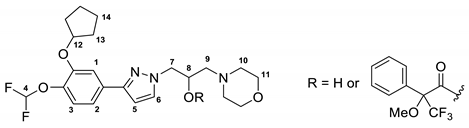
Insight into GEBR-32a: Chiral Resolution, Absolute Configuration and Enantiopreference in PDE4D Inhibition

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
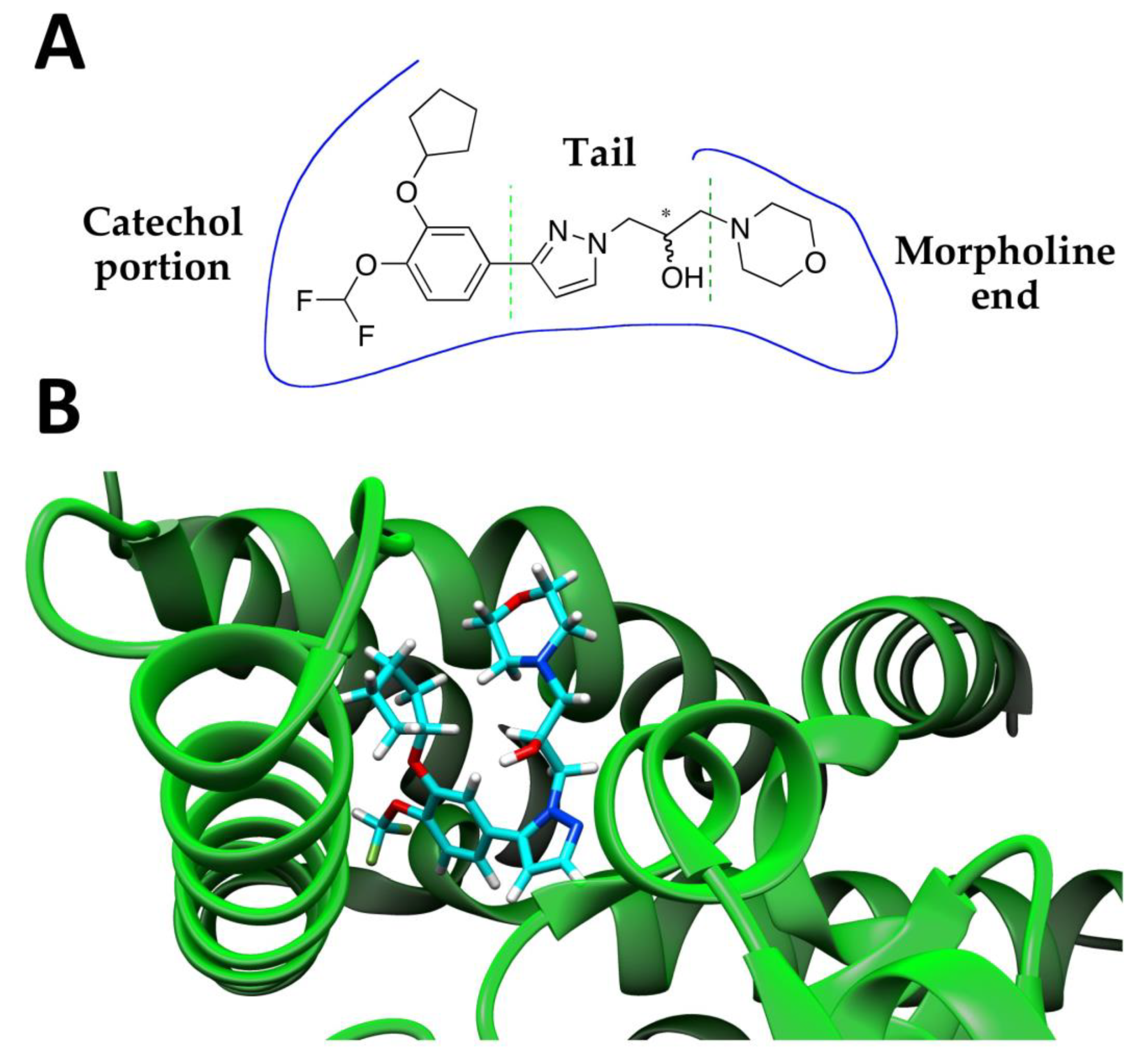
2. Results and Discussion
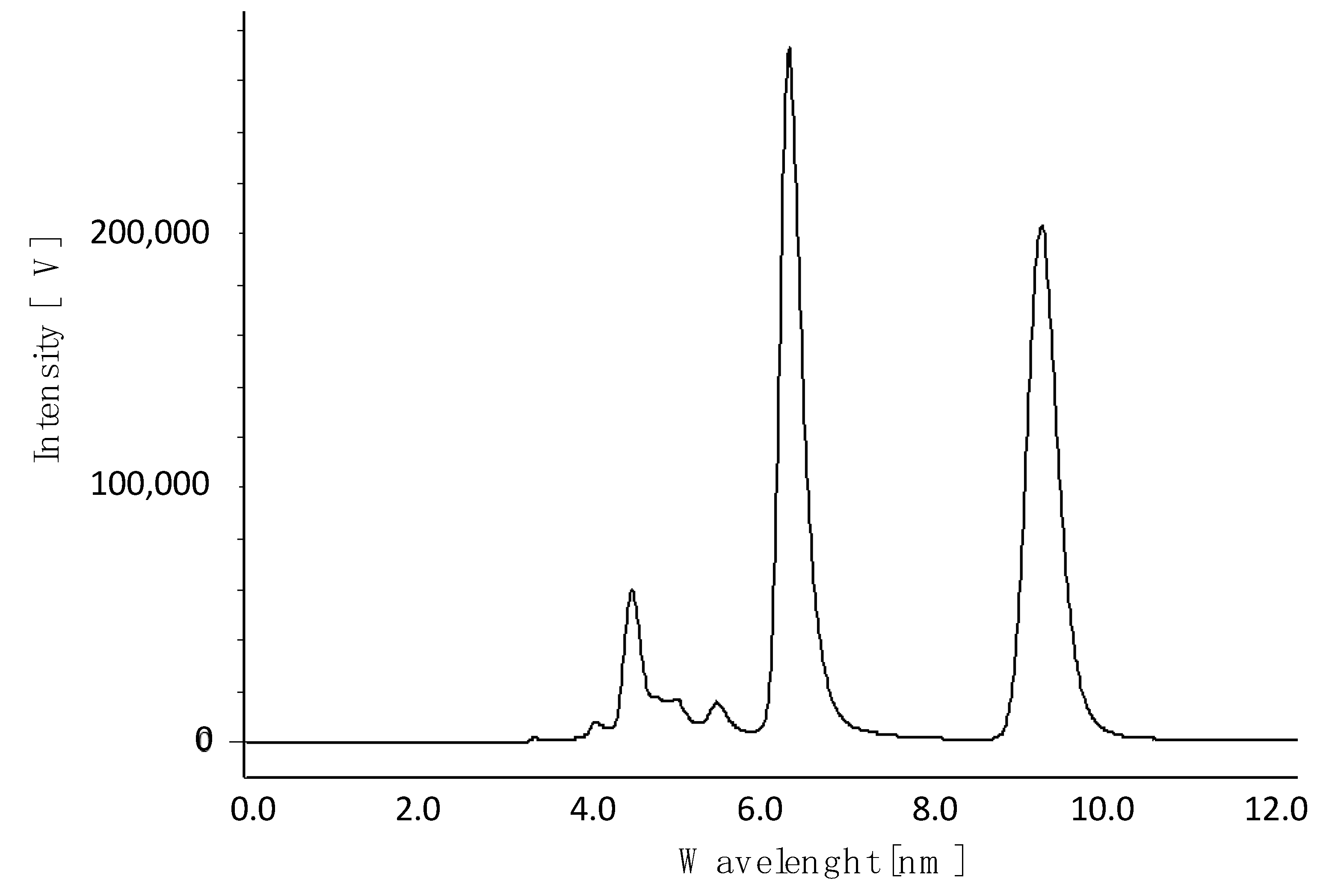
2.1. Chiral Chromatographic Resolution
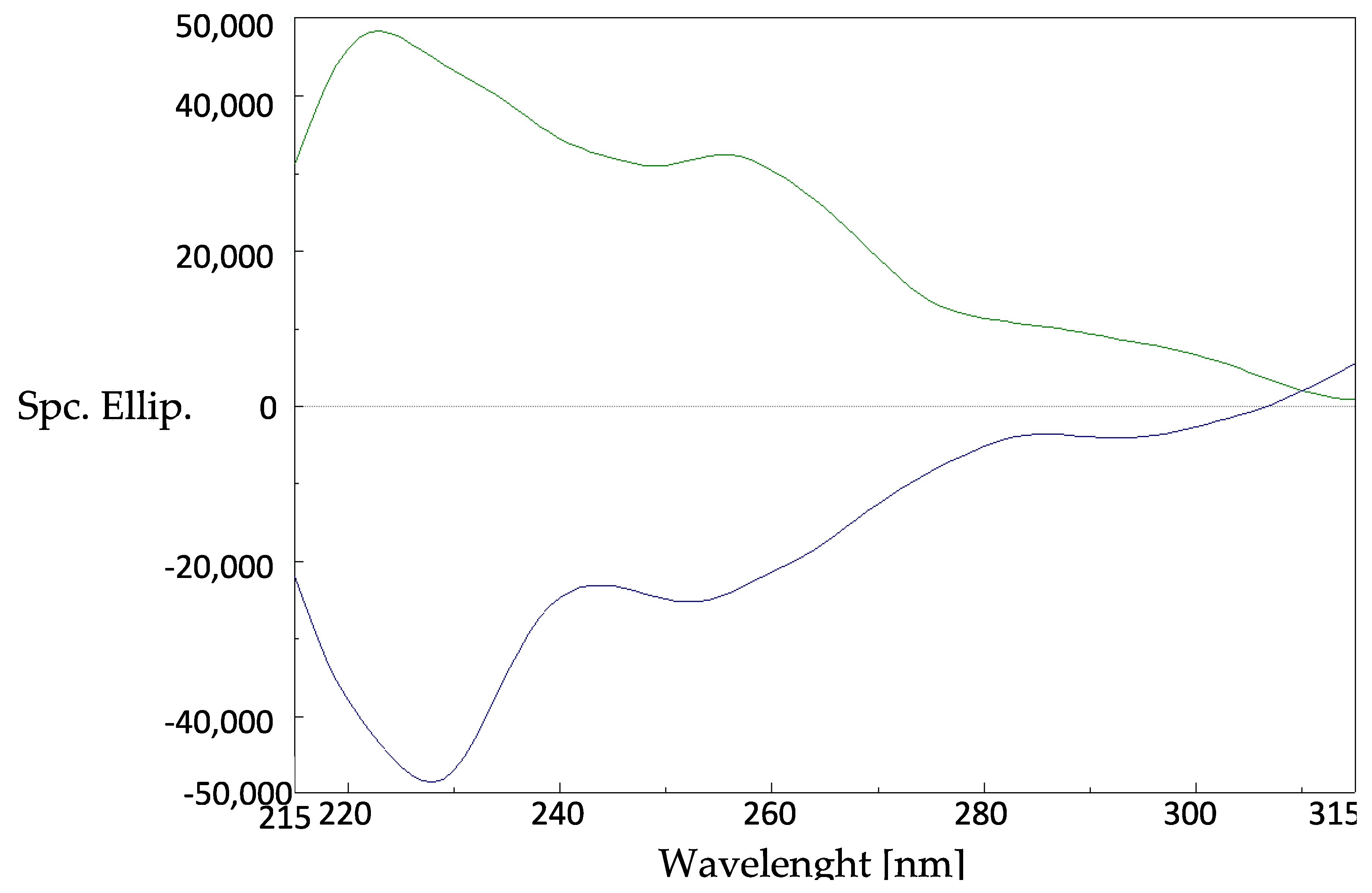
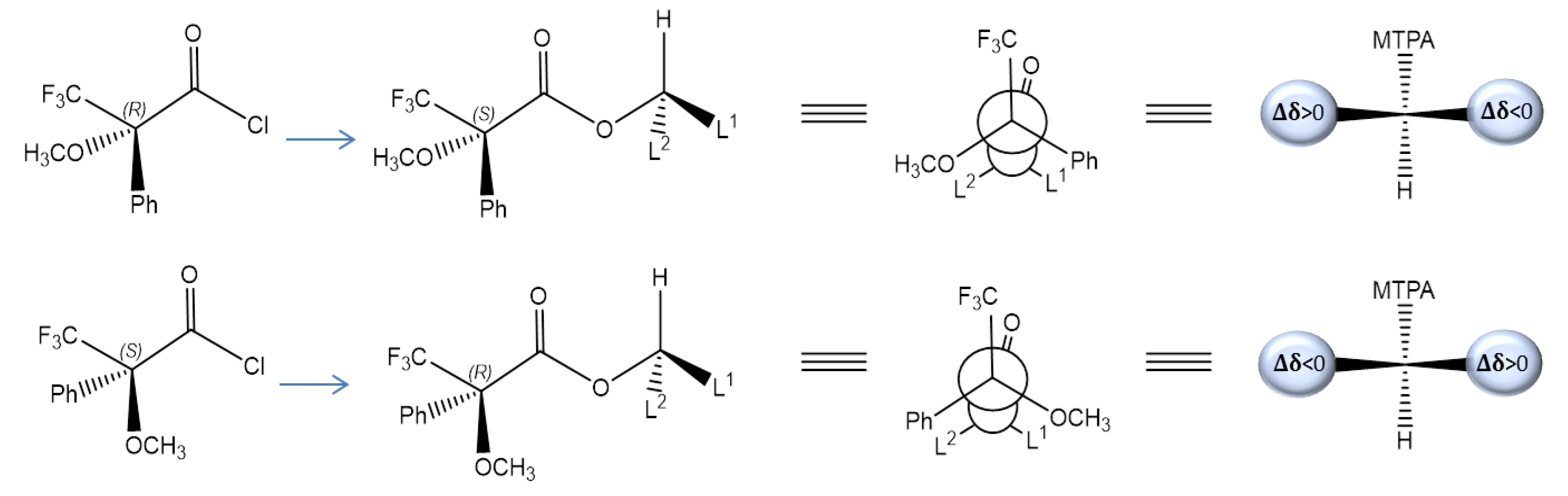
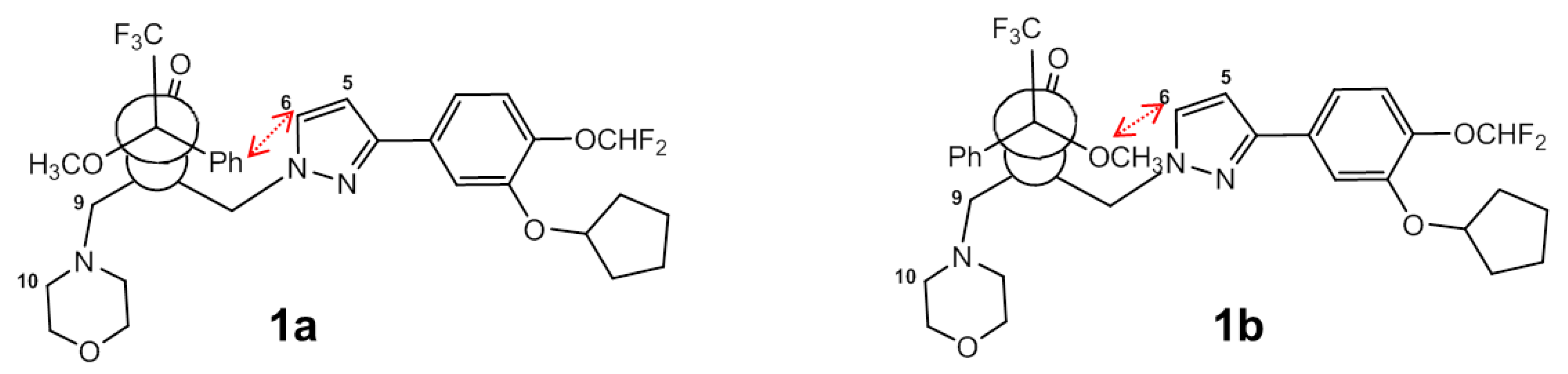
2.2. Assignment of the Absolute Configuration (AC)
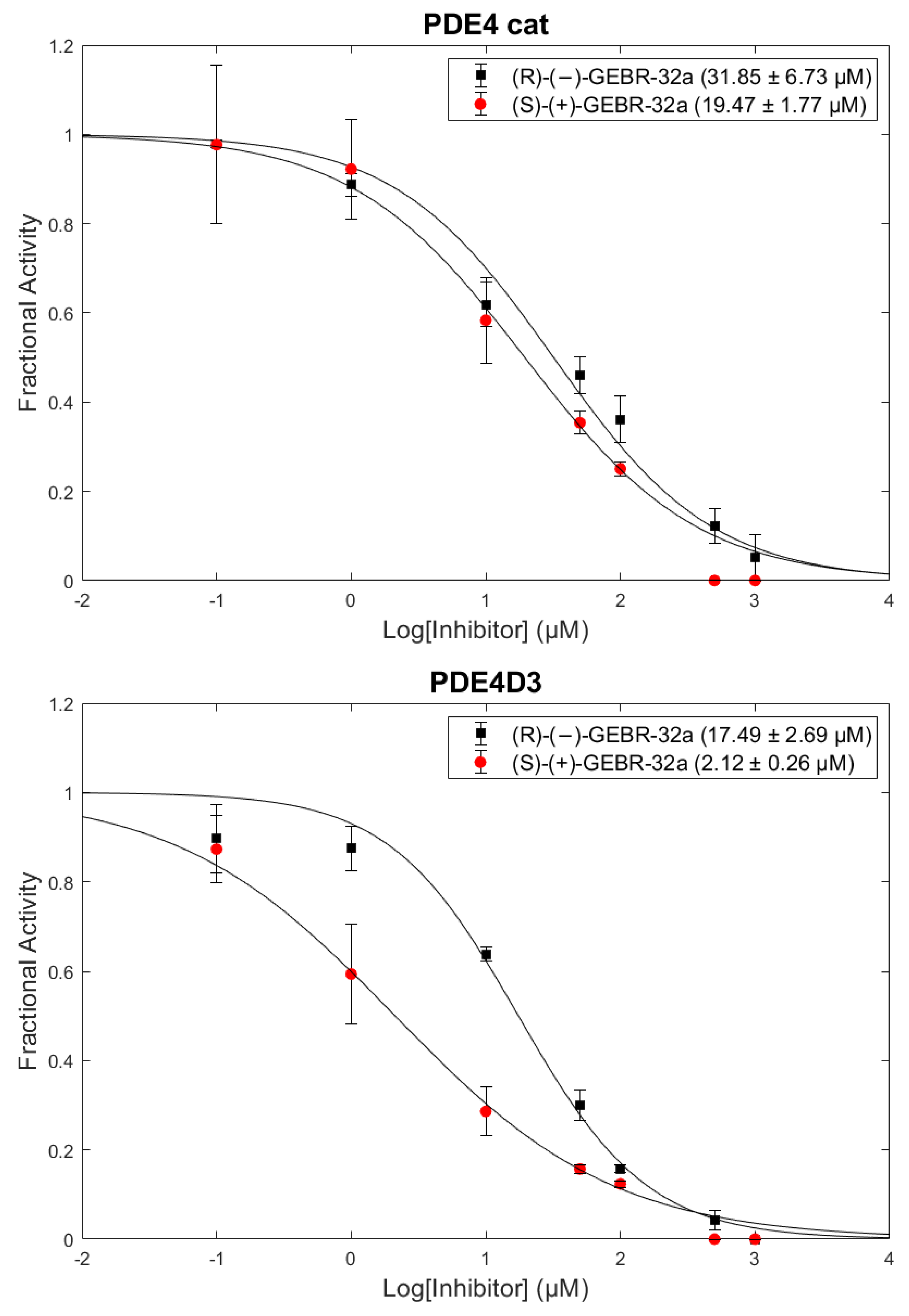
2.3. Enzymatic Activity
3. Materials and Methods
3.1. General
3.2. Chromatographic Resolution
3.3. Specific Rotation
3.4. Circular Dichroism
3.5. Absolute Configuration Assignment
3.6. NMR Analysis
3.7. Expression and Purification of PDE4D Catalytic Domain and PDE4D3
3.8. PDE4 Activity and Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzehimer’s Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H.M. The pharmacoeconomics of Alzheimer’s disease. Am. J. Manag. Care 2000, 6, 1139–1148. [Google Scholar]
- Colucci, L.; Bosco, M.; Fasanaro, A.M.; Gaeta, G.L.; Ricci, G.; Amenta, F. Alzheimer’s Disease Costs: What We Know and What We Should Take into Account. J. Alzheimer’s Dis. 2014, 42, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [PubMed]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi, S.M.; Talarek, S.; Listos, J.; Nabavi, S.F.; Devi, K.P.; de Oliveirad, M.R.; Tewari, D.; Argüelles, S.; Mehrzadi, S.; Hosseinzadeh, A.; et al. Phosphodiesterase inhibitors say NO to Alzheimer’s disease. Food anche Chem. Toxicol. 2019, 134, 110822–110840. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Brullo, C.; Prickaerts, J.; Arancio, O.; Villa, C.; Rebosio, C.; Calcagno, E.; Balbi, M.; van Hagen, B.T.J.; Argyrousi, E.K.; et al. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer’s disease. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Gulisano, W.; Tropea, M.R.; Arancio, O.; Palmeri, A.; Puzzo, D. Sub-efficacious doses of phosphodiesterase 4 and 5 inhibitors improve memory in a mouse model of Alzheimer’s disease. Neuropharmacology 2018, 138, 151–159. [Google Scholar] [CrossRef]
- Blokland, A.; Heckman, P.; Vanmierlo, T.; Schreiber, R.; Paes, D.; Prickaerts, J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol. Sci. 2019, 40, 971–985. [Google Scholar] [CrossRef]
- Prosdocimi, T.; Mollica, L.; Donini, S.; Semrau, M.S.; Lucarelli, A.P.; Aiolfi, E.; Cavalli, A.; Storici, P.; Alfei, S.; Brullo, C.; et al. Molecular Bases of PDE4D Inhibition by Memory-Enhancing GEBR Library Compounds. Biochemistry 2018, 57, 2876–2888. [Google Scholar] [CrossRef]
- Rossi, D.; Tarantino, M.; Rossino, G.; Rui, M.; Juza, M.; Collina, S. Approaches for multi-gram scale isolation of enantiomers for drug discovery. Expert Opin. Drug Discov. 2017, 12, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- Cosimelli, B.; Greco, G.; Laneri, S.; Novellino, E.; Sacchi, A.; Collina, S.; Rossi, D.; Cosconati, S.; Barresi, E.; Taliani, S.; et al. Studies on enantioselectivity of chiral 4-acetylamino-6-alkyloxy-2-alkylthiopyrimidines acting as antagonists of the human A3 adenosine receptor. Medchemcomm 2018, 9, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Rui, M.; Marra, A.; Pace, V.; Juza, M.; Rossi, D.; Collina, S. Novel enantiopure sigma receptor modulators: Quick (semi-)preparative chiral resolution via hplc and absolute configuration assignment. Molecules 2016, 21, 1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Marra, A.; Rui, M.; Brambilla, S.; Juza, M.; Collina, S. “Fit-for-purpose” development of analytical and (semi)preparative enantioselective high performance liquid and supercritical fluid chromatography for the access to a novel σ 1 receptor agonist. J. Pharm. Biomed. Anal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Nasti, R.; Collina, S.; Mazzeo, G.; Ghidinelli, S.; Longhi, G.; Memo, M.; Abbate, S. The role of chirality in a set of key intermediates of pharmaceutical interest, 3-aryl-substituted-γ-butyrolactones, evidenced by chiral HPLC separation and by chiroptical spectroscopies. J. Pharm. Biomed. Anal. 2017. [Google Scholar] [CrossRef]
- Rossi, D.; Nasti, R.; Marra, A.; Meneghini, S.; Mazzeo, G.; Longhi, G.; Memo, M.; Cosimelli, B.; Greco, G.; Novellino, E.; et al. Enantiomeric 4-Acylamino-6-alkyloxy-2 Alkylthiopyrimidines As Potential A3 Adenosine Receptor Antagonists: HPLC Chiral Resolution and Absolute Configuration Assignment by a Full Set of Chiroptical Spectroscopy. Chirality 2016, 28, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Gaggeri, R.; Rossi, D.; Christodoulou, M.S.; Passarella, D.; Leoni, F.; Azzolina, O.; Collina, S. Chiral Flavanones from Amygdalus lycioides Spach: Structural Elucidation and Identification of TNFalpha Inhibitors by Bioactivity-guided Fractionation. Molecules 2012, 1665–1674. [Google Scholar] [CrossRef]
- Gaggeri, R.; Rossi, D.; Collina, S.; Mannucci, B.; Baierl, M.; Juza, M. Quick development of an analytical enantioselective high performance liquid chromatography separation and preparative scale-up for the flavonoid naringenin. J. Chromatogr. A 2011, 1218, 5414–5422. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004, 104, 17–118. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. α-Methoxy-α-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem. 1969, 34, 2543–2549. [Google Scholar] [CrossRef]
- Fadia, Y.S.; Ashour, M.L.; Singa, A.N.B.; Wink, M. A Comprehensive Review of Bioactive Peptides from Marine Fungi and Their Biological Significance. Mar. Drugs 2019, 17, 559–583. [Google Scholar]
- Hub, L.; Mosher, H.S. α-Methoxy-α-trifluoromethylphenylacetic acid. Configuration by asymmetric synthesis. J. Org. Chem. 1970, 35, 3691–3694. [Google Scholar] [CrossRef]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of configuration and fluorine-19 chemical shifts of α-methoxy-α-trifluoromethylphenyl acetate derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Peterson, J.R.; Bickford, L.C.; Morgan, D.; Kim, A.S.; Ouerfelli, O.; Kirschner, M.W.; Rosen, M.K. Chemical Inhibition of N-WASP by stablization of a native autoinhibited conformation. Nat. Struct. Mol. Biol. 2004, 11, 747–755. [Google Scholar] [CrossRef]
- Vasile, F.; Civera, M.; Belvisi, L.; Potenza, D.; Tiana, G. Thermodynamically–weighted conformational ensemble of cyclic RGD peptidomimetics from NOE data. J. Phys. Chem. B 2016, 120, 7098–7107. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Panigada, M.; Siccardi, A.; Potenza, D.; Tiana, G. A Combined NMR-Computational Study of the Interaction between Influenza Virus Hemagglutinin and Sialic Derivatives from Human and Avian Receptors on the Surface of Transfected Cells. Int. J. Mol. Sci. 2018, 19, 1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef]
- Bieniossek, C.; Imasaki, T.; Takagi, Y.; Berger, I. MultiBac: Expanding the research toolbox for multiprotein complexes. Trends Biochem. Sci. 2012, 37, 49–57. [Google Scholar] [CrossRef]
- Chock, S.P.; Huang, C.Y. An optimized continuous assay for cAMP phosphodiesterase and calmodulin. Anal. Biochem. 1984, 138, 34–43. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds GEBR-32a are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mobile Phase a (v/v) | Flow | k1 | k2 | α | Rs |
|---|---|---|---|---|---|
| Hex/EtOH (90:10) | 1 mL/min | 3.05 | 3.07 | N/A | N/A |
| Hex/EtOH (80:20) | 1 mL/min | 2.47 | 3.02 | 1.22 | N/A |
| Hex/IPA (90:10) | 1 mL/min | 8.24 | 8.71 | 1.06 | N/A |
| Hex/IPA (80:20) | 1 mL/min | 3.10 | 3.55 | 1.15 | 0.77 |
| Hex/IPA (70:30) | 1 mL/min | 1.78 | 2.07 | 1.16 | 0.89 |
| Hex/IPA (50:50) | 1 mL/min | 1.07 | 1.23 | 1.15 | N/A |
| IPA | 0.5 mL/min | 0.87 | 1.56 | 1.79 | 2.61 |
| IPA/EtOH (50:50) | 0.5 mL/min | 3.10 | 3.38 | 1.09 | 1.27 |
| EtOH | 0.5 mL/min | 2.63 | 2.89 | 1.10 | 1.34 |
| EtOH/ MeOH (50:50) | 0.5 mL/min | 2.24 | 2.42 | 1.08 | 1.09 |
| MeOH | 0.5 mL/min | 0.48 | 0.53 | 1.10 | N/A |
| Cmpd | Rt | ee | (c 0.2%, MeOH) | Isolated Amount | Yield |
|---|---|---|---|---|---|
| (–)-GEBR-32a | 6.0 min | 99.5% | −18.6 | 30 mg | 43% |
| (+)-GEBR-32a | 8.4 min | 99.9% | +19.4 | 34 mg | 48% |
 | |||
| Proton (–)-GEBR-32a | 1a | 1b | Δδ = δS − δRa |
| H5 | 6.28 | 6.40 | −0.12 |
| H6 | 6.99 | 7.28 | −0.29 |
| H7 | 4.30–4.23 | 4.45–4.32 | −0.15/0.09 |
| H9 | 2.50 | 2.53 | −0.03 |
| H10 | 2.61–2.34 | 2.52–2.34 | 0.09 |
| H11 | 3.57 | 3.60 | −0.03 |
| Proton (+)-GEBR-32a | 2a | 2b | Δδ = δS − δR |
| H5 | 6.40 | 6.29 | 0.11 |
| H6 | 7.36 | 6.99 | 0.37 |
| H7 | 4.42–4.30 | 4.31–4.23 | 0.09/0.07 |
| H9 | 2.48 | 2.51 | −0.03 |
| H10 | 2.51–2.34 | 2.62–2.36 | −0.11/−0.02 |
| H11 | 3.59 | 3.61 | −0.02 |
| IC50 (µM) PDE4 cat | IC50 (µM) PDE4D3 | |
|---|---|---|
| (R)-(-)-GEBR-32a | 31.8 ± 6.7 | 15.5 ± 2.7 |
| (S)-(+)-GEBR-32a | 19.5 ± 1.8 | 2.1 ± 0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalloro, V.; Russo, K.; Vasile, F.; Pignataro, L.; Torretta, A.; Donini, S.; Semrau, M.S.; Storici, P.; Rossi, D.; Rapetti, F.; et al. Insight into GEBR-32a: Chiral Resolution, Absolute Configuration and Enantiopreference in PDE4D Inhibition. Molecules 2020, 25, 935. https://doi.org/10.3390/molecules25040935
Cavalloro V, Russo K, Vasile F, Pignataro L, Torretta A, Donini S, Semrau MS, Storici P, Rossi D, Rapetti F, et al. Insight into GEBR-32a: Chiral Resolution, Absolute Configuration and Enantiopreference in PDE4D Inhibition. Molecules. 2020; 25(4):935. https://doi.org/10.3390/molecules25040935
Chicago/Turabian StyleCavalloro, Valeria, Katia Russo, Francesca Vasile, Luca Pignataro, Archimede Torretta, Stefano Donini, Marta S. Semrau, Paola Storici, Daniela Rossi, Federica Rapetti, and et al. 2020. "Insight into GEBR-32a: Chiral Resolution, Absolute Configuration and Enantiopreference in PDE4D Inhibition" Molecules 25, no. 4: 935. https://doi.org/10.3390/molecules25040935