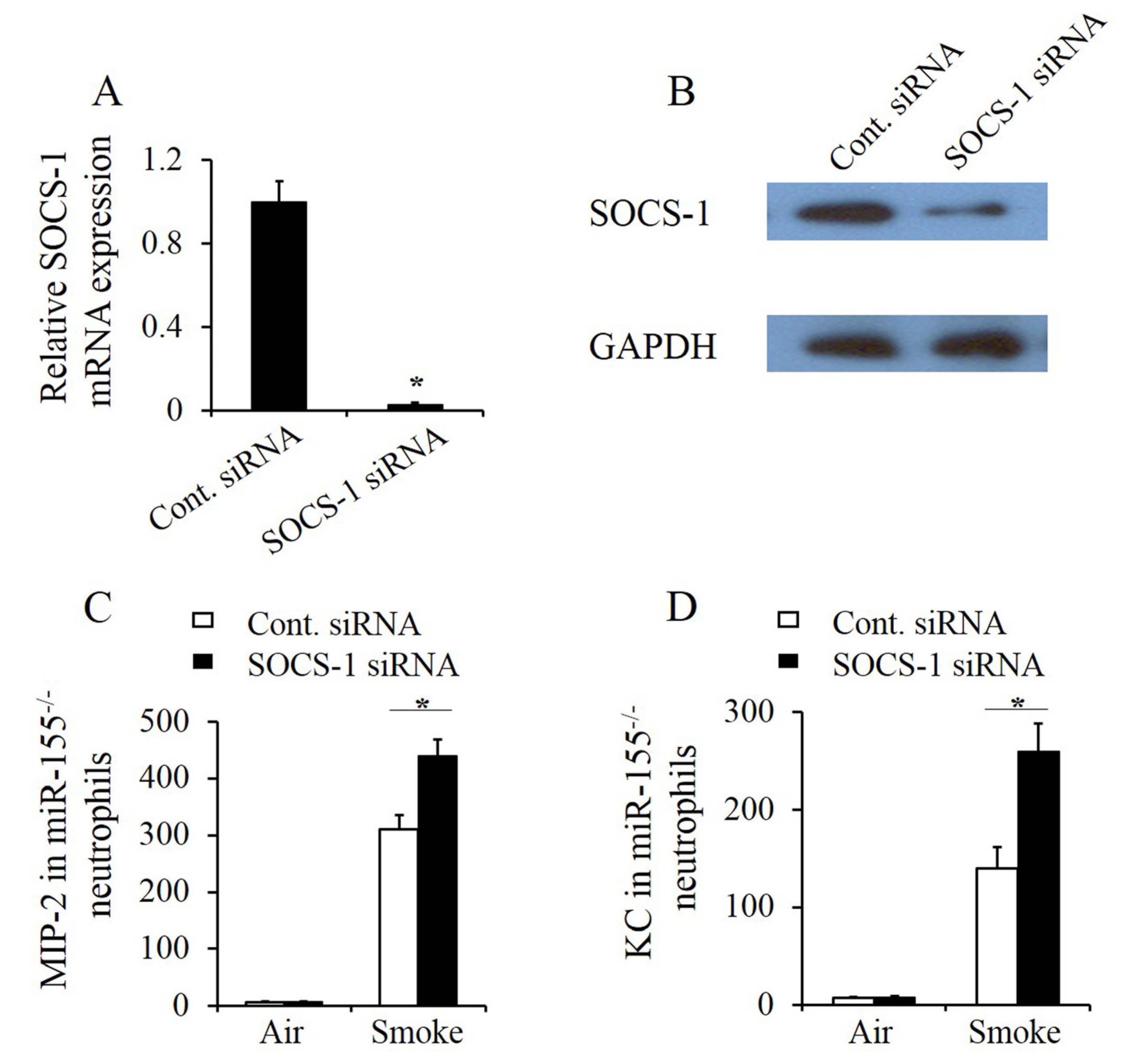
2.5. SOCS-1 Silencing Enhanced Smoke-Induced Inflammatory Response in Mir-155–/– Neutrophils
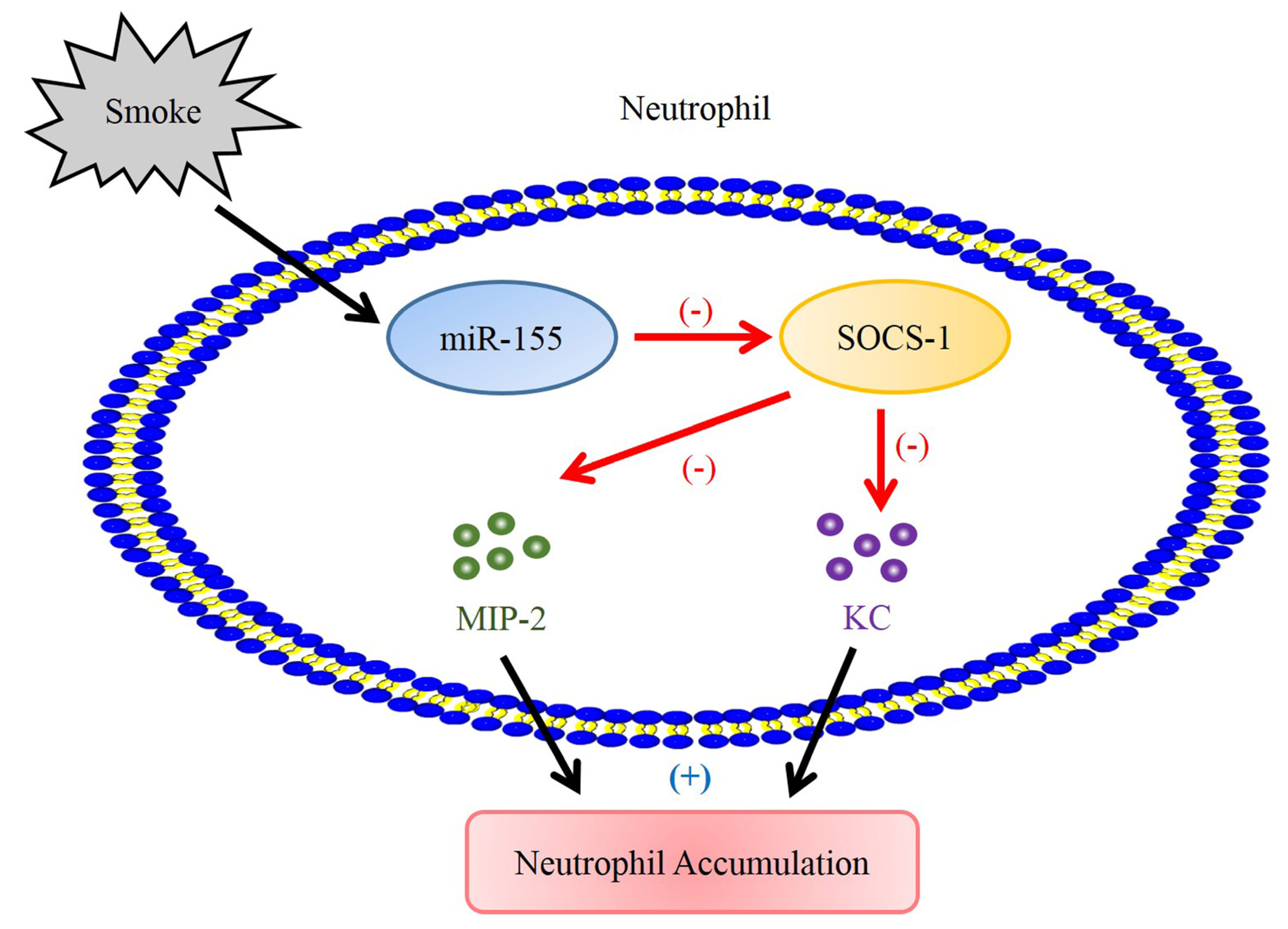
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Smoke Inhalation
4.3. Bronchoalveolar Lavage Fluid
4.4. Real-Time RT-PCR
4.5. Lung Histology
4.6. Lung Injury Score
4.7. ELISA and Myeloperoxidase Assay
4.8. Immunoblotting
4.9. Preparation of Neutrophils from Mouse Lung
4.10. Lung Neutrophils Smoke Exposure
4.11. Knockdown of SOCS-1
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Peck, M.; Molnar, J.; Swart, D. A global plan for burn prevention and care. Bull. World Health Organ. 2009, 87, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Kadri, S.S.; Miller, A.C.; Hohmann, S.; Bonne, S.; Nielsen, C.; Wells, C.; Gruver, C.; Quraishi, S.A.; Sun, J.; Cai, R.; et al. Risk factors for in-hospital mortality in smoke inhalation-associated acute lung injury: Data from 68 United States hospitals. Chest 2016, 150, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Syrkina, O.; Yu, L.; Dedaj, R.; Zhao, H.; Shiedlin, A.; Liu, Y.; Garg, H.; Quinn, D.A.; Hales, C.A. High MW hyaluronan inhibits smoke inhalation-induced lung injury and improves survival. Respirology 2010, 15, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, C.; Chen, X.; Shi, Q.; Su, W.; Zhao, H. SOCS-1 suppresses inflammation through inhibition of NALP3 inflammasome formation in smoke inhalation-induced acute lung injury. Inflammation 2018, 41, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, M.; Lan, Y.; Wang, C. Imbalance of Th17/Tregs in rats with smoke inhalation-induced acute lung injury. Sci. Rep.-UK 2016, 6. [Google Scholar] [CrossRef]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute respiratory distress syndrome. JAMA-J. Am. Med. Assoc. 2018, 319, 698. [Google Scholar] [CrossRef]
- Nakashima, T.; Yokoyama, A.; Onari, Y.; Shoda, H.; Haruta, Y.; Hattori, N.; Naka, T.; Kohno, N. Suppressor of cytokine signaling 1 inhibits pulmonary inflammation and fibrosis. J. Allergy Clin. Immun. 2008, 121, 1269–1276. [Google Scholar] [CrossRef]
- Galam, L.; Soundararajan, R.; Breitzig, M.; Rajan, A.; Yeruva, R.R.; Czachor, A.; Harris, F.; Lockey, R.F.; Kolliputi, N. SOCS-1 rescues IL-1beta-mediated suppression of epithelial sodium channel in mouse lung epithelial cells via ASK-1. Oncotarget 2016, 7, 29081–29091. [Google Scholar] [CrossRef]
- Kolliputi, N.; Waxman, A.B. IL-6 cytoprotection in hyperoxic acute lung injury occurs via suppressor of cytokine signaling-1-induced apoptosis signal-regulating kinase-1 degradation. Am. J. Resp. Cell Mol. 2009, 40, 314–324. [Google Scholar] [CrossRef]
- Galam, L.; Parthasarathy, P.T.; Cho, Y.; Cho, S.H.; Lee, Y.C.; Lockey, R.F.; Kolliputi, N. Adenovirus-mediated transfer of the SOCS-1 gene to mouse lung confers protection against hyperoxic acute lung injury. Free Radical Bio. Med. 2015, 84, 196–205. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.; Pattarayan, D.; Rajaguru, P.; Sudhakar Gandhi, P.S.; Thimmulappa, R.K. MicroRNA regulation of acute lung injury and acute respiratory distress syndrome. J. Cell. Physiol. 2016, 231, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Vaporidi, K.; Vergadi, E.; Kaniaris, E.; Hatziapostolou, M.; Lagoudaki, E.; Georgopoulos, D.; Zapol, W.M.; Bloch, K.D.; Iliopoulos, D. Pulmonary microRNA profiling in a mouse model of ventilator-induced lung injury. Am. J. Physiol.-Lung C. 2012, 303, L199–L207. [Google Scholar] [CrossRef]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef]
- Malmhäll, C.; Alawieh, S.; Lu, Y.; Sjöstrand, M.; Bossios, A.; Eldh, M.; Rådinger, M. MicroRNA-155 is essential for TH2-mediated allergen-induced eosinophilic inflammation in the lung. J. Allergy Clin. Immun. 2014, 133, 1429–1438. [Google Scholar] [CrossRef]
- Rao, R.; Nagarkatti, P.; Nagarkatti, M. Role of miRNA in the regulation of inflammatory genes in staphylococcal enterotoxin B-induced acute inflammatory lung injury and mortality. Toxicol. Sci. 2015, 144, 284–297. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Z.; Su, J.; Chen, W.; Wang, X.; Bai, S.; Zhang, J.; Yu, S. Macrophage micro-RNA-155 promotes lipopolysaccharide-induced acute lung injury in mice and rats. Am. J. Physiol.-Lung C. 2016, 311, L494–L506. [Google Scholar] [CrossRef]
- Guo, Z.; Wen, Z.; Qin, A.; Zhou, Y.; Liao, Z.; Liu, Z.; Liang, Y.; Ren, T.; Xu, L. Antisense oligonucleotide treatment enhances the recovery of acute Lung injury through IL-10–secreting M2-like macrophage-induced expansion of CD4+ regulatory T cells. J. Immunol. 2013, 190, 4337–4348. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. MicroRNA regulation of inflammatory responses. Annu. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Guedes, J.R.; Pereira, D.A.L.; Pedroso, D.L.M. MiR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology 2012, 135, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Nagarkatti, P.; Nagarkatti, M. Staphylococcal enterotoxin B-induced microRNA-155 targets SOCS1 to promote acute inflammatory lung injury. Infect. Immun. 2014, 82, 2971–2979. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zhang, Y.; Yang, X.; Yan, J.; Sun, Y.; Chen, Z.; Jiang, H. Isoflurane attenuates LPS-induced acute lung injury by targeting miR-155-HIF1-alpha. Front. Biosci.-Landmrk. 2015, 20, 139–156. [Google Scholar]
- O’Connell, R.M.; Kahn, M.E.; Kahn, D.; Gibson, W.S.J.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef]
- Yuan, Z.; Syed, M.; Panchal, D.; Joo, M.; Bedi, C.; Lim, S.; Onyuksel, H.; Rubinstein, I.; Colonna, M.; Sadikot, R.T. TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am. J. Physiol.-Lung C. 2016, 310, L426–L438. [Google Scholar] [CrossRef]
- Rajasekhar, M.; Olsson, A.M.; Steel, K.J.A.; Georgouli, M.; Ranasinghe, U.; Brender Read, C.; Frederiksen, K.S.; Taams, L.S. MicroRNA-155 contributes to enhanced resistance to apoptosis in monocytes from patients with rheumatoid arthritis. J. Autoimmun. 2017, 79, 53–62. [Google Scholar] [CrossRef]
- De Santis, R.; Liepelt, A.; Mossanen, J.C.; Dueck, A.; Simons, N.; Mohs, A.; Trautwein, C.; Meister, G.; Marx, G.; Ostareck-Lederer, A.; et al. MiR-155 targets Caspase-3 mRNA in activated macrophages. RNA Biol. 2015, 13, 43–58. [Google Scholar] [CrossRef]
- MatuteBello, G.; Liles, W.C.; Radella, F.; Steinberg, K.P.; Ruzinski, J.T.; Jonas, M.; Chi, E.Y.; Hudson, L.D.; Martin, T.R. Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Resp. Crit. Care 1997, 156, 1969–1977. [Google Scholar] [CrossRef]
- Gong, J.; Liu, H.; Wu, J.; Qi, H.; Wu, Z.; Shu, H.; Li, H.; Chen, L.; Wang, Y.; Li, B.; et al. Maresin 1 prevents lipopolysaccharide-induced neutrophil survival and accelerates resolution of acute lung injury. Shock 2015, 44, 371–380. [Google Scholar] [CrossRef]
- Shi, W.; Xu, C.; Hussain, M.; Wu, F.; Lu, M.; Wu, X.; Tang, L.; Wu, X.; Wu, J. Inhibition of myosin light-chain kinase enhances the clearance of lipopolysaccharide-induced lung inflammation possibly by accelerating neutrophil apoptosis. Shock 2017, 48, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.W.; Shi, L.; Xu, C.; Zhao, Y.; Liu, B.; Li, Y.; Shiedlin, A.; Xiang, C.; Shen, H.; Quinn, D.A.; et al. TLR4 through IFN-beta promotes low molecular mass hyaluronan-induced neutrophil apoptosis. J. Immunol. 2011, 186, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, C.; Ma, Y.; Zhu, K.; Chen, X.; Shi, Q.; Su, W.; Zhao, H. SOCS-1 ameliorates smoke inhalation-induced acute lung injury through inhibition of ASK-1 activity and DISC formation. Clin. Immunol. 2018, 191, 94–99. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011, 32, 461–469. [Google Scholar] [CrossRef]
- Su, V.Y.; Chiou, S.; Lin, C.; Chen, W.; Yu, W.; Chen, Y.; Chen, C.; Yang, K. Induced pluripotent stem cells reduce neutrophil chemotaxis via activating GRK2 in endotoxin-induced acute lung injury. Respirology 2017, 22, 1156–1164. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Foley, N.M.; Xu, Y.; Li, Y.P.; Pan, J.; Redmond, H.P.; Wang, J.H.; Wang, J. B7H3 ameliorates LPS-induced acute lung injury via attenuation of neutrophil migration and infiltration. Sci. Rep.-UK 2016, 6. [Google Scholar] [CrossRef]
- Eisenhardt, S.U.; Weiss, J.B.W.; Smolka, C.; Maxeiner, J.; Pankratz, F.; Bemtgen, X.; Kustermann, M.; Thiele, J.R.; Schmidt, Y.; Bjoern Stark, G.; et al. MicroRNA-155 aggravates ischemia–reperfusion injury by modulation of inflammatory cell recruitment and the respiratory oxidative burst. Basic Res. Cardiol. 2015, 110, 32. [Google Scholar] [CrossRef]
- Lee, Y.; Han, J.; Byun, J.; Park, H.; Park, E.; Chong, Y.H.; Cho, M.; Kang, J.L. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-κB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J. Leukocyte Biol. 2012, 91, 921–932. [Google Scholar] [CrossRef]
- Issa, R.; Xie, S.; Lee, K.; Stanbridge, R.D.; Bhavsar, P.; Sukkar, M.B.; Chung, K.F. GRO-α regulation in airway smooth muscle by IL-1β and TNF-α: Role of NF-κB and MAP kinases. Am. J. Physiol.-Lung C. 2006, 291, L66–L74. [Google Scholar] [CrossRef]
- Zhao, H.; Leu, S.W.; Shi, L.; Dedaj, R.; Zhao, G.; Garg, H.G.; Shen, L.; Lien, E.; Fitzgerald, K.A.; Shiedlin, A.; et al. TLR4 is a negative regulator in noninfectious lung inflammation. J. Immunol. 2010, 184, 5308–5314. [Google Scholar] [CrossRef]
- Qin, X.; Zhu, G.; Huang, L.; Zhang, W.; Huang, Y.; Xi, X. LL-37 and its analog FF/CAP18 attenuate neutrophil migration in sepsis-induced acute lung injury. J. Cell. Biochem. 2019, 120, 4863–4871. [Google Scholar] [CrossRef]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Xie, Y.; Zhang, L.; Zhao, H. MicroRNA-155 Participates in Smoke-Inhalation-Induced Acute Lung Injury through Inhibition of SOCS-1. Molecules 2020, 25, 1022. https://doi.org/10.3390/molecules25051022
Zhang Y, Xie Y, Zhang L, Zhao H. MicroRNA-155 Participates in Smoke-Inhalation-Induced Acute Lung Injury through Inhibition of SOCS-1. Molecules. 2020; 25(5):1022. https://doi.org/10.3390/molecules25051022
Chicago/Turabian StyleZhang, Yue, Yifang Xie, Leifang Zhang, and Hang Zhao. 2020. "MicroRNA-155 Participates in Smoke-Inhalation-Induced Acute Lung Injury through Inhibition of SOCS-1" Molecules 25, no. 5: 1022. https://doi.org/10.3390/molecules25051022
APA StyleZhang, Y., Xie, Y., Zhang, L., & Zhao, H. (2020). MicroRNA-155 Participates in Smoke-Inhalation-Induced Acute Lung Injury through Inhibition of SOCS-1. Molecules, 25(5), 1022. https://doi.org/10.3390/molecules25051022