
Direct Base-Assisted C‒H Cyclonickelation of 6-Phenyl-2,2′-bipyridine



Abstract
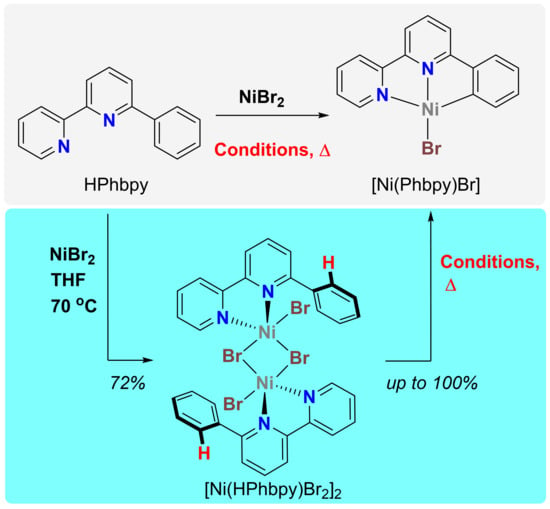
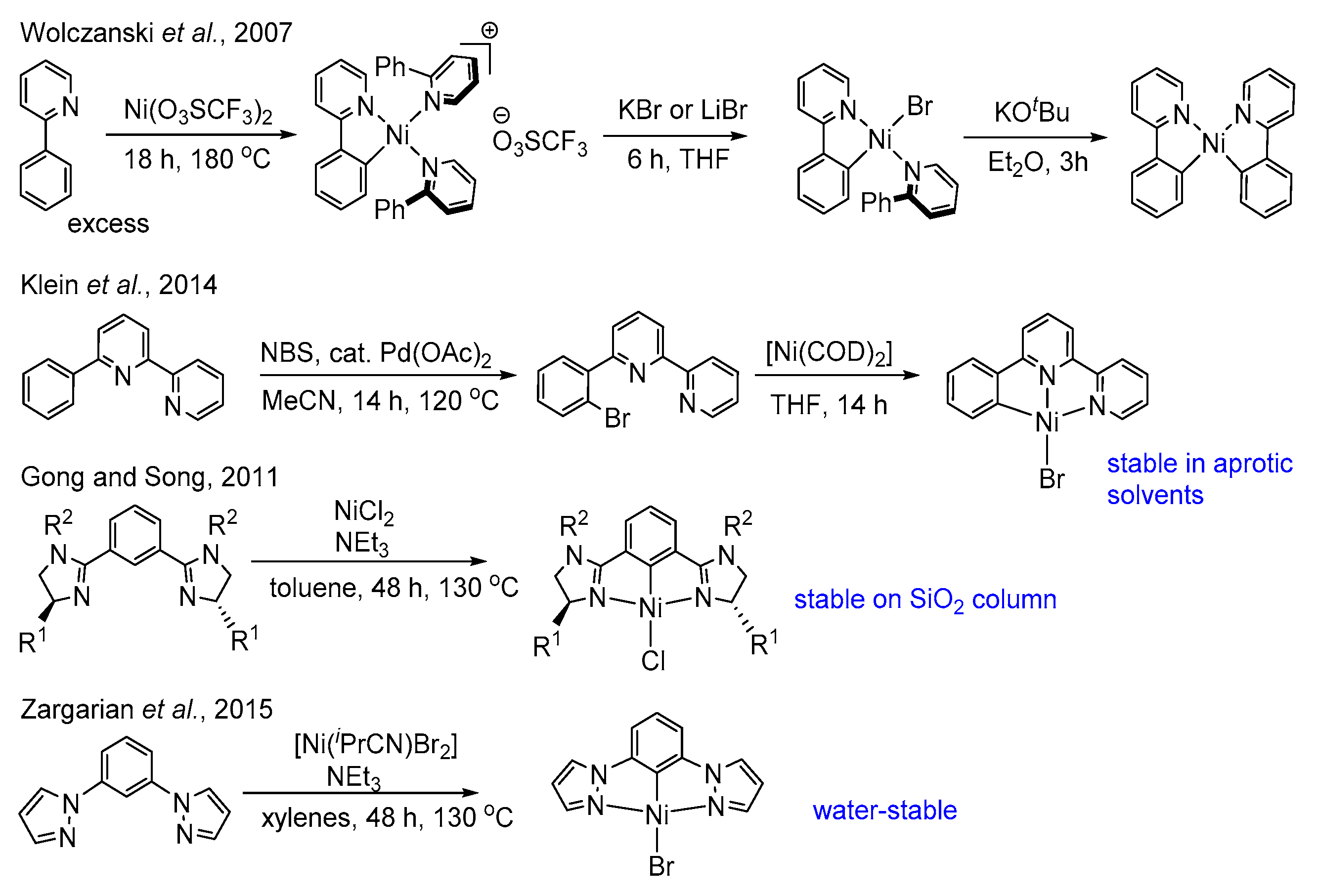
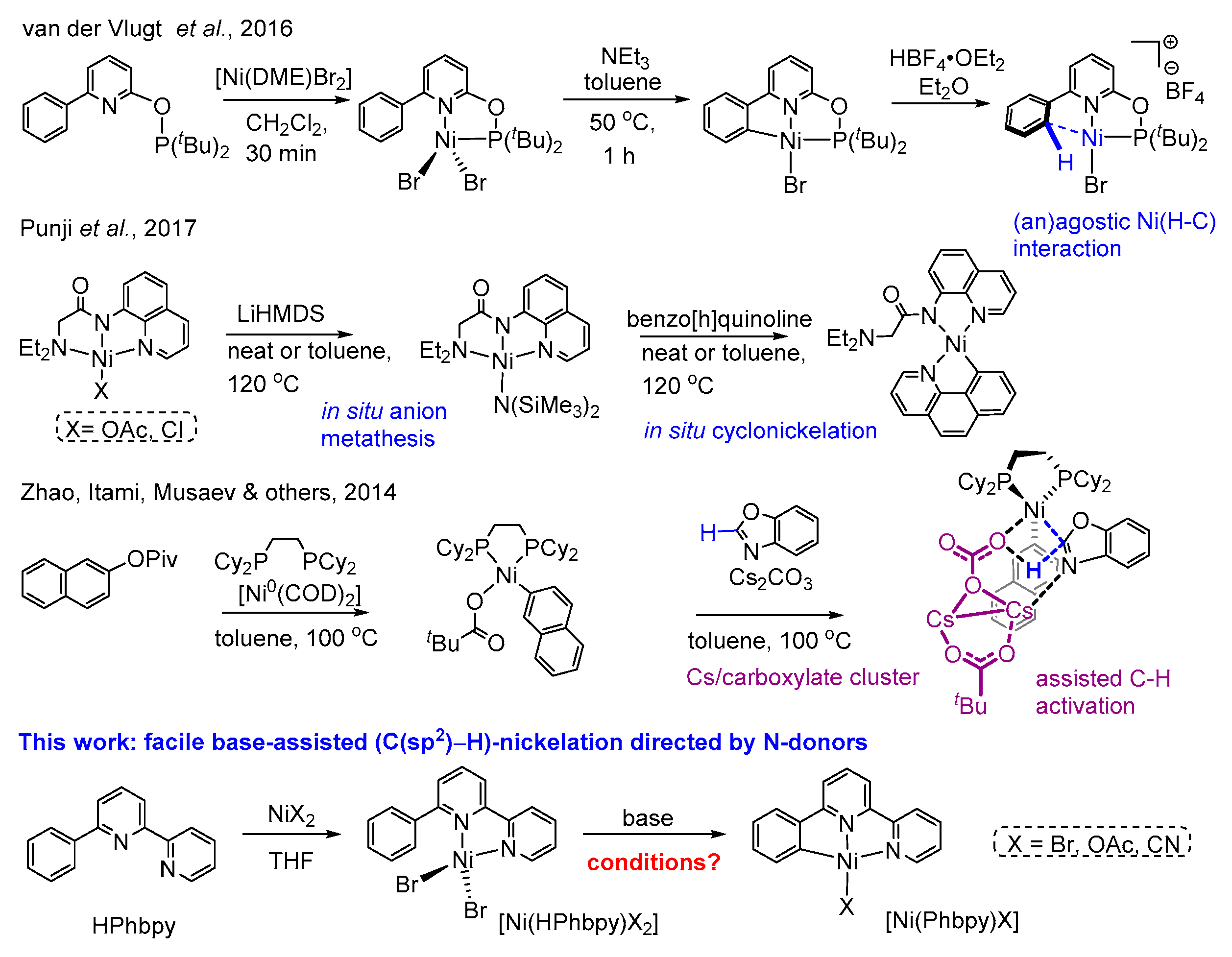
1. Introduction
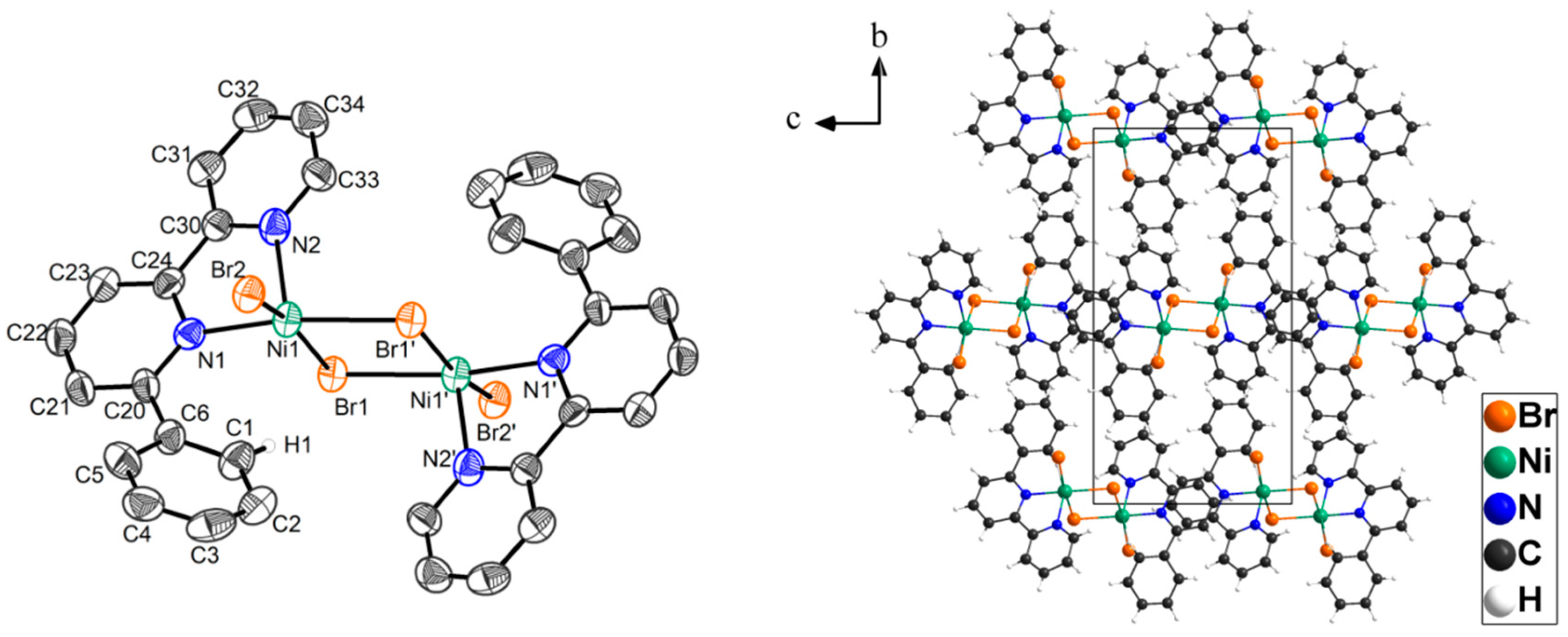
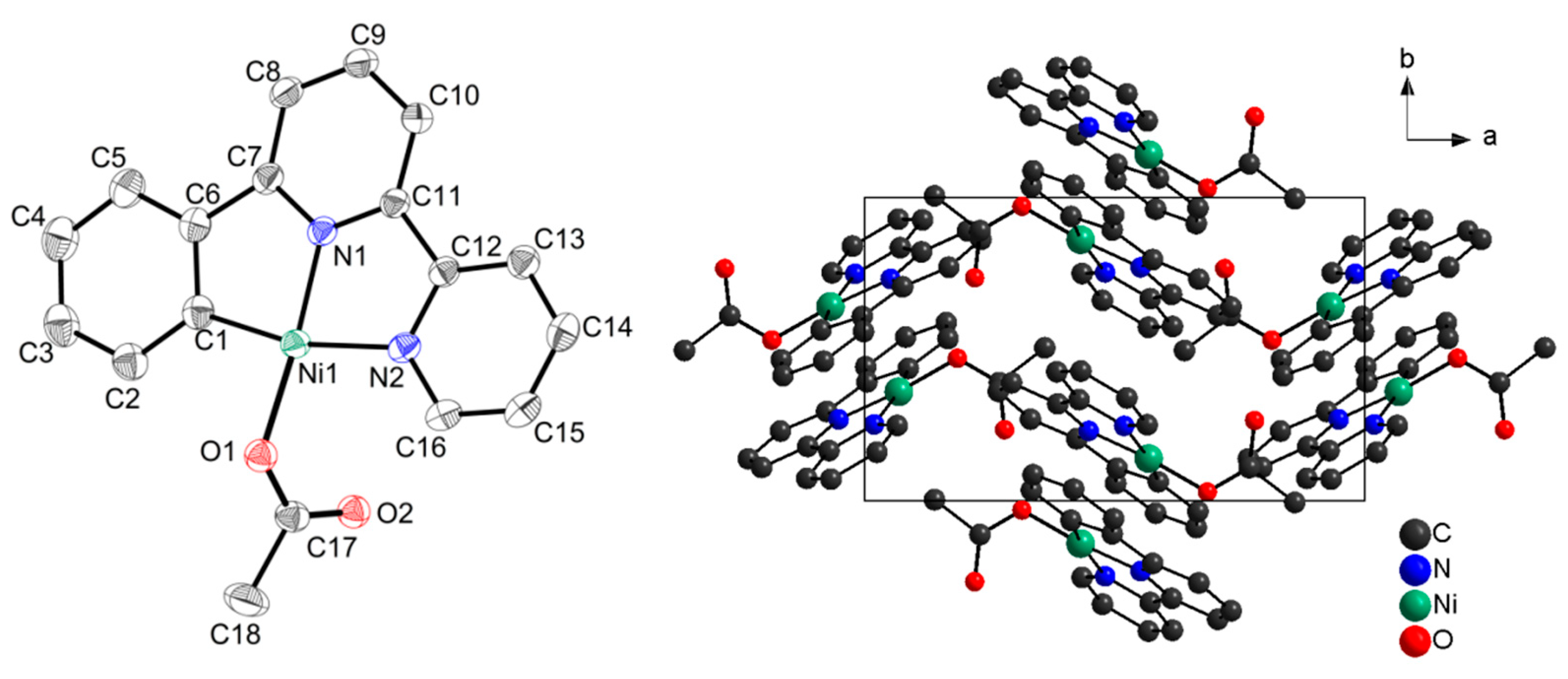
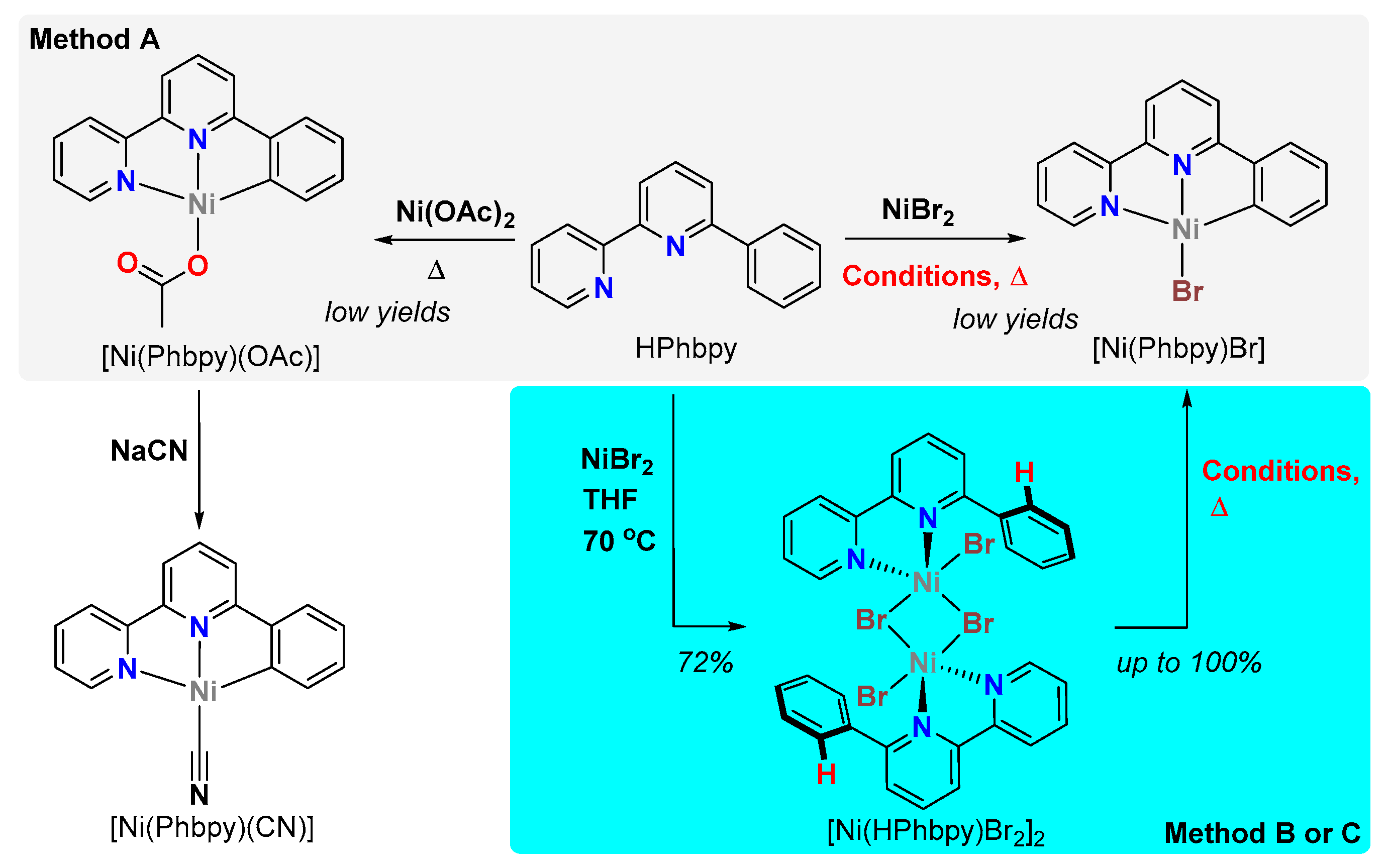
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cyclonickelation Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Labinger, J.A. Platinum-Catalyzed C−H Functionalization. Chem. Rev. 2017, 117, 8483–8496. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.C.; Siekman, R.W. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc. 1965, 87, 3272–3273. [Google Scholar] [CrossRef]
- Cope, A.C.; Friedrich, E.C. Electrophilic Aromatic Substitution Reactions by Platinum(II) and Palladium(II) Chlorides on N,N-Dimethylbenzylamines. J. Am. Chem. Soc. 1968, 90, 909–913. [Google Scholar] [CrossRef]
- Chassot, L.; Mueller, E.; Von Zelewsky, A. cis-Bis(2-phenylpyridine)platinum(II) (CBPPP): A Simple Molecular Platinum Compound. Inorg. Chem. 1984, 23, 4249–4253. [Google Scholar] [CrossRef]
- Constable, E.C.; Henney, R.P.G.; Leese, T.A.; Tocher, D.A. Cyclometallation Reactions of 6-Phenyl-2,2’-bipyridine; a Potential C,N,N-Donor Analogue of 2,2’:6’,2”-Terpyridine. Crystal and Molecular Structure of Dichlorobis(6-phenyl-2,2’-bipyridine)ruthenium(II). J. Chem. Soc., Dalton Trans. 1990, 443–449. [Google Scholar] [CrossRef]
- Constable, E.C.; Henney, R.P.G.; Leese, T.A.; Tocher, D.A. Cyclopalladated and Cycloplatinated Complexes of 6-Phenyl-2,2′-bipyridine: Platinum-Platinum Interactions in the Solid State. J. Chem. Soc. Chem. Commun. 1990, 513–515. [Google Scholar] [CrossRef]
- Berenguer, J.R.; Lalinde, E.; Moreno, M.T. Luminescent cyclometalated-pentafluorophenyl PtII, PtIV and heteropolynuclear complexes. Coord. Chem. Rev. 2018, 366, 69–90. [Google Scholar] [CrossRef]
- Tang, M.-C.; Chan, A.K.-W.; Chan, M.-Y.; Yam, V.W.-W. Platinum and Gold Complexes for OLEDs. Top. Curr. Chem. (Z) 2016, 374, 1–43. [Google Scholar] [CrossRef]
- Kalinowski, J.; Fattori, V.; Cocchi, M.; Williams, J.A.G. Light-emitting devices based on organometallic platinum complexes as emitters. Coord. Chem. Rev. 2011, 255, 2401–2425. [Google Scholar] [CrossRef]
- Williams, J.A.G. Metal complexes of pincer ligands: Excited states, photochemistry, and luminescence. Top. Organomet. Chem. 2011, 40, 89–130. [Google Scholar]
- Yang, S.; Meng, F.; Wu, X.; Yin, Z.; Liu, X.; You, C.; Wang, Y.; Su, S.; Zhu, W. Dinuclear platinum(II) complex dominated by a zig-zag-type cyclometalated ligand: A new approach to realize high-efficiency near infrared emission. J. Mater. Chem. C 2018, 6, 5769–5777. [Google Scholar] [CrossRef]
- Sivchik, V.; Sarker, R.K.; Liu, Z.-Y.; Chung, K.-Y.; Grachova, E.V.; Karttunen, A.J.; Chou, P.-T.; Koshevoy, I.O. Improvement of the Photophysical Performance of Platinum-Cyclometalated Complexes in Halogen-Bonded Adducts. Chem. Eur. J. 2018, 24, 11475–11484. [Google Scholar] [CrossRef]
- Soellner, J.; Strassner, T. The “Enders Triazole” Revisited: Highly Efficient, Blue Platinum(II) Emitters. Organometallics 2018, 37, 1821–1824. [Google Scholar] [CrossRef]
- Schulze, B.; Friebe, C.; Jäger, M.; Görls, H.; Birckner, E.; Winter, A.; Schubert, U.S. PtII Phosphors with Click-Derived 1,2,3-Triazole-Containing Tridentate Chelates. Organometallics 2018, 37, 145–155. [Google Scholar] [CrossRef]
- Sesolis, H.; Chan, C.K.-M.; Gontard, G.; Fu, H.L.-K.; Yam, V.W.-W.; Amouri, H. Dinuclear (N∧C∧N) Pincer Pt(II) Complexes with Bridged Organometallic Linkers: Synthesis, Structures, Self-Aggregation, and Photophysical Properties. Organometallics 2017, 36, 4794–4801. [Google Scholar] [CrossRef]
- Schneider, L.; Sivchik, V.; Chung, K.-Y.; Chen, Y.-T.; Karttunen, A.J.; Chou, P.-T.; Koshevoy, I.O. Cyclometalated Platinum(II) Cyanometallates: Luminescent Blocks for Coordination Self-Assembly. Inorg. Chem. 2017, 56, 4459–4467. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tong, G.S.M.; Wan, Q.; Cheng, G.; Tong, W.-Y.; Ang, W.-H.; Kwong, W.-L.; Che, C.-M. Highly phosphorescent platinum(II) emitters: Photophysics, materials and biological applications. Chem. Sci. 2016, 7, 1653–1673. [Google Scholar] [CrossRef]
- Solomatina, A.I.; Chelushkin, P.S.; Abakumova, T.O.; Zhemkov, V.A.; Kim, M.; Bezprozvanny, I.; Gurzhiy, V.V.; Melnikov, A.S.; Anufrikov, Y.A.; Koshevoy, I.O.; et al. Reactions of Cyclometalated Platinum(II) [Pt(N∧C)(PR3)Cl] Complexes with Imidazole and Imidazole-Containing Biomolecules: Fine-Tuning of Reactivity and Photophysical Properties via Ligand Design. Inorg. Chem. 2019, 58, 204–217. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, Q.; Zheng, W.; Wang, Z.; Lin, Y.; Zhang, E.; Lü, S.; Xiang, J.; Zhao, Y.; Wang, F. Luminescent cyclometallated platinum(II) complexes: Highly promising EGFR/DNA probes and dual-targeting anticancer agents. Inorg. Chem. Front. 2018, 5, 413–424. [Google Scholar] [CrossRef]
- Chow, P.-K.; Cheng, G.; Tong, G.S.M.; Ma, C.; Kwok, W.-M.; Ang, W.-H.; Chung, C.Y.-S.; Yang, C.; Wang, F.; Che, C.-M. Highly luminescent palladium(II) complexes with sub-millisecond blue to green phosphorescent excited states. Photocatalysis and highly efficient PSF-OLEDs. Chem. Sci. 2016, 7, 6083–6098. [Google Scholar] [CrossRef]
- Fleetham, T.; Li, G.; Li, J. Phosphorescent Pt(II) and Pd(II) Complexes for Efficient, High-Color-Quality, and Stable OLEDs. Adv. Mater. 2017, 29, 1601861. [Google Scholar] [CrossRef]
- Hartwell, G.E.; Lawrence, R.V.; Smas, M.J. The Formation of Palladium(II)- and Platinum(II)-Carbon Bonds by Proton Abstraction from Benzo[h]quinoline and 8-Methylquinoline. J. Chem. Soc., Chem. Commun. 1970, 912. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C‒H Bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Thu, H.-Y.; Yu, W.-Y.; Che, C.-M. Intermolecular Amidation of Unactivated sp2 and sp3 C‒H Bonds via Palladium-Catalyzed Cascade C‒H Activation/Nitrene Insertion. J. Am. Chem. Soc. 2006, 128, 9048–9049. [Google Scholar] [CrossRef] [PubMed]
- Christman, W.E.; Morrow, T.J.; Arulsamy, N.; Hulley, E.B. Absolute Estimates of PdII(η2-Arene) C−H Acidity. Organometallics 2018, 37, 2706–2715. [Google Scholar] [CrossRef]
- Kleiman, J.P.; Dubeck, M. The preparation of cyclopentadienyl [o-(phenylazo)phenyl]nickel. J. Am. Chem. Soc. 1963, 85, 1544–1545. [Google Scholar] [CrossRef]
- Grove, D.M.; van Koten, G.; Ubbels, H.J.C.; Zoet, R. Organonickel(II) Complexes of the Terdentate Monoanionic Ligand o,o’-Bis[(dimethylamino)methyl]phenyl (N-C-N). Syntheses and the X-ray Crystal Structure of the Stable Nickel(II) Formate [Ni(N-C-N)O2CH]. Organometallics 1984, 3, 1003–1009. [Google Scholar] [CrossRef]
- Volpe, E.C.; Chadeayne, A.R.; Wolczanski, P.T.; Lobkovsky, E.B. Heterolytic CH-bond activation in the synthesis of Ni{(2-aryl-κC2)pyridine-κN}2 and derivatives. J. Organomet. Chem. 2007, 692, 4774–4783. [Google Scholar] [CrossRef]
- Klein, A.; Rausch, B.; Kaiser, A.; Vogt, N.; Krest, A. The cyclometalated nickel complex [(Phbpy)NiBr] (Phbpy– = 2,2′-bipyridine-6-phen-2-yl)—Synthesis, spectroscopic and electrochemical studies. J. Organomet. Chem. 2014, 774, 86–93. [Google Scholar] [CrossRef]
- Sandleben, A.; Vogt, N.; Hörner, G.; Klein, A. On the Redox Series of Cyclometalated Nickel Complexes [Ni((R)Ph(R’)bpy)Br]+/0/‒/2‒ (H‒(R)Ph(R’)bpy = substituted 6-phenyl-2,2′-bipyridine). Organometallics 2018, 37, 3332–3341. [Google Scholar] [CrossRef]
- Shao, D.-D.; Niu, J.-L.; Hao, X.-Q.; Gong, J.-F.; Song, M.-P. Neutral and cationic chiral NCN pincer nickel(II) complexes with 1,3-bis(2′-imidazolinyl)benzenes: Synthesis and characterization. Dalton Trans. 2011, 40, 9012–9019. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, J.P.; Vabre, B.; Moungang-Soumé, B.; Zargarian, D. Synthesis and Reactivities of New NCN-Type Pincer Complexes of Nickel. Organometallics 2015, 34, 133–145. [Google Scholar] [CrossRef]
- Yang, C.; Wu, W.-D.; Zhao, L.; Wang, M.-X. Macrocyclic Aryl−Nickel(II) Complexes: Synthesis, Structure, and Reactivity Studies. Organometallics 2015, 34, 5167–5174. [Google Scholar] [CrossRef]
- Jongbloed, L.S.; García-López, D.; van Heck, R.; Siegler, M.A.; Carbó, J.J.; van der Vlugt, J.I. Arene C(sp2)‒H Metalation at NiII Modeled with a Reactive PONCPh Ligand. Inorg. Chem. 2016, 55, 8041–8047. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, R.A.; Soni, V.; Punji, B. Expeditious and Solvent-Free Nickel-Catalyzed C−H Arylation of Arenes and Indoles. ChemSusChem 2017, 10, 2242–2248. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Muto, K.; Yamaguchi, J.; Zhao, C.; Itami, K.; Musaev, D.G. Key Mechanistic Features of Ni-Catalyzed C−H/C−O Biaryl Coupling of Azoles and Naphthalen-2-yl Pivalates. J. Am. Chem. Soc. 2014, 136, 14834–14844. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef]
- Davies, D.L.; Macgregor, S.A.; McMullin, C.L. Computational Studies of Carboxylate-Assisted C-H Activation and Functionalization at Group 8-10 Transition Metal Centres. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef]
- Alharis, R.A.; McMullin, C.L.; Davies, D.L.; Singh, K.; Macgregor, S.A. Understanding Electronic Effects on Carboxylate-Assisted C–H Activation at Ruthenium: The Importance of Kinetic and Thermodynamic Control. Faraday Discuss. 2019, 220, 386–403. [Google Scholar] [CrossRef]
- Alharis, R.A.; McMullin, C.L.; Davies, D.L.; Singh, K.; Macgregor, S.A. The Importance of Kinetic and Thermodynamic Control when Assessing Mechanisms of Carboxylate-Assisted C−H Activation. J. Am. Chem. Soc. 2019, 141, 8896–8906. [Google Scholar] [CrossRef]
- Beattie, D.D.; Grunwald, A.C.; Perse, T.; Schafer, L.L.; Love, J.A. Understanding Ni(II)-Mediated C(sp3)–H Activation: Tertiary Ureas as Model Substrates. J. Am. Chem. Soc. 2018, 140, 12602–12610. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed]
- Kommagalla, Y.; Chatani, N. Cobalt(II)-catalyzed CH functionalization using an N,N′-bidentate directing group. Coord. Chem. Rev. 2017, 350, 117–135. [Google Scholar] [CrossRef]
- Tang, H.; Zhou, B.; Huang, X.-R.; Wang, C.; Yao, J.; Chen, H. Origins of Selective C(sp2)−H Activation Using Transition Metal Complexes with N,N-Bidentate Directing Groups: A Combined Theoretical−Experimental Study. ACS Catal. 2014, 4, 649–656. [Google Scholar] [CrossRef]
- Feth, M.P.; Klein, A.; Bertagnolli, H. Investigation of the ligand exchange behaviour of square planar nickel(II) complexes by X-ray absorption spectroscopy and X-ray diffraction. Eur. J. Inorg. Chem. 2003, 839–852. [Google Scholar] [CrossRef]
- Sun, H.-Y.; Gorelsky, S.E.; Stuart, D.R.; Campeau, L.-C.; Fagnou, K. Mechanistic Analysis of Azine N-Oxide Direct Arylation: Evidence for a Critical Role of Acetate in the Pd(OAc)2 Precatalyst. J. Org. Chem. 2010, 75, 8180–8189. [Google Scholar] [CrossRef]
- Garica-Cuadrado, D.; de Mendoza, P.; Braga, A.A.C.; Maseras, F.; Echavarren, A.M. Proton-Abstraction Mechanism in the Palladium-Catalyzed Intramolecular Arylation: Substituent Effects. J. Am. Chem. Soc. 2007, 129, 6880–6886. [Google Scholar] [CrossRef]
- Raamata, E.; Kaupmeesa, K.; Ovsjannikova, G.; Trummalb, A.; Kütta, A.; Saamea, J.; Koppela, I.; Kaljuranda, I.; Lippinga, L.; Rodimaa, T.; et al. Acidities of strong neutral Brønsted acids in different media. J. Phys. Org. Chem. 2013, 26, 162–170. [Google Scholar] [CrossRef]
- Yang, P.; Yang, Y.; Zhang, C.; Yang, X.J.; Hu, H.M.; Gao, Y.; Wu, B. Synthesis, structure, and catalytic ethylene oligomerization of nickel(II) and cobalt(II) complexes with symmetrical and unsymmetrical 2,9-diaryl-1,10-phenanthroline ligands. Inorg. Chim. Acta 2009, 362, 89–96. [Google Scholar] [CrossRef]
- Roy, P.; Bour, J.R.; Kampf, J.W.; Sanford, M.S. Catalytically Relevant Intermediates in the Ni-Catalyzed C(sp2)–H and C(sp3)–H Functionalization of Aminoquinoline Substrates. J. Am. Chem. Soc. 2019, 141, 17382–17387. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; p. 455. [Google Scholar]
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Millvalley, CA, USA, 2010; p. 9. [Google Scholar]
- Pattanayak, P.; Pratihar, J.L.; Patra, D.; Burrows, A.; Mohan, M.; Chattopadhyay, S.; Lal Pratihar, J.; Patra, D.; Burrows, A.; Mohan, M.; et al. Regiospecific ortho-aromatic hydroxylation via cyclonickelation using hydrogen peroxide and other oxygen donors: Synthesis of metalloazosalophens. Eur. J. Inorg. Chem. 2007, 2007, 4263–4271. [Google Scholar] [CrossRef]
- Sripothongnak, S.; Barone, N.; Ziegler, C.J. C–H bond activation and ring oxidation in nickel carbahemiporphyrazines. Chem. Commun. 2009, 30, 4584–4586. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick. G.M. SHELXL 2016. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122.
- X-RED32 1.31 & X-SHAPE; Version 1.06; STOE & Cie GmbH: Darmstadt, Germany, 2006.
- APEX2—Software Suite for Crystallographic Programs; Bruker AXS, Inc.: Madison, WI, USA, 2010.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C: Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS-2008/1—Bruker AXS Area Detector Scaling and Absorption Correction; Bruker AXS, Inc.: Madison, WI, USA, 2008. [Google Scholar]
Sample Availability: Samples of the compounds [Ni(Phbpy)Br], [Ni(HPhbpy)Br2]2, and [Ni(Phbpy)(OAc)] are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | NiX2 | Base (eq.) | Solvent | Time (h) | T (°C) | Yield (%) | Analysis | |
|---|---|---|---|---|---|---|---|---|
| 1 | A | NiBr2 | no base | toluene | 60 | 111 g | 0 | visual b |
| 2 | A | NiBr2 | NEt3 (11.5) | toluene | 48 | 111 g | 1.5 | 1H NMR c |
| 3 | A | NiBr2 | NaHCO3 (2) | toluene/MeOH | 3 | 111 g | 2.5 | 1H NMR |
| 4 | A | NiBr2 | Na2CO3 (4) | solid | 1 | 170 | 0 | visual b |
| 5 | B | NiBr2 | Cs2CO3 (2) | toluene | 66 | 111 g | trace | 1H NMR |
| 6 | B | NiBr2 | Na2CO3 (3) | toluene | 90 | 111 g | 19 | 1H NMR |
| 7 | C | [Ni(HPhbpy)Br2] | NaOtBu (2) | diethyl ether | 16 | 23 | 0 | 1H NMR |
| 8 | C | [Ni(HPhbpy)Br2] | KOAc/K2CO3 (2/4) | toluene | 16 | 111 g | 6 | 1H NMR |
| 9 | C | [Ni(HPhbpy)Br2] | KOAc/K2CO3 (2/4) | toluene | 62 | 111 g | 28 | 1H NMR |
| 10 | A | Ni(OAc)2.4H2O | - | toluene/EtOH//DME | 15 | 111 | 5 d | XRD |
| 11 | A | Ni(OAc)2 | - | solid | 1 | 250 | 0 e | visual b |
| 12 | A | Ni(OAc)2 | - | solid | 1 | 180 | 4 f | XRD |
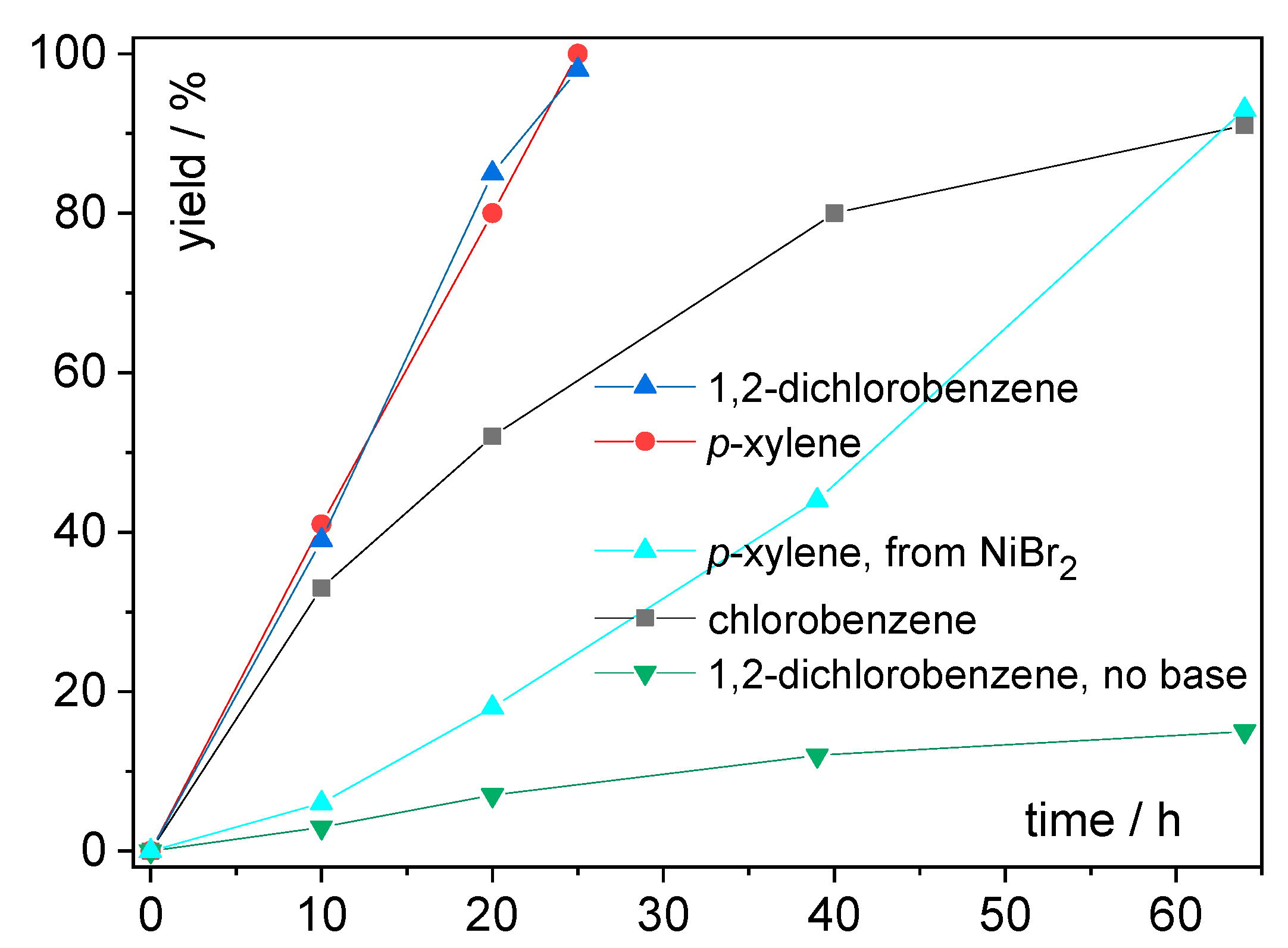
| Entry | Solvent | Time (h) | Tbat (°C) | Tb.p. (°C) | Yield (%) | |
|---|---|---|---|---|---|---|
| 13 | toluene | 72 | 120 | 111 | 0.099 | 43 |
| 14 | chlorobenzene | 64 | 160 | 131 | 0.188 | 91 |
| 15 | 1,2-dichlorobenzene | 25 | 190 | 180 | 0.225 | 98 |
| 16 | p-xylene | 25 | 160 | 138 | 0.074 | 100 |
| 17 | benzonitrile | 20 | 200 | 190 | 0.333 | 0 |
| 18 | p-xylene c (from NiBr2) | 64 | 160 | 138 | 0.074 | 93 |
| 19 | 1,2-dichlorobenzene d (no base) | 64 | 190 | 180 | 0.225 | 15 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, N.; Sivchik, V.; Sandleben, A.; Hörner, G.; Klein, A. Direct Base-Assisted C‒H Cyclonickelation of 6-Phenyl-2,2′-bipyridine. Molecules 2020, 25, 997. https://doi.org/10.3390/molecules25040997
Vogt N, Sivchik V, Sandleben A, Hörner G, Klein A. Direct Base-Assisted C‒H Cyclonickelation of 6-Phenyl-2,2′-bipyridine. Molecules. 2020; 25(4):997. https://doi.org/10.3390/molecules25040997
Chicago/Turabian StyleVogt, Nicolas, Vasily Sivchik, Aaron Sandleben, Gerald Hörner, and Axel Klein. 2020. "Direct Base-Assisted C‒H Cyclonickelation of 6-Phenyl-2,2′-bipyridine" Molecules 25, no. 4: 997. https://doi.org/10.3390/molecules25040997
APA StyleVogt, N., Sivchik, V., Sandleben, A., Hörner, G., & Klein, A. (2020). Direct Base-Assisted C‒H Cyclonickelation of 6-Phenyl-2,2′-bipyridine. Molecules, 25(4), 997. https://doi.org/10.3390/molecules25040997