Synthesis and Anticancer Cytotoxicity of Azaaurones Overcoming Multidrug Resistance

 ,
,  ,
,  ,
, 
Abstract
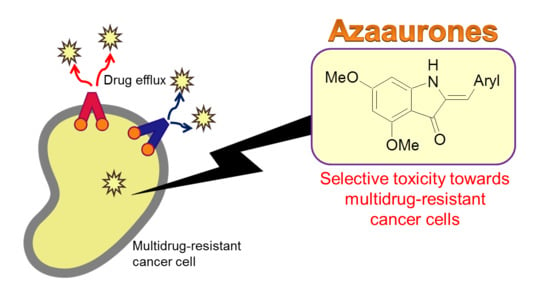
1. Introduction
2. Results and Discussion
2.1. Synthesis of Azaaurones
2.2. Structure–Activity Relationship Study
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of Azaaurones 1–13
3.3. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Borst, P. Cancer drug pan-resistance: Pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012, 2, 120066. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Szakács, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef]
- Lorendeau, D.; Dury, L.; Nasr, R.; Boumendjel, A.; Teodor, E.; Gutschow, M.; Falson, P.; Di Pietro, A.; Baubichon-Cortay, H. MRP1-dependent Collateral Sensitivity of Multidrug-resistant Cancer Cells: Identifying Selective Modulators Inducing Cellular Glutathione Depletion. Curr. Med. Chem. 2017, 24, 1186–1213. [Google Scholar] [CrossRef]
- Efferth, T.; Saeed, M.E.M.; Kadioglu, O.; Seo, E.J.; Shirooie, S.; Mbaveng, A.T.; Nabavi, S.M.; Kuete, V. Collateral sensitivity of natural products in drug-resistant cancer cells. Biotechnol. Adv. 2019. [Google Scholar] [CrossRef]
- Dankó, B.; Tóth, S.; Martins, A.; Vágvölgyi, M.; Kúsz, N.; Molnár, J.; Chang, F.R.; Wu, Y.C.; Szakács, G.; Hunyadi, A. Synthesis and SAR Study of Anticancer Protoflavone Derivatives: Investigation of Cytotoxicity and Interaction with ABCB1 and ABCG2 Multidrug Efflux Transporters. ChemMedChem 2017, 12, 850–859. [Google Scholar] [CrossRef]
- Füredi, A.; Tóth, S.; Szebényi, K.; Pape, V.F.; Türk, D.; Kucsma, N.; Cervenak, L.; Tóvári, J.; Szakács, G. Identification and Validation of Compounds Selectively Killing Resistant Cancer: Delineating Cell Line-Specific Effects from P-Glycoprotein-Induced Toxicity. Mol. Cancer Ther. 2017, 16, 45–56. [Google Scholar] [CrossRef]
- Türk, D.; Hall, M.D.; Chu, B.F.; Ludwig, J.A.; Fales, H.M.; Gottesman, M.M.; Szakács, G. Identification of compounds selectively killing multidrug-resistant cancer cells. Cancer Res. 2009, 69, 8293–8301. [Google Scholar] [CrossRef]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.W. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Kiemlian Kwee, J. Yin and Yang of Polyphenols in Cancer Prevention: A Short Review. Anticancer Agents Med. Chem. 2016, 16, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Pape, V.F.S.; Tóth, S.; Füredi, A.; Szebényi, K.; Lovrics, A.; Szabó, P.; Wiese, M.; Szakács, G. Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance. Eur. J. Med. Chem. 2016, 117, 335–354. [Google Scholar] [CrossRef]
- Hall, M.D.; Salam, N.K.; Hellawell, J.L.; Fales, H.M.; Kensler, C.B.; Ludwig, J.A.; Szakács, G.; Hibbs, D.E.; Gottesman, M.M. Synthesis, activity, and pharmacophore development for isatin-beta-thiosemicarbazones with selective activity toward multidrug-resistant cells. J. Med. Chem. 2009, 52, 3191–3204. [Google Scholar] [CrossRef]
- Heffeter, P.; Pape, V.F.S.; Enyedy, E.A.; Keppler, B.K.; Szakács, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef]
- Harker, W.G.; Sikic, B.I. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Res. 1985, 45, 4091–4096. [Google Scholar]
- Homolya, L.; Holló, M.; Müller, M.; Mechetner, E.B.; Sarkadi, B. A new method for a quantitative assessment of P-glycoprotein-related multidrug resistance in tumour cells. Br. J. Cancer 1996, 73, 849–855. [Google Scholar] [CrossRef]
- Boumendjel, A. Aurones: A subclass of flavones with promising biological potential. Curr. Med. Chem. 2003, 10, 2621–2630. [Google Scholar] [CrossRef]
- Zwergel, C.; Gaascht, F.; Valente, S.; Diederich, M.; Bagrel, D.; Kirsch, G. Aurones: Interesting natural and synthetic compounds with emerging biological potential. Nat. Prod. Commun. 2012, 7, 389–394. [Google Scholar] [CrossRef]
- Souard, F.; Okombi, S.; Beney, C.; Chevalley, S.; Valentin, A.; Boumendjel, A. 1-Azaaurones derived from the naturally occurring aurones as potential antimalarial drugs. Bioorg. Med. Chem. 2010, 18, 5724–5731. [Google Scholar] [CrossRef]
- Carrasco, M.P.; Machado, M.; Gonçalves, L.; Sharma, M.; Gut, J.; Lukens, A.K.; Wirth, D.F.; André, V.; Duarte, M.T.; Guedes, R.C.; et al. Probing the Azaaurone Scaffold against the Hepatic and Erythrocytic Stages of Malaria Parasites. ChemMedChem 2016, 11, 2194–2204. [Google Scholar] [CrossRef]
- Campaniço, A.; Carrasco, M.P.; Njoroge, M.; Seldon, R.; Chibale, K.; Perdigão, J.; Portugal, I.; Warner, D.F.; Moreira, R.; Lopes, F. Azaaurones as Potent Antimycobacterial Agents Active against MDR- and XDR-TB. ChemMedChem 2019, 14, 1537–1546. [Google Scholar] [CrossRef]
- Baiceanu, E.; Nguyen, K.-A.; Gonzalez-Lobato, L.; Nasr, R.; Baubichon-Cortay, H.; Loghin, F.; Le Borgne, M.; Chow, L.; Boumendjel, A.; Peuchmaur, M.; et al. 2-Indolylmethylenebenzofuranones as first effective inhibitors of ABCC2. Eur. J. Med. Chem. 2016, 122, 408–418. [Google Scholar] [CrossRef]
- Rodríguez-García, C.; Sánchez-Quesada, C.; Gaforio, J.J. Dietary Flavonoids as cancer chemopreventive agents: An updated review of human studies. Antioxidants 2019, 8, 137. [Google Scholar] [CrossRef]
- Strobl, C.D.; Schaffer, S.; Haug, T.; Völkl, S.; Peter, K.; Singer, K.; Böttcher, M.; Mougiakakos, D.; Mackensen, A.; Aigner, M. Selective PRMT5 Inhibitors Suppress Human CD8+ T Cells by Upregulation of p53 and Impairment of the AKT Pathway Similar to the Tumor Metabolite MTA. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef]
- Rádai, Z.; Windt, T.; Nagy, V.; Füredi, A.; Kiss, N.Z.; Ranđelović, I.; Tóvári, J.; Keglevich, G.; Szakács, G.; Tóth, S. Synthesis and anticancer cytotoxicity with structural context of an α-hydroxyphosphonate based compound library derived from substituted benzaldehydes. New J. Chem. 2019, 43, 14028–14035. [Google Scholar] [CrossRef]
- Pape, V.F.; Türk, D.; Szabó, P.; Wiese, M.; Enyedy, E.A.; Szakács, G. Synthesis and characterization of the anticancer and metal binding properties of novel pyrimidinylhydrazone derivatives. J. Inorg. Biochem. 2015, 144, 18–30. [Google Scholar] [CrossRef]
Sample Availability: Samples of these compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | Aryl | IC50 (MES-SA) | IC50 (Dx5) | SR 1 |
| 1 |  | 147.2 ± 32.8 | 36.6 ± 14.5 | 4.0 |
| 2 |  | 22.7 ± 1.4 | 10.8 ± 1.7 | 2.1 |
| 3 |  | 43.9 ± 3.6 | 13.7 ± 3.0 | 3.2 |
| 4 |  | 17.8 ± 4.1 | 3.7 ± 0.6 | 4.8 |
| 5 |  | 17.1 ± 1.7 | 5.8 ± 1.3 | 2.9 |
| 6 |  | 11.9 ± 1.6 | 3.4 ± 0.2 | 3.8 |
| 7 |  | 61.9 ± 10.0 | 13.1 ± 5.6 | 2.7 |
| 8 |  | 158.6 ± 43.7 | 32.1 ± 13.3 | 4.9 |
| 9 |  | 170.8 ± 4.3 | 131.7 ± 13.8 | 1.3 |
| 10 |  | 9.2 ± 2.3 | 19.5 ± 3.5 | 0.5 |
| 11 |  | 44.5 ± 12.8 | 27.4 ± 7.8 | 1.6 |
| 12 |  | 30.4 ± 2.0 | 15.6 ± 1.4 | 1.9 |
| 13 |  | 169.9 ± 30.2 | 44.2 ± 16.0 | 3.8 |
| Doxorubicin | 0.36 ± 0.11 | 4.15 ± 0.38 | 0.087 | |
| NSC57969 | 4.63 ± 1.31 | 0.59 ± 0.10 | 7.9 | |
 | |||
|---|---|---|---|
| Compound | IC50 (MES-SA) | IC50 (Dx5) | SR |
| Genistein | 53.6 ± 4.63 | 40.3 ± 1.8 | 1.3 |
| Kaempferide | 57.0 ± 2.52 | 39.3 ± 3.93 | 1.5 |
| Apigenin | 36.6 ± 1.72 | 20.3 ± 1.68 | 1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, S.; Szepesi, Á.; Tran-Nguyen, V.-K.; Sarkadi, B.; Német, K.; Falson, P.; Di Pietro, A.; Szakács, G.; Boumendjel, A. Synthesis and Anticancer Cytotoxicity of Azaaurones Overcoming Multidrug Resistance. Molecules 2020, 25, 764. https://doi.org/10.3390/molecules25030764
Tóth S, Szepesi Á, Tran-Nguyen V-K, Sarkadi B, Német K, Falson P, Di Pietro A, Szakács G, Boumendjel A. Synthesis and Anticancer Cytotoxicity of Azaaurones Overcoming Multidrug Resistance. Molecules. 2020; 25(3):764. https://doi.org/10.3390/molecules25030764
Chicago/Turabian StyleTóth, Szilárd, Áron Szepesi, Viet-Khoa Tran-Nguyen, Balázs Sarkadi, Katalin Német, Pierre Falson, Attilio Di Pietro, Gergely Szakács, and Ahcène Boumendjel. 2020. "Synthesis and Anticancer Cytotoxicity of Azaaurones Overcoming Multidrug Resistance" Molecules 25, no. 3: 764. https://doi.org/10.3390/molecules25030764
APA StyleTóth, S., Szepesi, Á., Tran-Nguyen, V.-K., Sarkadi, B., Német, K., Falson, P., Di Pietro, A., Szakács, G., & Boumendjel, A. (2020). Synthesis and Anticancer Cytotoxicity of Azaaurones Overcoming Multidrug Resistance. Molecules, 25(3), 764. https://doi.org/10.3390/molecules25030764