The Effect of Mg, Fe(II), and Al Doping on CH4: Adsorption and Diffusion on the Surface of Na-Kaolinite (001) by Molecular Simulations


Abstract
1. Introduction
2. Simulation Details
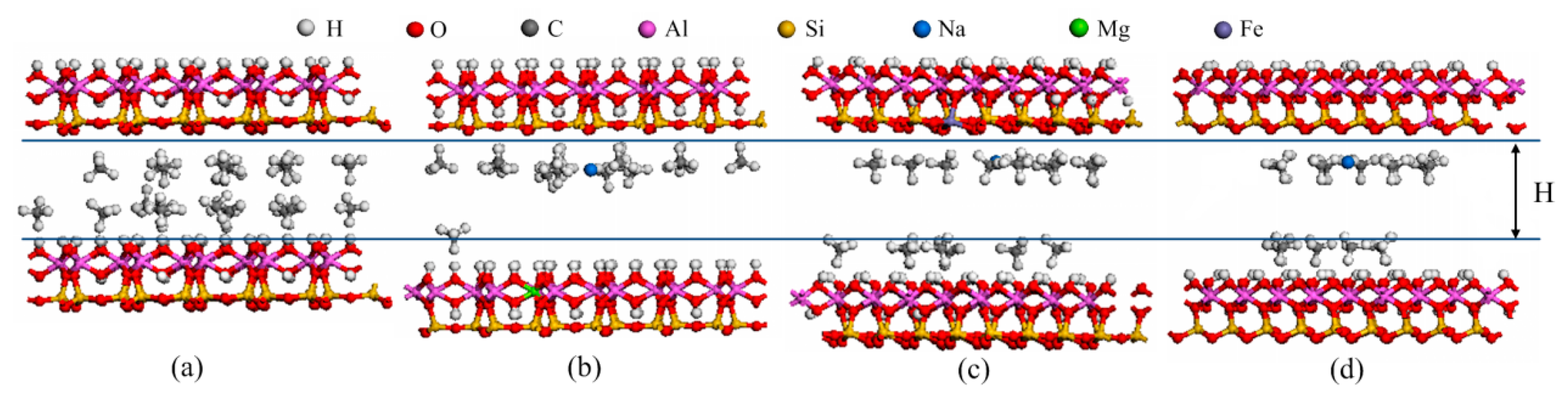
2.1. Models
2.2. Grand Canonical Monte Carlo Simulation
2.3. Molecular Dynamic Simulation
3. Results and Discussion
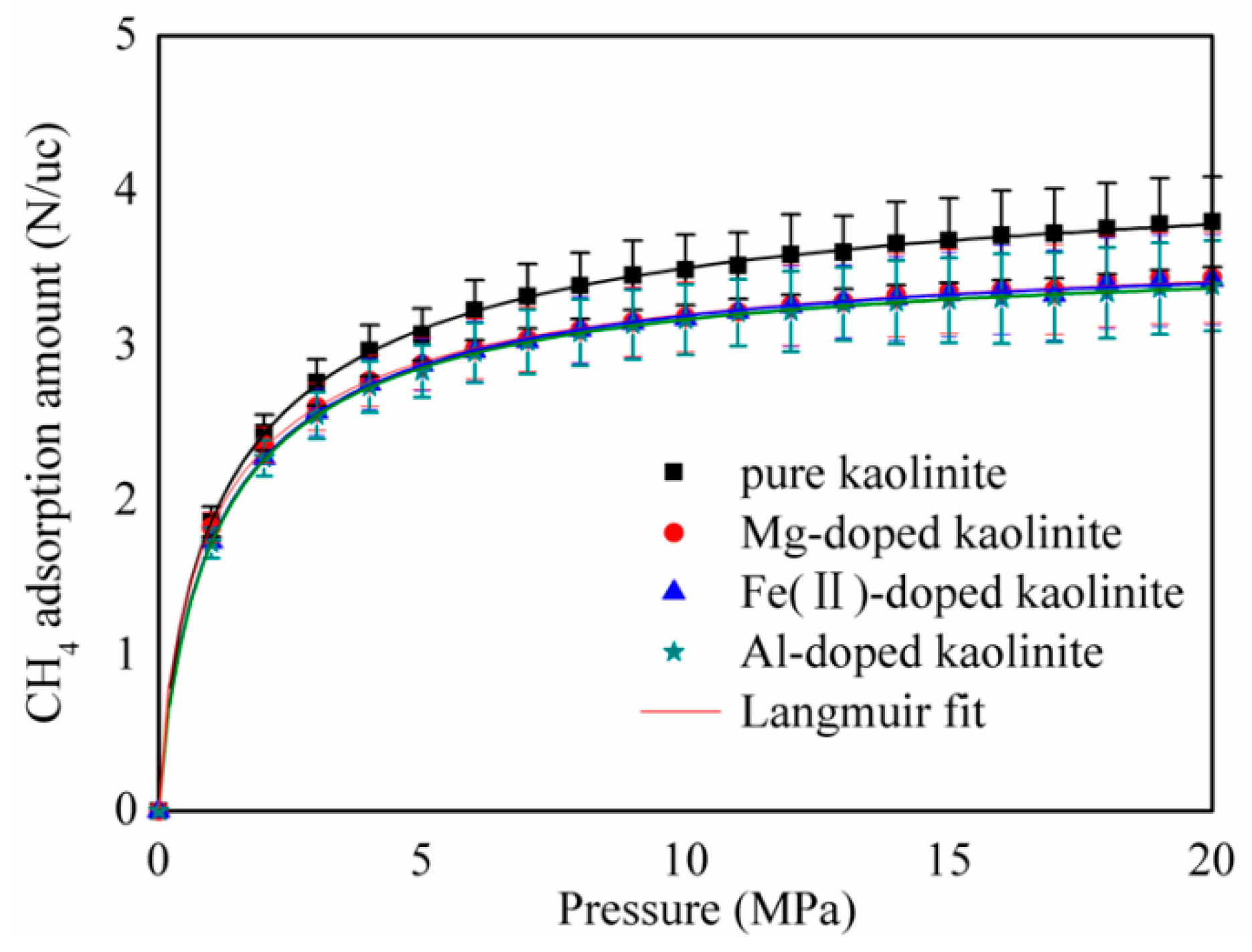
3.1. CH4 Adsorption Amount
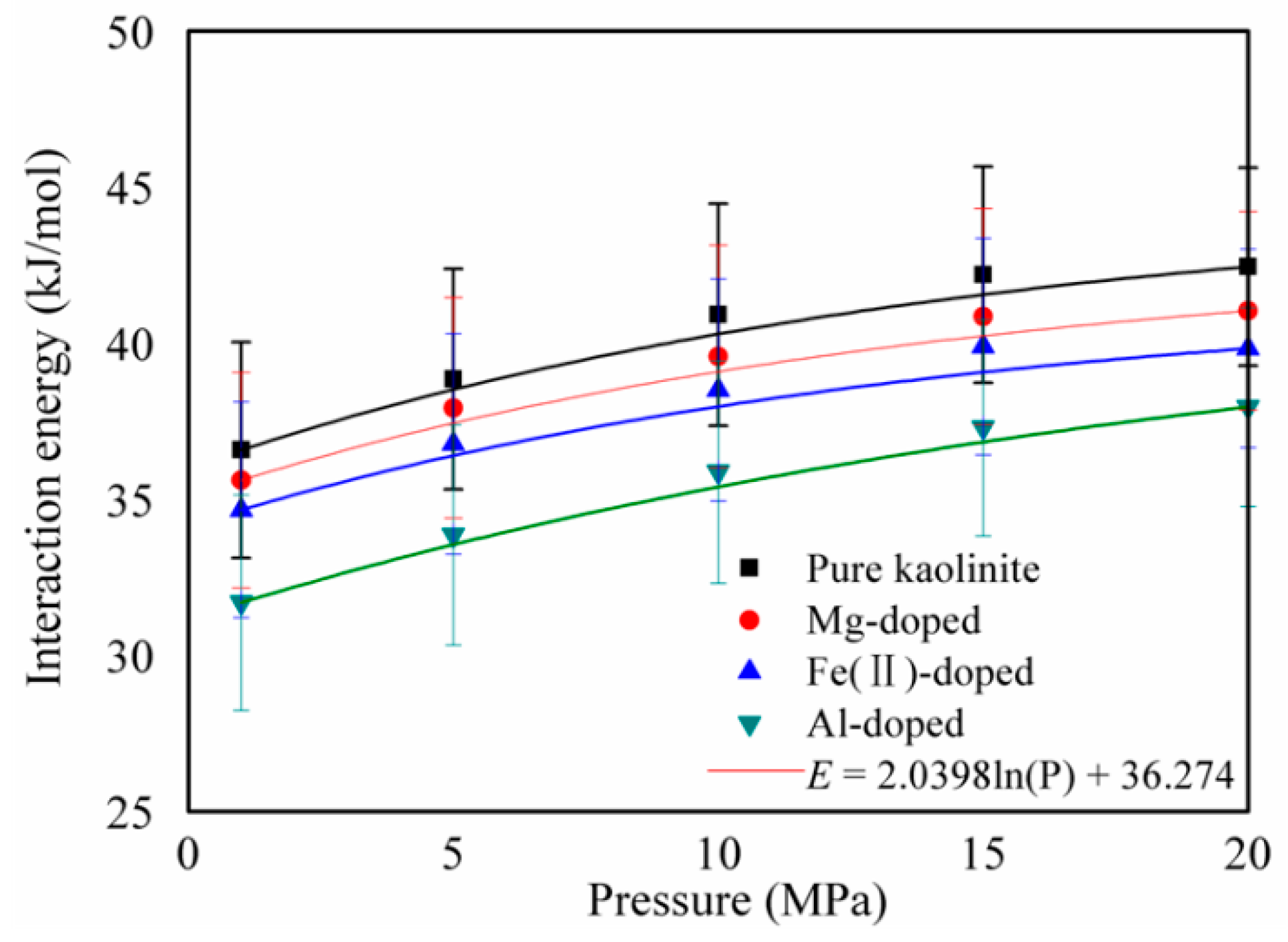
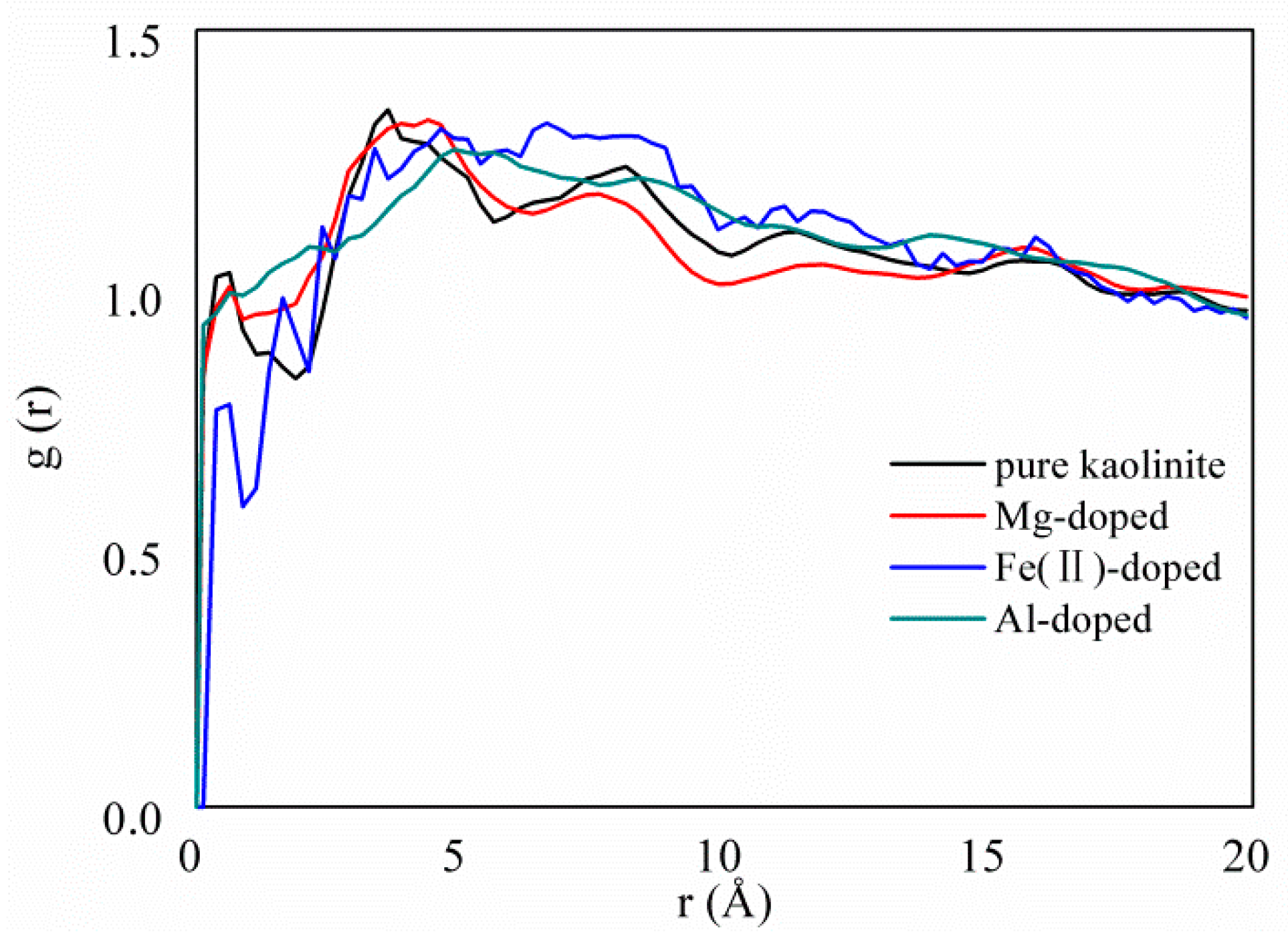
3.2. Specific Surface Area and Interaction Energy
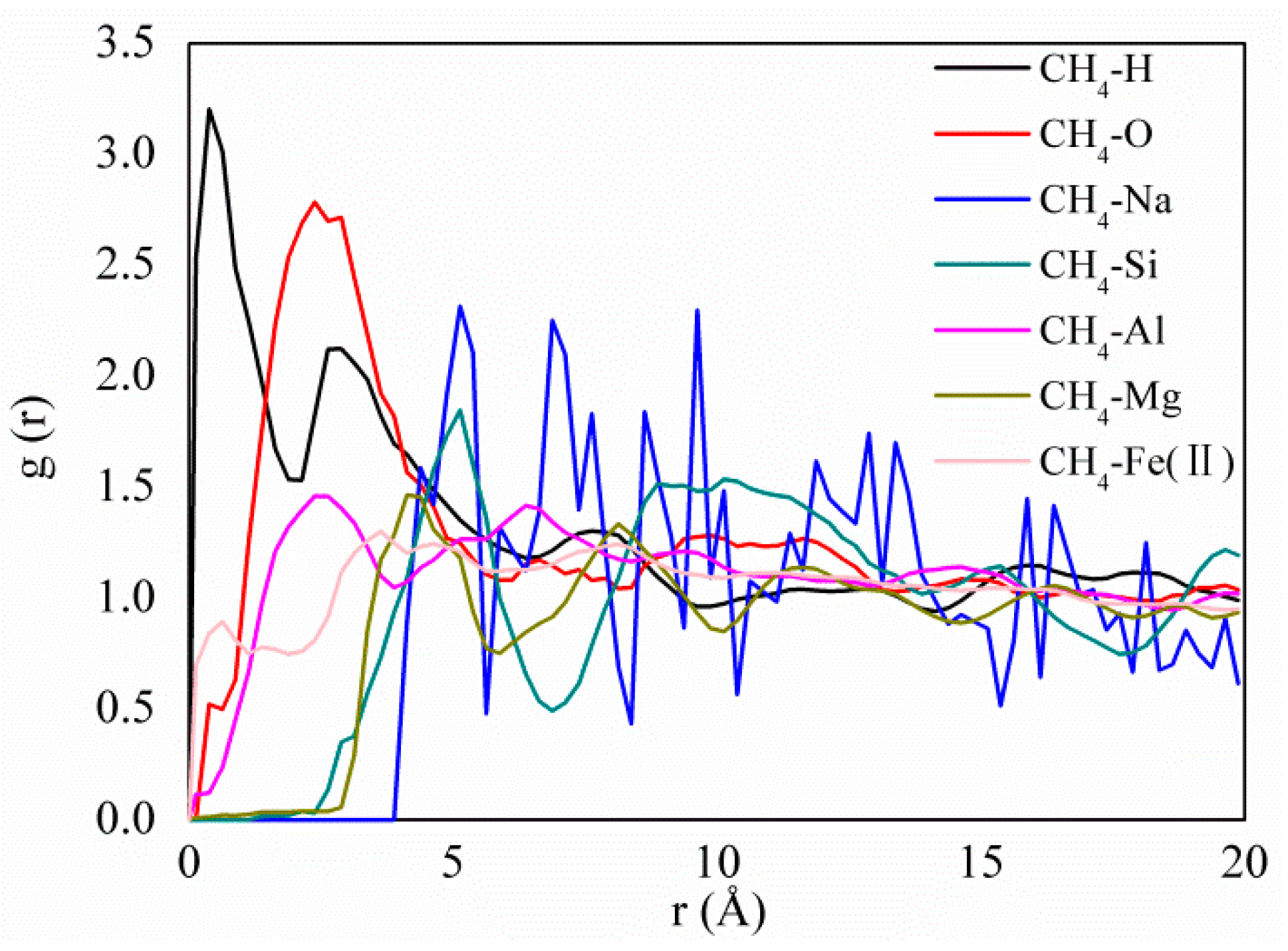
3.3. Adsorption Strength and Adsorption Site
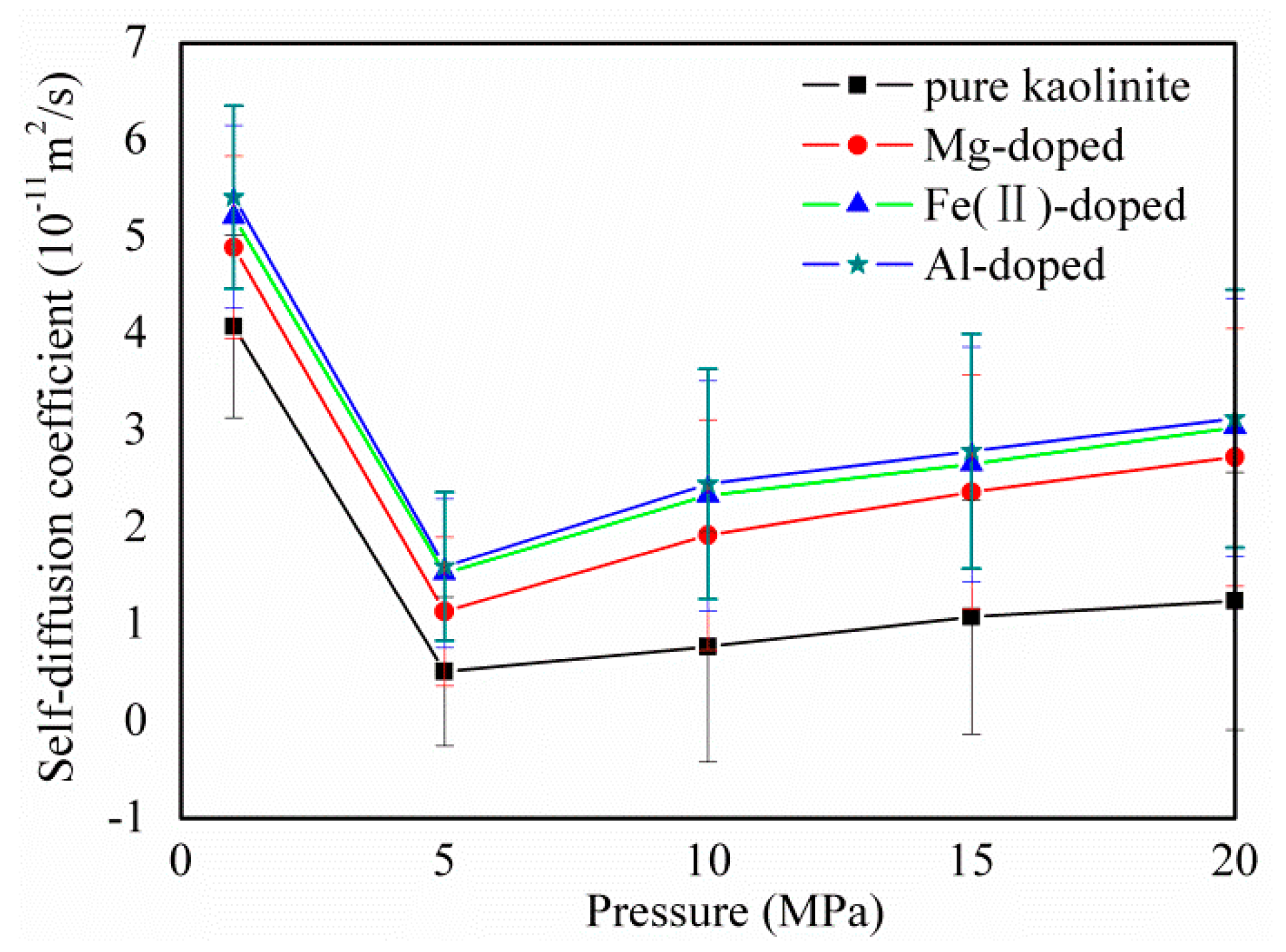
3.4. CH4 diffusion Coefficients
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vengosh, A.; Jackson, R.B.; Warner, N.; Darrah, T.H.; Kondash, A. A critical review of the risks to water resources from unconventional shale gas development and hydraulic fracturing in the United States. Environ. Sci. Technol. 2014, 48, 8334–8348. [Google Scholar] [CrossRef]
- Zou, C.; Dong, D.; Wang, S.; Li, J.; Li, X.; Wang, Y.; Li, D.; Cheng, K. Geological characteristics and resource potential of shale gas in China. Pet. Explor. Dev. 2010, 37, 641–653. [Google Scholar] [CrossRef]
- Weijermars, R. Economic appraisal of shale gas plays in continental Europe. Appl. Energy 2013, 106, 100–115. [Google Scholar] [CrossRef]
- Huo, R.; Duan, K.B. Conditions of Shale Gas Accumulation and Exploration Practices in China. Adv. Mater. Res. 2014, 978, 161–164. [Google Scholar] [CrossRef]
- Yuan, W.; Pan, Z.; Li, X.; Yang, Y.; Zhao, C.; Connell, L.D.; Li, S.; He, J. Experimental study and modelling of methane adsorption and diffusion in shale. Fuel 2014, 117, 509–519. [Google Scholar] [CrossRef]
- Zhao, L.; Ward, C.R.; French, D.; Graham, I.T.; Dai, S.; Yang, C.; Xie, P.; Zhang, S. Origin of a kaolinite-nh4-illite-pyrophyllite-chlorite assemblage in a marine-influenced anthracite and associated strata from the Jincheng coalfield, Qinshui basin, Northern China. Int. J. Coal Geol. 2018, 185, 61–78. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, D.; Pan, Z.; Yao, Y.; Li, C. Mineral occurrence and its impact on fracture generation in selected Qinshui basin coals: An experimental perspective. Int. J. Coal Geol. 2015, 150, 35–50. [Google Scholar] [CrossRef]
- Ji, L.; Zhang, T.; Milliken, K.L.; Qu, J.; Zhang, X. Experimental investigation of main controls to methane adsorption in clay-rich rocks. Appl. Geochem. 2012, 27, 2533–2545. [Google Scholar] [CrossRef]
- Liu, D.; Yuan, P.; Liu, H.; Li, T.; Tan, D.; Yuan, W.; He, H. High-pressure adsorption of methane on Montmorillonite, Kaolinite and Illite. Appl. Clay Sci. 2013, 85, 25–30. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, J.; Kang, T. Monte Carlo simulations of methane adsorption on kaolinite as a function of pore size. J. Nat. Gas Sci. Eng. 2018, 49, 410–416. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, J.; Kang, T. Molecular simulation of methane adsorption and its effect on kaolinite swelling as functions of pressure and temperature. Mol. Simul. 2018, 44, 789–796. [Google Scholar] [CrossRef]
- Zhang, B.; Kai, W.; Kang, T.; Kang, G.; Zhao, G. Effect of the basal spacing on ch4 diffusion in kaolinite. Chem. Phys. Lett. 2019, 732, 136639. [Google Scholar] [CrossRef]
- Plançon, A.; Giese, R.; Snyder, R.; Drits, V.; Bookin, A. Stacking faults in the kaolin-group minerals: Defect structures of kaolinite. Clays Clay Miner. 1989, 37, 203–210. [Google Scholar] [CrossRef]
- Frost, R.L.; Horváth, E.; Makó, É.; Kristóf, J. Modification of low-and high-defect kaolinite surfaces: Implications for kaolinite mineral processing. J. Colloid Interface Sci. 2004, 270, 337–346. [Google Scholar] [CrossRef]
- Zhang, J.; Clennell, M.; Dewhurst, D.; Liu, K. Combined monte carlo and molecular dynamics simulation of methane adsorption on dry and moist coal. Fuel 2014, 122, 186–197. [Google Scholar] [CrossRef]
- Mignon, P.; Ugliengo, P.; Sodupe, M.; Hernandez, E.R. Ab initio molecular dynamics study of the hydration of Li+, Na+ and K+ in a montmorillonite model. Influence of isomorphic substitution. Phys. Chem. Chem. Phys. 2010, 12, 688–697. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, J.; Kang, T. Effect of water on methane adsorption on the kaolinite (0 0 1) surface based on molecular simulations. Appl. Surf. Sci. 2018, 439, 792–800. [Google Scholar] [CrossRef]
- He, M.-C.; Zhao, J. Effects of Mg, Ca, and Fe(II) doping on the kaolinite (001) surface with H2O adsorption. Clays Clay Miner. 2012, 60, 330–337. [Google Scholar] [CrossRef]
- Hong, H.; Min, X.; Zhou, Y. Orbital calculations of kaolinite surface: On substitution of Al3+ for Si4+ in the tetrahedral sites. J. Wuhan Univ. Technol.-Mater. Sci. Ed. 2007, 22, 661–666. [Google Scholar] [CrossRef]
- Rand, B.; Melton, I.E. Particle interactions in aqueous kaolinite suspensions: I. Effect of ph and electrolyte upon the mode of particle interaction in homoionic sodium kaolinite suspensions. J. Colloid Interface Sci. 1977, 60, 308–320. [Google Scholar] [CrossRef]
- Hu, H.; Li, X.; Fang, Z.; Wei, N.; Li, Q. Small-molecule gas sorption and diffusion in coal: Molecular simulation. Energy 2010, 35, 2939–2944. [Google Scholar] [CrossRef]
- Mosher, K.; He, J.; Liu, Y.; Rupp, E.; Wilcox, J. Molecular simulation of methane adsorption in micro-and mesoporous carbons with applications to coal and gas shale systems. Int. J. Coal Geol. 2013, 109, 36–44. [Google Scholar] [CrossRef]
- Peng, D.-Y.; Robinson, D.B. A new two-constant equation of state. Ind. Eng. Chem. Fundam. 1976, 15, 59–64. [Google Scholar] [CrossRef]
- Coles, C.A.; Yong, R.N. Aspects of kaolinite characterization and retention of pb and cd. Appl. Clay Sci. 2002, 22, 39–45. [Google Scholar] [CrossRef]
- Yariv, S.; Lapides, I.; Nasser, A.; Lahav, N.; Brodsky, I.; Michaelian, K. Infrared study of the intercalation of potassium halides in kaolinite. Clays Clay Miner. 2000, 48, 10–18. [Google Scholar] [CrossRef]
- Yeh, I.-C.; Berkowitz, M.L. Ewald summation for systems with slab geometry. J. Chem. Phys. 1999, 111, 3155–3162. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. Dreiding: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Xiang, Z.; Cao, D. Porous covalent–organic materials: Synthesis, clean energy application and design. J. Mater. Chem. A 2013, 1, 2691–2718. [Google Scholar] [CrossRef]
- Fan, H.B.; Yuen, M.M. Material properties of the cross-linked epoxy resin compound predicted by molecular dynamics simulation. Polymer 2007, 48, 2174–2178. [Google Scholar] [CrossRef]
- Accelrys, I. Materials Studio; Accelrys Software Inc.: San Diego, CA, USA, 2010. [Google Scholar]
- Lan, Q.; Bassi, A.S.; Zhu, J.-X.J.; Margaritis, A. A modified langmuir model for the prediction of the effects of ionic strength on the equilibrium characteristics of protein adsorption onto ion exchange/affinity adsorbents. Chem. Eng. J. 2001, 81, 179–186. [Google Scholar] [CrossRef]
- Sun, B.; Kayal, S.; Chakraborty, A. Study of HKUST (Copper benzene-1,3,5-tricarboxylate, Cu-BTC MOF)-1 metal organic frameworks for CH4 adsorption: An experimental Investigation with GCMC (grand canonical Monte-carlo) simulation. Energy 2014, 76, 419–427. [Google Scholar] [CrossRef]
- Frost, H.; Düren, T.; Snurr, R.Q. Effects of surface area, free volume, and heat of adsorption on hydrogen uptake in metal− organic frameworks. J. Phys. Chem. B 2006, 110, 9565–9570. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Yao, J. Effect of surface chemistry for CH4/CO2 adsorption in kerogen: A molecular simulation study. J. Nat. Gas Sci. Eng. 2016, 31, 738–746. [Google Scholar] [CrossRef]
- Thomas, K.M. Hydrogen adsorption and storage on porous materials. Catal. Today 2007, 120, 389–398. [Google Scholar] [CrossRef]
- Kumar, K.; Kumar, A. Adsorptive separation of carbon dioxide from flue gas using mesoporous MCM-41: A molecular simulation study. Korean J. Chem. Eng. 2018, 35, 535–547. [Google Scholar] [CrossRef]
- Akkermans, R.L.; Spenley, N.A.; Robertson, S.H. Monte carlo methods in materials studio. Mol. Simul. 2013, 39, 1153–1164. [Google Scholar] [CrossRef]
- Yang, J.Z.; Liu, Q.L.; Wang, H.T. Analyzing adsorption and diffusion behaviors of ethanol/water through silicalite membranes by molecular simulation. J. Membr. Sci. 2007, 291, 1–9. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, B.; Kang, T. Effect of geological depths on CH4 adsorption, diffusion and swelling in kaolinite by molecular simulations. Energy Fuels 2020, 34, 1620–1626. [Google Scholar] [CrossRef]
- García-Serrano, L.; Flores-Sandoval, C.; Zaragoza, I. Theoretical study of the adsorption of isobutane over h-mordenite zeolite by ab initio and dft methods. J. Mol. Catal. A Chem. 2003, 200, 205–212. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Clennell, M.; Dewhurst, D.; Pervukhina, M. Molecular simulation of CO2–CH4 competitive adsorption and induced coal swelling. Fuel 2015, 160, 309–317. [Google Scholar] [CrossRef]
- Chen, G.; Lu, S.; Zhang, J.; Xue, Q.; Han, T.; Xue, H.; Tian, S.; Li, J.; Xu, C.; Pervukhina, M. Research of CO2 and N2 adsorption behavior in k-illite slit pores by gcmc method. Sci. Rep. 2016, 6, 37579. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, J.; Lu, S.; Pervukhina, M.; Liu, K.; Xue, Q.; Tian, H.; Tian, S.; Li, J.; Clennell, M. Adsorption behavior of hydrocarbon on illite. Energy Fuels 2016, 30, 9114–9121. [Google Scholar] [CrossRef]
- Muniz-Miranda, F.; Lodesani, F.; Tavanti, F.; Presti, D.; Malferrari, D.; Pedone, A. Supercritical CO2 confined in palygorskite and sepiolite minerals: A classical molecular dynamics investigation. J. Phys. Chem. C 2016, 120, 26945–26954. [Google Scholar] [CrossRef]
- Soper, A. The radial distribution functions of water and ice from 220 to 673 k and at pressures up to 400 mpa. Chem. Phys. 2000, 258, 121–137. [Google Scholar] [CrossRef]
- Czímerová, A.; Bujdák, J.; Dohrmann, R. Traditional and novel methods for estimating the layer charge of smectites. Appl. Clay Sci. 2006, 34, 2–13. [Google Scholar] [CrossRef]
- An, J.-H.; Dultz, S. Adsorption of tannic acid on chitosan-montmorillonite as a function of ph and surface charge properties. Appl. Clay Sci. 2007, 36, 256–264. [Google Scholar] [CrossRef]
- Einstein, A. Investigations on the theory of the brownian movement. Ann. D. Phys. 1905, 17, 549. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, B.; Kang, T. Monte carlo and molecular dynamic simulations of CH4 diffusion in kaolinite as functions of pressure and temperature. J. Nat. Gas Sci. Eng. 2018, 54, 65–71. [Google Scholar] [CrossRef]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | VL (molecules/uc) | PL (MPa) | Correlation Coefficient R2 |
|---|---|---|---|
| Kaolinite (001) | 3.91 | 0.95 | 0.992 |
| Mg-doped | 3.54 | 1.01 | 0.991 |
| Fe(II)-doped | 3.51 | 1.05 | 0.996 |
| Al-doped | 3.50 | 1.06 | 0.997 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Zhang, B.; Kang, T. The Effect of Mg, Fe(II), and Al Doping on CH4: Adsorption and Diffusion on the Surface of Na-Kaolinite (001) by Molecular Simulations. Molecules 2020, 25, 1001. https://doi.org/10.3390/molecules25041001
Wang K, Zhang B, Kang T. The Effect of Mg, Fe(II), and Al Doping on CH4: Adsorption and Diffusion on the Surface of Na-Kaolinite (001) by Molecular Simulations. Molecules. 2020; 25(4):1001. https://doi.org/10.3390/molecules25041001
Chicago/Turabian StyleWang, Kai, Bin Zhang, and Tianhe Kang. 2020. "The Effect of Mg, Fe(II), and Al Doping on CH4: Adsorption and Diffusion on the Surface of Na-Kaolinite (001) by Molecular Simulations" Molecules 25, no. 4: 1001. https://doi.org/10.3390/molecules25041001
APA StyleWang, K., Zhang, B., & Kang, T. (2020). The Effect of Mg, Fe(II), and Al Doping on CH4: Adsorption and Diffusion on the Surface of Na-Kaolinite (001) by Molecular Simulations. Molecules, 25(4), 1001. https://doi.org/10.3390/molecules25041001