Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence



Abstract
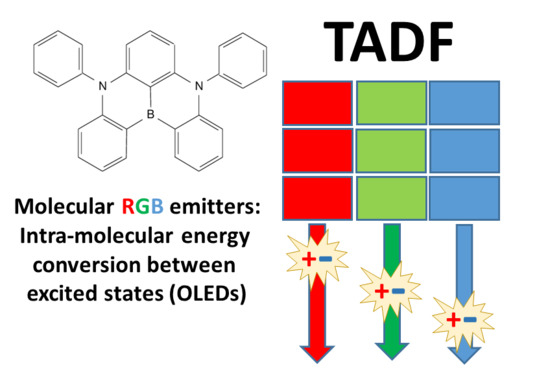
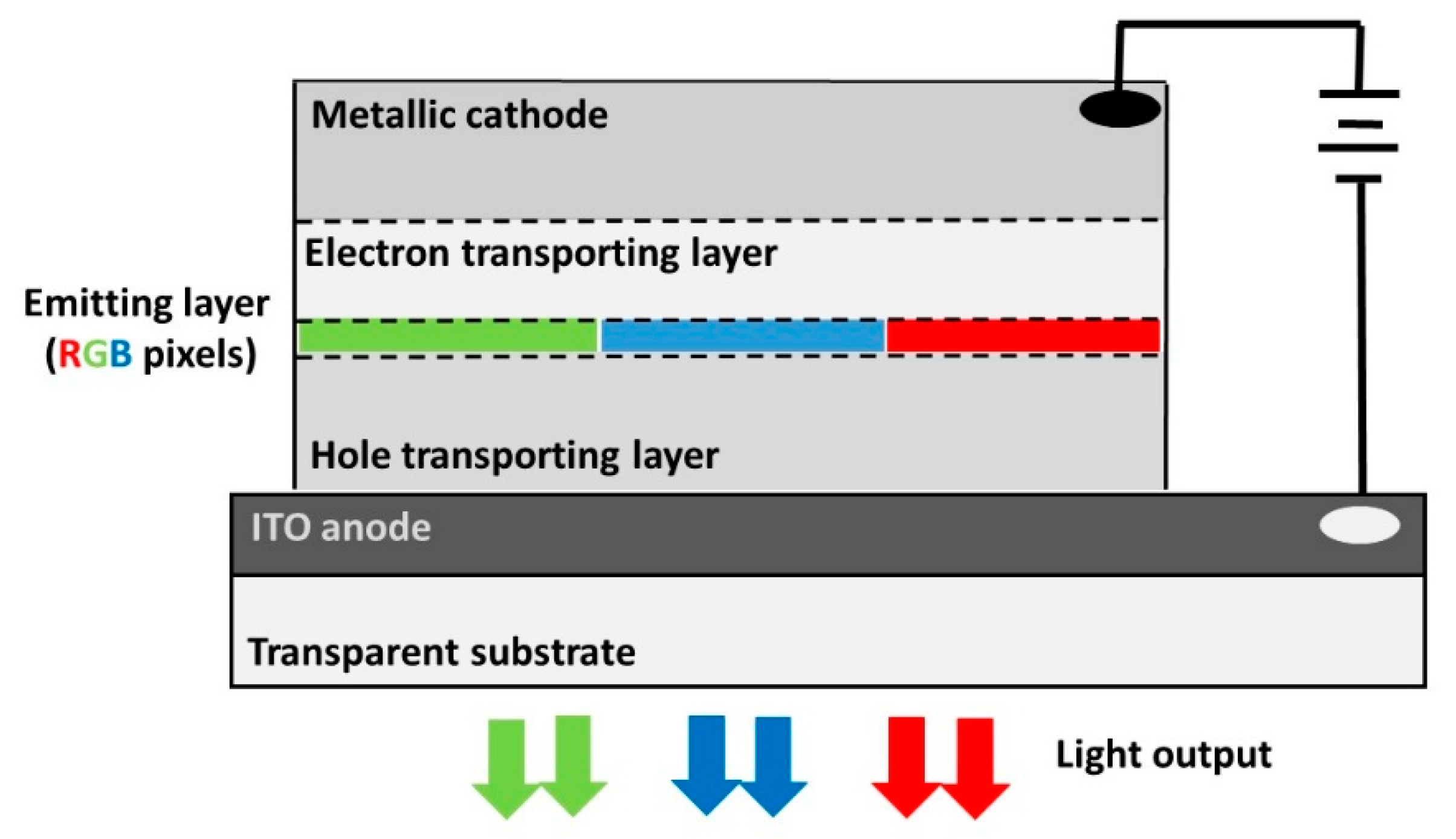
1. Introduction: The Quest for New Energy Conversion Mechanisms for Efficient Light Emission
2. Current Achievements and Challenges for Theoretical Methods at the Molecular Scale
3. Understanding the Grounds of a TD-DFT Calculation
- (i)
- All excited-state calculations are done converging first the ground-state eigenvalues and orbitals, and then using those orbitals and the associated density to calculate all TD-DFT-related magnitudes, which thus precludes the incorporation of an explicit dependence on time in the kernel —a fact known as the adiabatic approximation;
- (ii)
- The use of complex mathematical expressions for the integrand of modern semilocal Exc[ρ] functionals (i.e., GGA or meta-GGA) makes it difficult to obtain their second functional derivative analytically, and often the simplest expression known as the local density approximation (LDA) is used instead to speed up the calculations. The good performance of the adiabatic LDA (ALDA) approximation in finite systems has discouraged the development of more elaborate ways to incorporate the time dependence [24].
- (iii)
- Again, the use of a semilocal Exc[ρ] functional, depending only on and its successive derivatives, precludes the incorporation of an explicit spatial dependence in .
4. Beyond a TD-DFT Treatment of TADF
5. Case Study: 2CzPN and DABNA-1/TABNA Compounds
6. Summary and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Endo, A.; Sato, K.; Yoshimura, K.; Kawada, A.; Miyazaki, H.; Adachi, C. Efficient up-conversion of triplet excitons into a singlet state and its application for organic light emitting diodes. Appl. Phys. Lett. 2011, 98, 2011–2014. [Google Scholar] [CrossRef]
- Yang, X.; Xu, X.; Zhou, G. Recent advances of the emitters for high performance deep-blue organic light-emitting diodes. J. Mater. Chem. C 2015, 3, 915–944. [Google Scholar] [CrossRef]
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. [Google Scholar] [CrossRef]
- Wong, M.Y.; Zysman-Colman, E. Purely organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 2017, 29, 1605444. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M.R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 2018, 3, 18020. [Google Scholar] [CrossRef]
- Godumala, M.; Choi, S.; Cho, M.J.; Choi, D.H. Thermally activated delayed fluorescence blue dopants and hosts: From the design strategy to organic light-emitting diode applications. J. Mater. Chem. C. 2016, 4, 11355–11381. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef]
- Köhler, A.; Beljonne, D. The singlet-triplet exchange energy in conjugated polymers. Adv. Funct. Mater. 2004, 14, 11–18. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Olivier, Y.; Sancho-García, J.C.; Muccioli, L.; D’Avino, G.; Beljonne, D. Computational design of thermally activated delayed fluorescence materials: The challenges ahead. J. Phys. Chem. Lett. 2018, 9, 6149–6163. [Google Scholar] [CrossRef]
- Etherington, M.K.; Gibson, J.; Higginbotham, H.F.; Penfold, T.J.; Monkman, A.P. Revealing the spin–vibronic coupling mechanism of thermally activated delayed fluorescence. Nat. Commun. 2016, 7, 13680. [Google Scholar] [CrossRef]
- Gibson, J.; Monkman, A.P.; Penfold, T.J. The importance of vibronic coupling for efficient reverse intersystem crossing in thermally activated delayed fluorescence molecules. ChemPhysChem 2016, 17, 2956–2961. [Google Scholar] [CrossRef]
- Olivier, Y.; Yurash, B.; Muccioli, L.; D’Avino, G.; Mikhnenko, O.; Sancho-García, J.C.; Adachi, C.; Nguyen, T.-Q.; Beljonne, D. Nature of the singlet and triplet excitations mediating thermally activated delayed fluorescence. Phys. Rev. Mater. 2017, 1, 075602. [Google Scholar] [CrossRef]
- Olivier, Y.; Moral, M.; Muccioli, L.; Sancho-García, J.-C. Dynamic nature of excited states of donor–acceptor TADF materials for OLEDs: How theory can reveal structure–property relationships. J. Mater. Chem. C. 2017, 5, 5718–5729. [Google Scholar] [CrossRef]
- De Silva, P.; Kim, C.A.; Zhu, T.; Van Voorhis, T. Extracting design principles for efficient thermally activated delayed fluorescence (TADF) from a simple four-state model. Chem. Mater. 2019, 31, 6995–7006. [Google Scholar] [CrossRef]
- Li, X.-L.; Cai, X.; Ali, M.-U.; Su, S.-J.; Meng, H. Highly efficient thermally activated delayed fluorescence yellow organic light-emitting diodes with a low efficiency roll-off. J. Mater. Chem. C 2019, 7, 8063–8069. [Google Scholar] [CrossRef]
- Moral, M.; Muccioli, L.; Son, W.J.; Olivier, Y.; Sancho-García, J.C. Theoretical rationalization of the singlet–triplet gap in OLEDs materials: Impact of charge-transfer character. J. Chem. Theory Comput. 2015, 11, 168–177. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys. 2011, 13, 16987–16998. [Google Scholar] [CrossRef]
- Mewes, S.A.; Plasser, F.; Krylov, A.; Dreuw, A. Benchmarking excited-state calculations using exciton properties. J. Chem. Theory Comput. 2018, 14, 710–725. [Google Scholar] [CrossRef]
- Kümmel, S. Charge-Transfer Excitations: A Challenge for Time-Dependent Density Functional Theory That Has Been Met. Adv. Ener. Mater. 2017, 7, 1700440. [Google Scholar] [CrossRef]
- Penfold, T.J. On predicting the excited-state properties of thermally activated delayed fluorescence emitters. J. Phys. Chem. C 2015, 119, 13535–13544. [Google Scholar] [CrossRef]
- Sun, H.; Zhong, C.; Brédas, J.L. Reliable prediction with tuned range-separated functionals of the singlet–triplet gap in organic emitters for thermally activated delayed fluorescence. J. Chem. Theory Comput. 2015, 11, 3851–3858. [Google Scholar] [CrossRef]
- Maitra, N.T.; Burke, K. Demonstration of initial-state dependence in time-dependent density-functional theory. Phys. Rev. A 2001, 63, 042501. [Google Scholar] [CrossRef]
- Zimmerman, P.M.; Bell, F.; Casanova, D.; Head-Gordon, M. Mechanism for singlet fission in pentacene and tetracene: From single exciton to two triplets. J. Am. Chem. Soc. 2011, 133, 19944–19952. [Google Scholar] [CrossRef]
- Tamura, H.; Huix-Rotllant, M.; Burghardt, I.; Olivier, Y.; Beljonne, D. First-principles quantum dynamics of singlet fission: Coherent versus thermally activated mechanisms governed by molecular π stacking. Phys. Rev. Lett. 2015, 115, 107401. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure blue thermally activated delayed fluorescence molecules: Efficient HOMO-LUMO separation by the multiple resonance effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Nakatsuka, S.; Gotoh, H.; Kinoshita, K.; Yasuda, N.; Hatakeyama, T. Divergent synthesis of heteroatom-centered 4,8,12-triazatriangulenes. Angew. Chem. Int. Ed. 2017, 56, 5087–5090. [Google Scholar] [CrossRef]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-shot multiple borylation toward BN-doped nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Pershin, A.; Hall, D.; Lemaur, V.; Sancho-García, J.C.; Muccioli, L.; Zysman-Colman, E.; Beljonne, D.; Olivier, Y. Highly emissive excitons with reduced exchange energy in thermally activated delayed fluorescent molecules. Nat. Commun. 2019, 10, 597. [Google Scholar] [CrossRef]
- Christiansen, O.; Koch, H.; Jørgensen, P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. [Google Scholar] [CrossRef]
- Hellweg, A.; Grün, S.A.; Hättig, C. Benchmarking the performance of spin-component scaled CC2 in ground and electronically excited states. Phys. Chem. Chem. Phys. 2008, 10, 4119–4127. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Assessment of TD-DFT methods and of various spin scaled CIS(D) and CC2 versions for the treatment of low-lying valence excitations of large organic dyes. J. Chem. Phys. 2010, 132, 184103. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Rico, R.J.; Oumi, M.; Lee, T.J. A doubles correction to electronic excited states from configuration interaction in the space of single substitutions. Chem. Phys. Lett. 1994, 219, 21–29. [Google Scholar] [CrossRef]
- Wormit, M.; Rehn, D.R.; Harbach, P.H.; Wenzel, J.; Krauter, C.M.; Epifanovsky, E.; Dreuw, A. Investigating excited electronic states using the algebraic diagrammatic construction (ADC) approach of the polarisation propagator. Mol. Phys. 2014, 112, 774–784. [Google Scholar] [CrossRef]
- Angeli, C.; Pastore, M.; Cimiraglia, R. New perspectives in multireference perturbation theory: The n-electron valence state approach. Theor. Chem. Acc. 2007, 117, 743–754. [Google Scholar] [CrossRef]
- Comeau, D.C.; Bartlett, R.J. The equation-of-motion coupled-cluster method. Applications to open-and closed-shell reference states. Chem. Phys. Lett. 1993, 207, 414–423. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Lyskov, I.; Kleinschmidt, M.; Marian, C.M. Redesign of the DFT/MRCI Hamiltonian. J. Chem. Phys. 2016, 144, 034104. [Google Scholar] [CrossRef]
- Jacquemin, D.; Duchemin, I.; Blase, X. 0–0 energies using hybrid schemes: Benchmarks of TD-DFT, CIS (D), ADC (2), CC2, and BSE/GW formalisms for 80 real-life compounds. J. Chem. Theory Comput. 2015, 11, 5340–5359. [Google Scholar] [CrossRef]
- Lin, L.; Fan, J.; Cai, L.; Wang, C.K. Excited state dynamics of new-type thermally activated delayed fluorescence emitters: Theoretical view of light-emitting mechanism. Mol. Phys. 2018, 116, 19–28. [Google Scholar] [CrossRef]
- Northey, T.; Penfold, T.J. The intersystem crossing mechanism of an ultrapure blue organoboron emitter. Org. Electron. 2018, 59, 45–48. [Google Scholar] [CrossRef]
- Chen, X.K.; Kim, D.; Brédas, J.L. Thermally activated delayed fluorescence (TADF) path toward efficient electroluminescence in purely organic materials: Molecular level insight. Acc. Chem. Res. 2018, 51, 2215–2224. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A. A practicable real-space measure and visualization of static electron-correlation effects. Angew. Chem. Int. Ed. 2015, 54, 12308–12313. [Google Scholar] [CrossRef]
- Bauer, C.A.; Hansen, A.; Grimme, S. The fractional occupation number weighted density as a versatile analysis tool for molecules with a complicated electronic structure. Chem. Eur. J. 2017, 23, 6150–6164. [Google Scholar] [CrossRef]
- Loos, P.F.; Boggio-Pasqua, M.; Scemama, A.; Caffarel, M.; Jacquemin, D. Reference energies for double excitations. J. Chem. Theory Comput. 2019, 15, 1939–1956. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Method | E(S1) | E(T1) | ΔEST | fosc (S0 ← S1) |
|---|---|---|---|---|---|
| 2CzPN | TDA-PBE0 | 3.01 | 2.67 | 0.34 | 0.08 |
| SCS-CC2 a | 3.65 | 3.30 | 0.35 | 0.12 | |
| NEVPT2 | 3.33 | 3.02 | 0.32 | 0.20 | |
| Experimental b | 2.94 | 2.63 | 0.31 | -- | |
| DABNA-1 | TDA-PBE0 | 3.25 | 2.69 | 0.56 | 0.25 |
| SCS-CC2 a | 3.25 | 3.10 | 0.15 | 0.31 | |
| NEVPT2 | 3.04 | 2.95 | 0.09 | 0.21 | |
| Experimental c | 2.74 | 2.59 | 0.14 | -- | |
| TABNA | TDA-PBE0 | 3.69 | 3.12 | 0.57 | 0.12 |
| SCS-CC2 a | 3.67 | 3.50 | 0.17 | 0.13 | |
| NEVPT2 | 3.16 | 2.99 | 0.18 | 0.32 | |
| Experimental d | 3.11 | 2.90 | 0.21 | -- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz-Rodrigo, J.; Olivier, Y.; Sancho-García, J.-C. Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence. Molecules 2020, 25, 1006. https://doi.org/10.3390/molecules25041006
Sanz-Rodrigo J, Olivier Y, Sancho-García J-C. Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence. Molecules. 2020; 25(4):1006. https://doi.org/10.3390/molecules25041006
Chicago/Turabian StyleSanz-Rodrigo, Javier, Yoann Olivier, and Juan-Carlos Sancho-García. 2020. "Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence" Molecules 25, no. 4: 1006. https://doi.org/10.3390/molecules25041006
APA StyleSanz-Rodrigo, J., Olivier, Y., & Sancho-García, J.-C. (2020). Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence. Molecules, 25(4), 1006. https://doi.org/10.3390/molecules25041006