
Tricyclic Nucleobase Analogs and Their Ribosides as Substrates and Inhibitors of Purine-Nucleoside Phosphorylases III. Aminopurine Derivatives

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
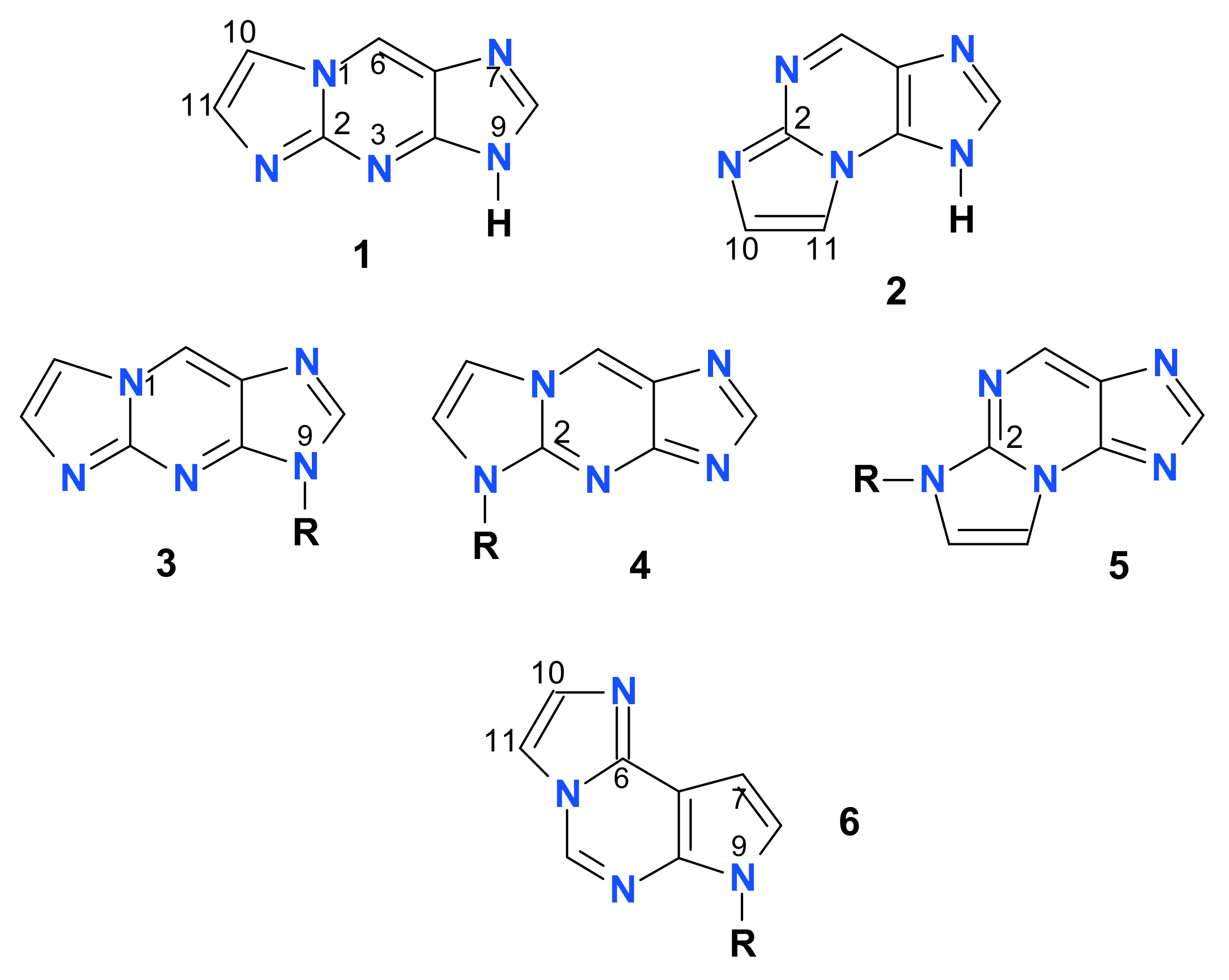
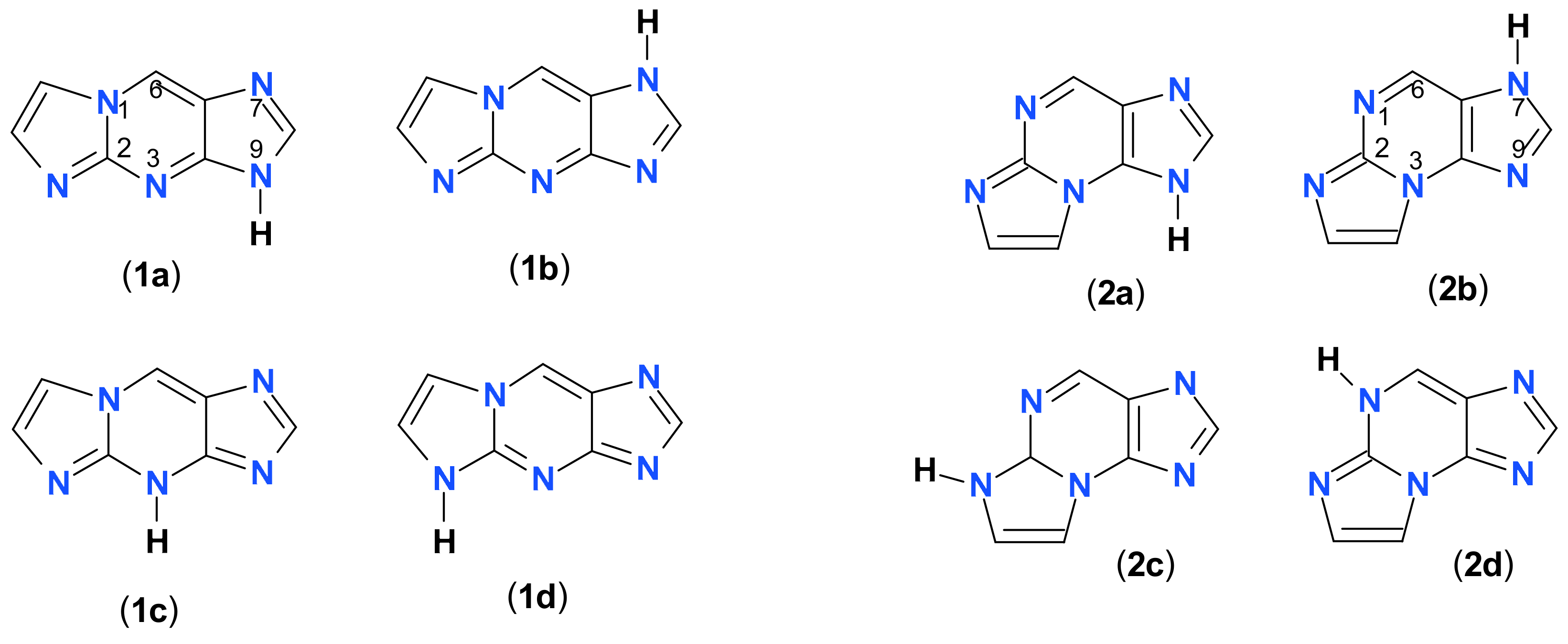
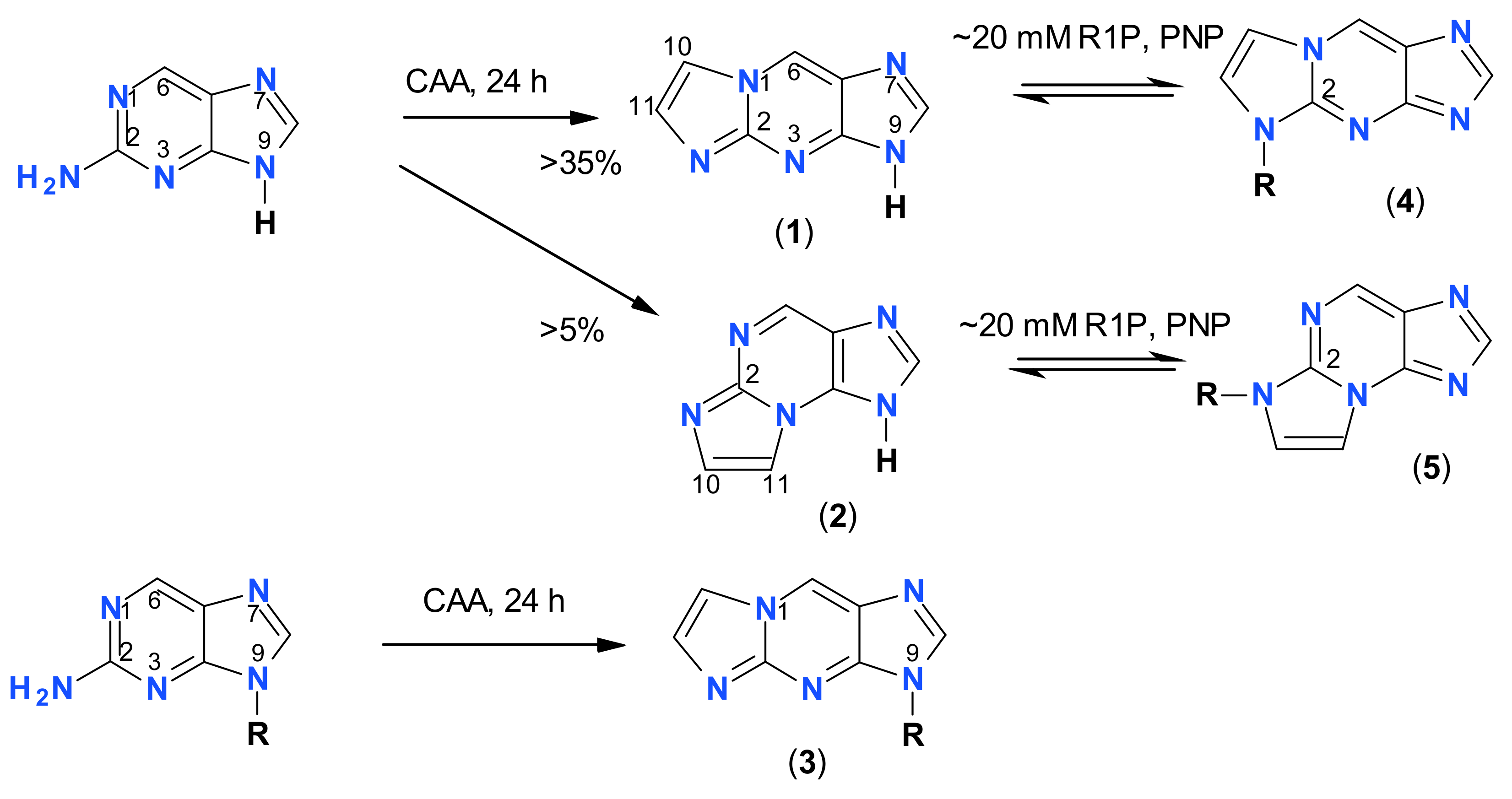
2.1. Reaction of Chloroacetaldehyde with 2-Aminopurine and Its Riboside
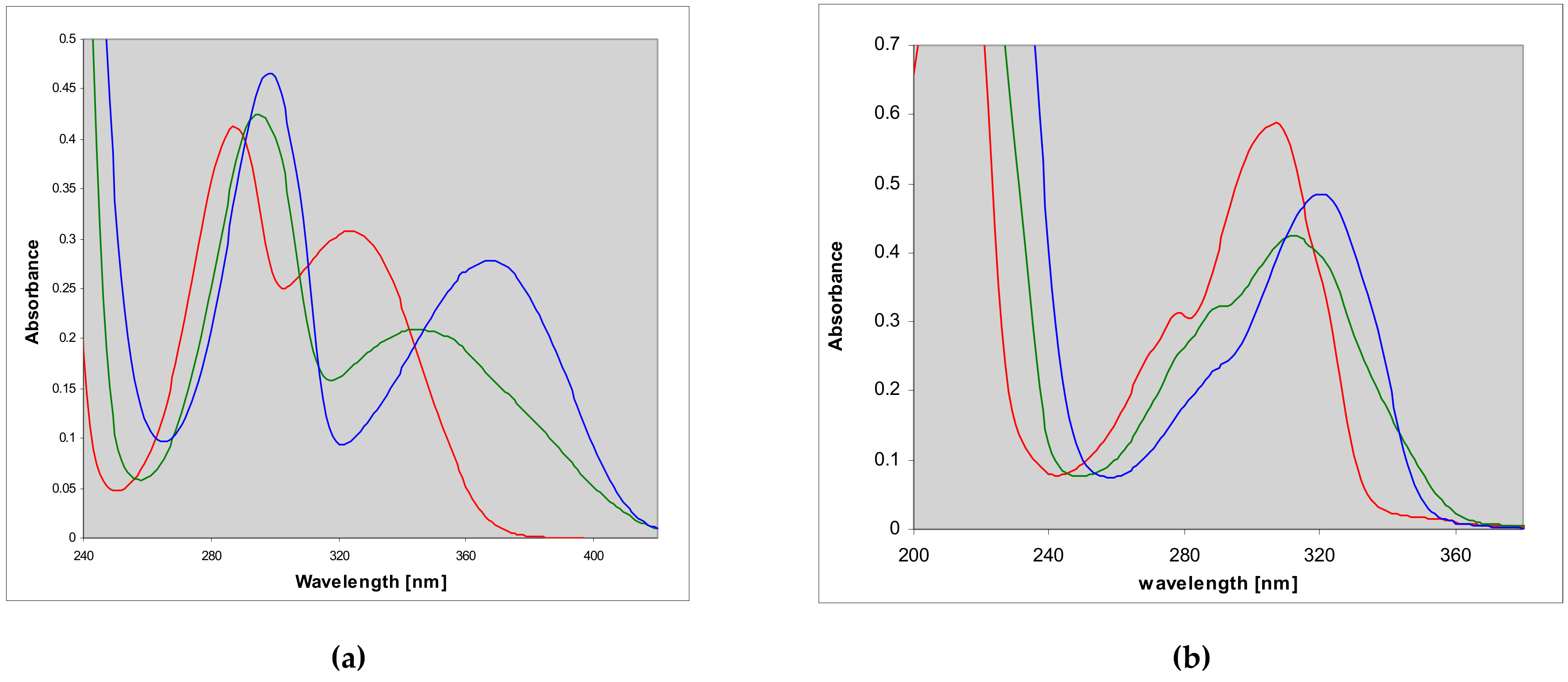
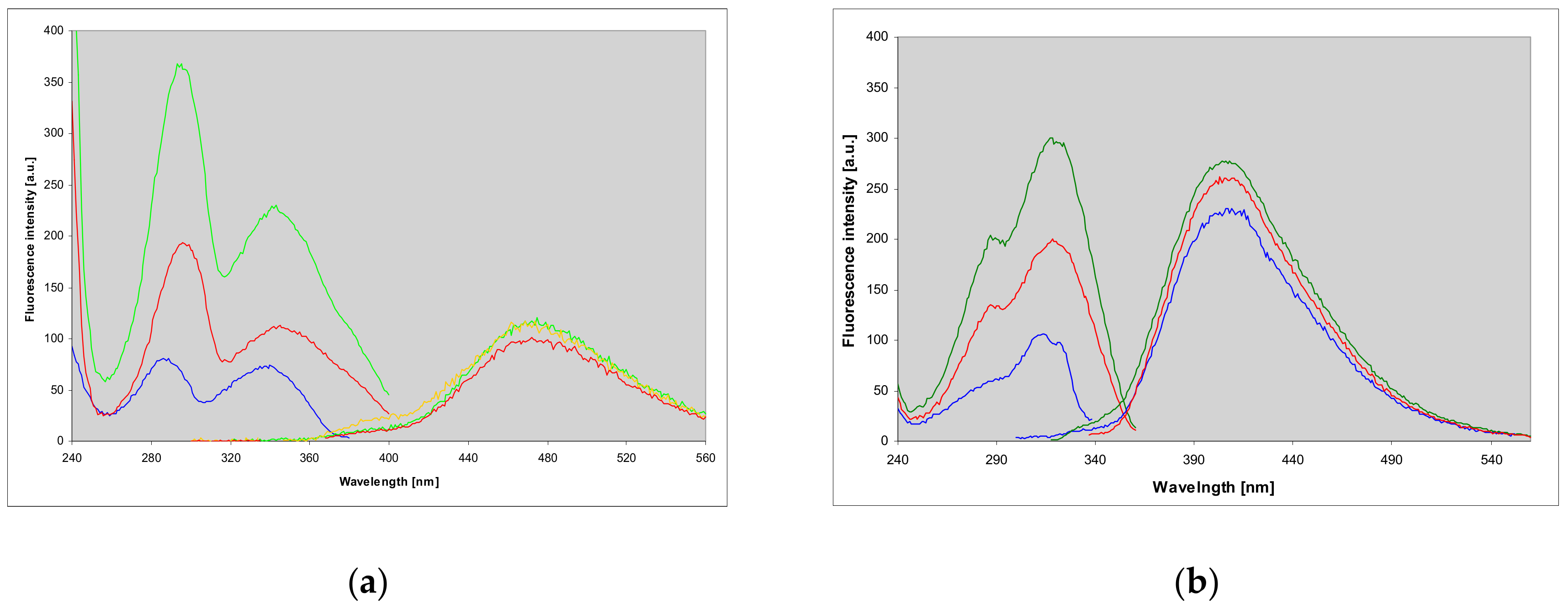
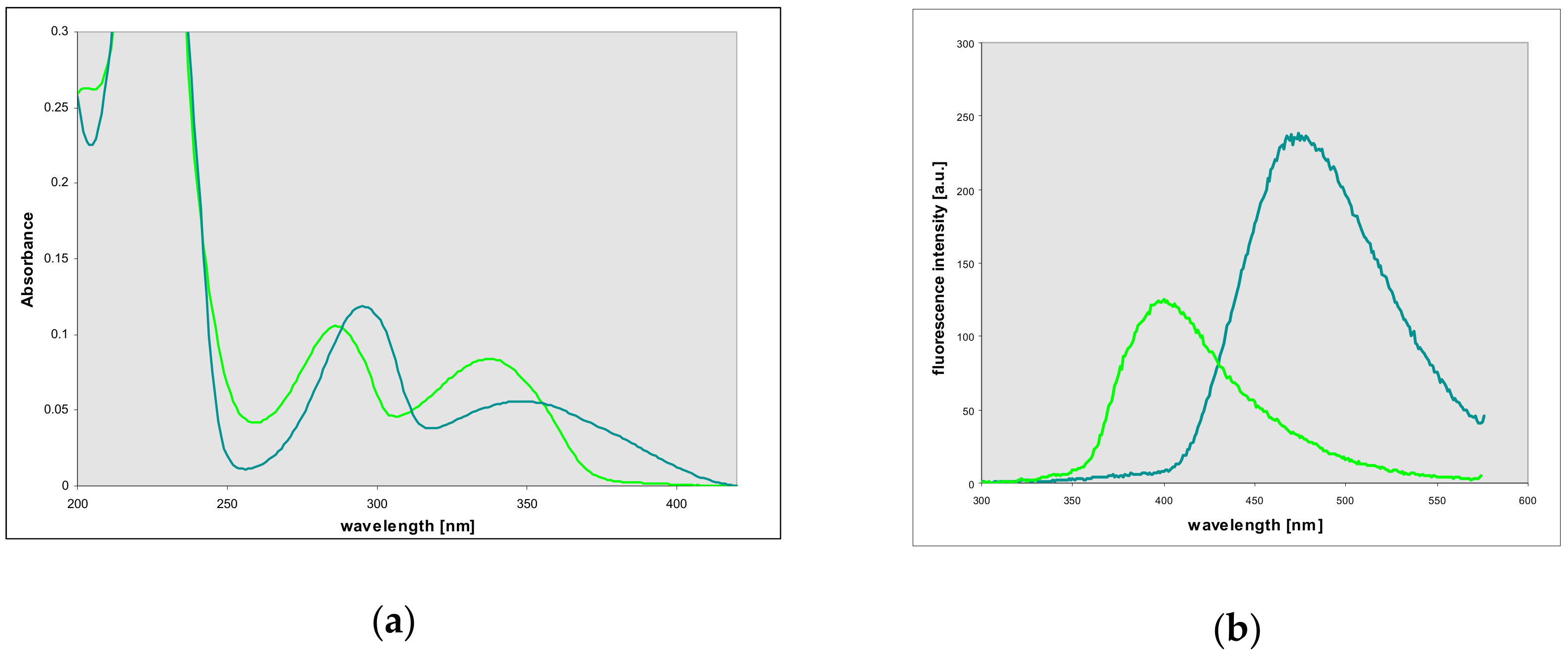
2.2. Properties of Two Isomers of Etheno-2-Aminopurine
2.3. Enzymatic Ribosylation of the Etheno-2-Aminopurine Isomers Using Various Forms of PNP
2.4. Properties and Identification of the Enzymatically Produced Ribosides
2.5. Phosphorolysis of The Ribosides with Various Forms of PNP
2.6. Properties of 1,N2-Etheno-Tubercidine
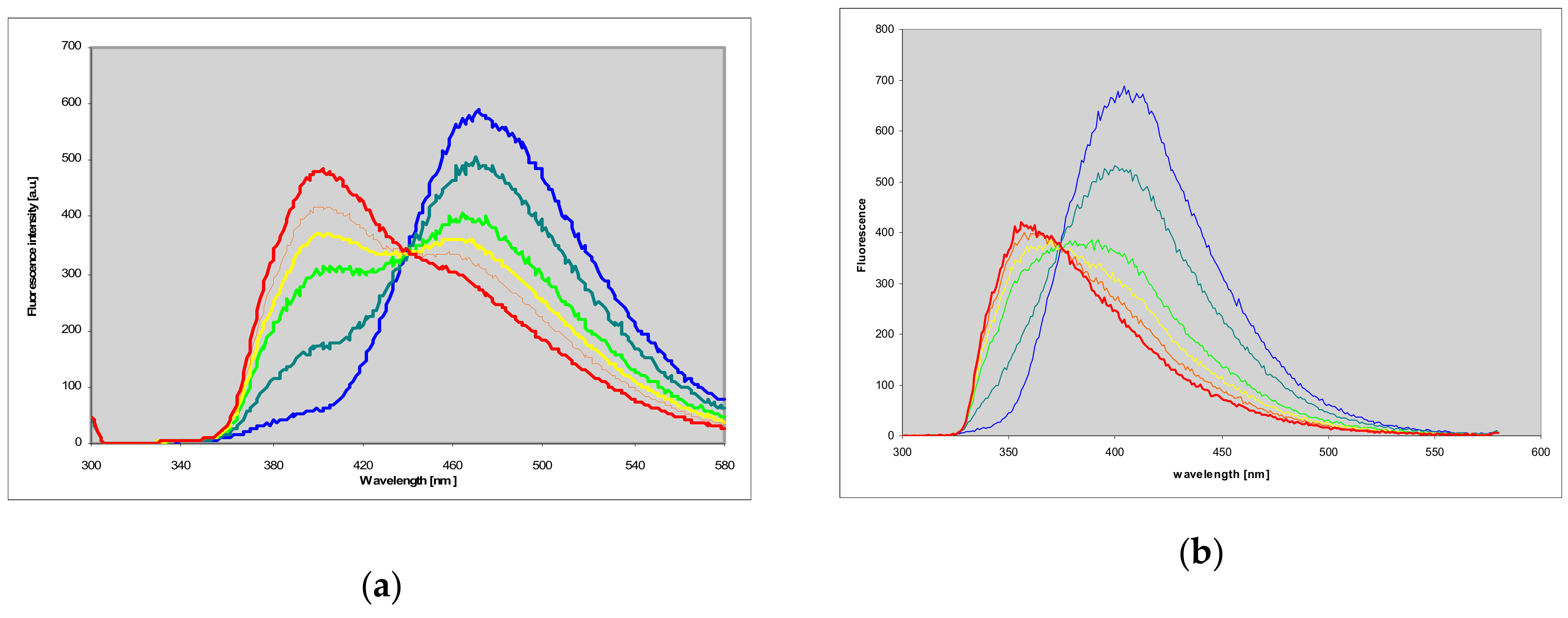
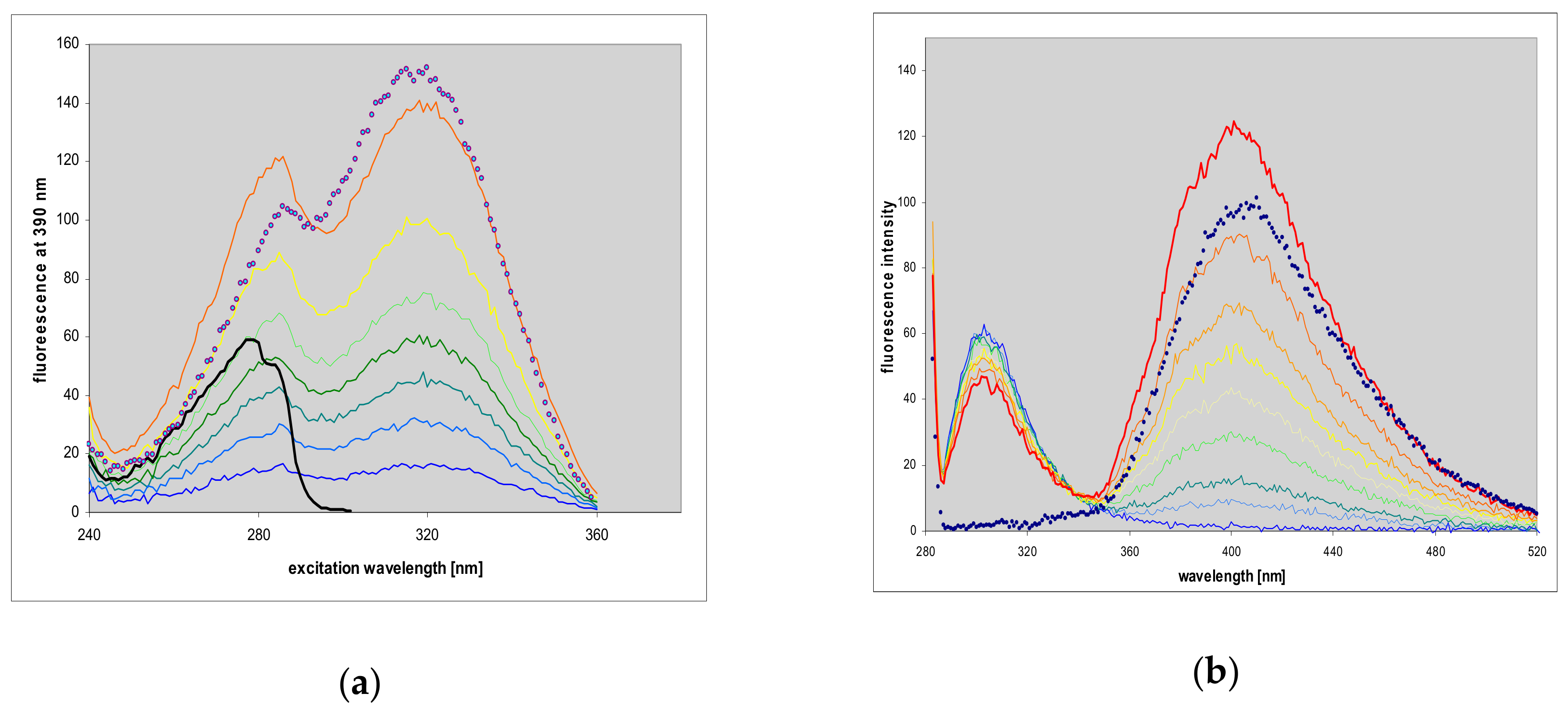
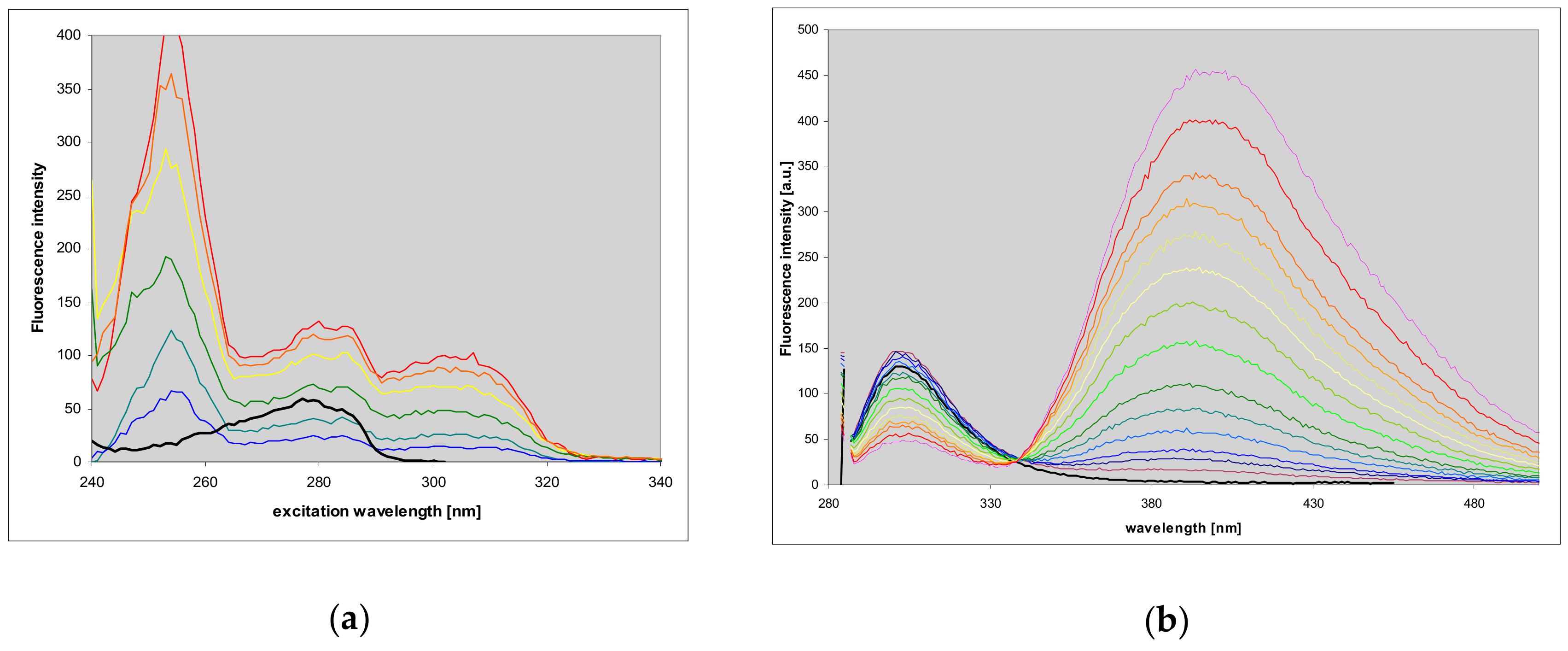
2.7. Fluorescence of Enzyme-Ligand Complexes
3. Discussion
3.1. Fluorescent Isomers of the Etheno-2-Aminopurine
3.2. Enzymatic Syntheses of the Tri-Cyclic Ribosides and Their Properties
3.3. Fluorescent Complexes
4. Materials and Methods
4.1. Chemicals
4.2. Spectral Measurements
4.3. Mass Spectrometry
4.4. Enzymes and Enzymatic Reactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leonard, N.J. Adenylates: Bound and unbound. Biopolymers 1985, 24, 9–28. [Google Scholar] [CrossRef]
- Leonard, N.J. Etheno-substituted nucleotides and coenzymes: Fluorescence and biological activity. Crit. Rev. Biochem. 1984, 15, 125–199. [Google Scholar] [CrossRef] [PubMed]
- Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent analogs of biomolecular building blocks: Design, properties, and applications. Chem. Rev. 2010, 110, 2579–2619. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chan, K.M.; Kool, E.T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. [Google Scholar] [CrossRef]
- Kauffman, R.F.; Lardy, H.A.; Barrio, J.R.; Barrio, C.G.; Leonard, N.J. Dimensional probes of enzyme binding-sites of adenine-nucleotides-interaction of lin-benzo-adenosine 5′-diphosphate and triphosphate with mitochondrial ATP-synthetase, purified ATPase, and adenine-nucleotide carrier. Biochemistry 1978, 17, 3686–3692. [Google Scholar] [CrossRef] [PubMed]
- Scopes, D.I.C.; Barrio, J.R.; Leonard, N.J. Defined dimensional changes in enzyme cofactors-fluorescent stretched-out analogs of adenine-nucleotides. Science 1977, 195, 296–298. [Google Scholar] [CrossRef]
- Golankiewicz, B.; Ostrowski, T. Tricyclic nucleoside analogues as antiherpes agents. Antivir Res. 2006, 71, 134–140. [Google Scholar]
- Jahnz-Wechmann, Z.; Framski, G.R.; Januszczyk, P.A.; Boryski, J. Bioactive fused heterocycles: Nucleoside analogs with an additional ring. Eur. J. Med. Chem. 2015, 97, 388–396. [Google Scholar] [CrossRef]
- Jahnz-Wechmann, Z.; Framski, G.R.; Januszczyk, P.A.; Boryski, J. Base-modified nucleosides: Etheno derivatives. Front. Chem. 2016, 4, 19–29. [Google Scholar] [CrossRef]
- Singer, B.; Kuśmierek, J.T. Chemical mutagenesis. Annu. Rev. Biochem. 1982, 51, 655–693. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair and mutagenesis. Env. Mol Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Bzowska, A.; Kulikowska, E.; Shugar, D. Purine nucleoside phosphorylases: Properties, functions, and clinical aspects. Pharmacol. Therap. 2000, 88, 349–425. [Google Scholar] [CrossRef]
- El Kouni, M.H. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol. Ther. 2003, 99, 283–309. [Google Scholar] [CrossRef]
- Grunebaum, E.; Cohen, A.; Roifman, C.M. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr. Op. Allergy Clin. Immunol. 2013, 13, 630–638. [Google Scholar] [CrossRef]
- Parker, W.B.; Sorscher, E.J. Use of E. coli purine nucleoside phosphorylase in the treatment of solid tumors. Curr. Pharm Des. 2017, 23, 7003–7024. [Google Scholar] [CrossRef]
- Doskocil, J.; Holy, A. Specificity of purine nucleoside phosphorylase from Escherichia coli. Collect. Czech. Chem. Commun. 1977, 42, 370–383. [Google Scholar] [CrossRef]
- Wierzchowski, J.; Stachelska-Wierzchowska, A.; Wielgus-Kutrowska, B.; Bzowska, A. 1,N6-ethenoadenine and other fluorescent nucleobase analogues as substrates for purine-nucleoside phosphorylases: Spectroscopic and kinetic studies. Curr. Pharm. Des. 2017, 23, 6972–6990. [Google Scholar]
- Mikhailopulo, A.I.; Miroshnikov, A.I. Biologically important nucleosides: Modern trends in biotechnology and application. Mendeleev Comm. 2011, 21, 57–68. [Google Scholar] [CrossRef]
- Kamel, S.; Yehia, H.; Neubauer, P.; Wagner, A. 1 Enzymatic Synthesis of Nucleoside Analogues by Nucleoside Phosphorylases 1. In Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives; Fernandez-Lucas, J., Camarasa-Rius, M.J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; Chapter 1; pp. 1–28. [Google Scholar]
- Yehia, H.; Kamel, S.; Paulick, K.; Neubauer, P.; Wagner, A. Substrate Spectra of Nucleoside Phosphorylases and their Potential in the Production of Pharmaceutically Active Compounds. Curr. Pharm. Des. 2017, 23, 6913–6935. [Google Scholar] [CrossRef]
- Stachelska-Wierzchowska, A.; Wierzchowski, J.; Bzowska, A.; Wielgus-Kutrowska, B. Tricyclic nitrogen base, 1,N6-Ethenoadenine, and its Ribosides, as Substrates for Purine-Nucleoside Phosphorylases: Spectroscopic and Kinetic Studies. Nucleosides Nucleotides Nucleic Acids 2018, 37, 89–101. [Google Scholar] [CrossRef]
- Stachelska-Wierzchowska, A.; Wierzchowski, J.; Wielgus-Kutrowska, B.; Bzowska, A. Tri-cyclic nucleobase analogs and their ribosides as substrates of purine-nucleoside phosphorylases. II. Guanine and isoguanine derivatives. Molecules 2019, 24, 1493. [Google Scholar] [CrossRef] [PubMed]
- Virta, P.; Holmstrom, T.; Roslund, M.U.; Mattjus, P.; Kronberg, L.; Sjoholm, R. New nucleoside analogs from 2-amino-9-(β-d-ribofuranosyl)purine. Org. Biomol. Chem. 2004, 2, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Bhat, G.A.; Schram, K.H.; Townsend, L.B. The synthesis of certain fluorescent imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine nucleoside derivatives related to epsilon-adenosine. J. Carbohydr. Nucleosides Nucleotides 1980, 7, 333–345. [Google Scholar]
- Seela, F.; Schweinberger, E.; Kuiying, X.; Sirivolu, V.R.; Rosemeyer, H.; Becker, E.-M. 1,N6-Etheno-2′-deoxytubercidin and pyrrolo-C: Synthesis, base pairing, and fluorescence properties of 7-deazapurine nucleosides and oligonucleotides. Tetrahedron 2007, 63, 3471–3482. [Google Scholar] [CrossRef]
- Evans, G.B.; Schramm, V.L.; Tyler, P.C. The Immucillins: Design, synthesis and application of transition-state analogues. Curr. Med. Chem. 2015, 22, 3897–3909. [Google Scholar] [CrossRef]
- Schramm, V.L. Transition states, analogues, and drug development. Acs Chem. Biol. 2013, 8, 71–81. [Google Scholar]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids–IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. J. Biomol. Nmr 1998, 12, 1–23. [Google Scholar] [CrossRef]
- Stoeckler, J.D.; Poirot, A.F.; Smith, R.M.; Parks, R.E., Jr.; Ealick, S.E.; Takabayashi, K.; Erion, M.D. Purine nucleoside phosphorylase. 3. Reversal of purine base specificity by site-directed mutagenesis. Biochemistry 1997, 36, 11749–11756. [Google Scholar] [CrossRef]
- Kamel, S.; Thiele, I.; Neubauer, P.; Wagner, A. Thermophilic nucleoside phosphorylases: Their properties, characteristics and applications. Biochim. Biopys. Acta-Proteins Proteom. 2020, 1868, 140304. [Google Scholar] [CrossRef]
- Kierdaszuk, B.; Modrak-Wójcik, A.; Wierzchowski, J.; Shugar, D. Formycin A and its N-methyl analogues, specific inhibitors of E. coli purine nucleoside phosphorylase: Induced tautomeric shift on binding to enzyme, and enzyme → ligand fluorescence resonance energy transfer. Biochim. Biophys. Acta 2000, 1476, 109–128. [Google Scholar] [CrossRef]
- Włodarczyk, J.; Galitonov, G.S.; Kierdaszuk, B. Identification of the tautomeric form of formycin A in its complex with Escherichia coli purine nucleoside phosphorylase based on the effect of enzyme-ligand binding on fluorescence and phosphorescence. Eur. Biophys. J. Biophys. Lett. 2004, 33, 377–385. [Google Scholar] [CrossRef]
- Patra, A.; Su, Y.; Zhang, Q.Q.; Johnson, K.M.; Guengerich, F.P.; Egli, M. Structural and Kinetic Analysis of Miscoding Opposite the DNA Adduct 1,N6-Ethenodeoxyadenosine by Human Translesion DNA Polymerase. J. Biol. Chem. 2016, 291, 14134–14145. [Google Scholar] [CrossRef]
- Calabretta, A.; Leumann, C.J. Base pairing and miscoding properties of 1,N6-ethenoadenine- and 3,N4-ethenocytosine-containing RNA oligonucleotides. Biochemistry 2013, 52, 1990–1997. [Google Scholar] [CrossRef]
- Shugar, D.; Psoda, A. Tautomerism of purines and pyrimidines. In Landoldt-Bornstein Numerical Data and Functional Relationships in Science and Technology; Springer: Berlin/Heidelberg, Germany, 1990; Volume 1, pp. 308–344. [Google Scholar]
- Shcherbakova, I.; Elguero, J.; Katritzky, A.R. Tautomerism of heterocycles: Condensed five-six, five-five, and six-six ring systems with heteroatoms in both rings. Adv. Heterocycl. Chem. 2000, 77, 51–113. [Google Scholar]
- Wierzchowski, J.; Antosiewicz, J.M.; Shugar, D. 8-Azapurines as isosteric purine fluorescent probes for nucleic acid and enzymatic research. Mol. Biosyst. 2014, 10, 2756–2774. [Google Scholar] [CrossRef]
- Wierzchowski, J. Excited-state proton transfer and phototautomerism in nucleobase and nucleoside analogs: A mini-review. Nucleosides Nucleotides Nucleic Acids 2014, 33, 626–644. [Google Scholar] [CrossRef]
- Stachelska-Wierzchowska, A.; Wierzchowski, J.; Wielgus-Kutrowska, B.; Bzowska, A.; Mikleuševic, G. Enzymatic synthesis of highly fluorescent 8-azapurine ribosides using purine-nucleoside phosphorylase reverse reaction: Variable ribosylation sites. Molecules 2013, 18, 12587–12598. [Google Scholar] [CrossRef]
- Stachelska-Wierzchowska, A.; Wierzchowski, J.; Bzowska, A.; Wielgus-Kutrowska, B. Site-selective ribosylation of fluorescent nucleobase analogs using purine-nucleoside phosphorylase as a catalyst: Effects of point mutations. Molecules 2016, 21, 44. [Google Scholar] [CrossRef]
- Narczyk, M.; Bertoša, B.; Papa, L.; Vuković, V.; Leščić-Ašler, I.; Wielgus-Kutrowska, B.; Bzowska, A.; Luić, M.; Štefanić, Z. Helicobacter pylori purine nucleoside phosphorylase shows new distribution patterns of open and closed active site conformations and unusual biochemical features. FEBS J. 2018, 285, 1305–1325. [Google Scholar] [CrossRef]
- Bzowska, A.; Luić, M.; Schroder, W.; Shugar, D.; Saenger, W.; Koellner, G. Calf spleen purine nucleoside phosphorylase: Purification, sequence and crystal structure of its complex with N(7)-acycloguanosine inhibitor. FEBS Lett. 1995, 367, 214–218. [Google Scholar] [CrossRef]
- Bzowska, A.; Kulikowska, E.; Poopeiko, N.E.; Shugar, D. Kinetics of phosphorolysis of 3-(β-d-ribofuranosyl)adenine and 3-(β-d-ribofuranosyl)hypoxanthine, non-conventional substrates of purine--nucleoside phosphorylase. Eur. J. Biochem. 1996, 239, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Stachelska-Wierzchowska, A.; Wierzchowski, J. Fluorescence of the tri-cyclic adenine and isoguanine derivatives and their ribosides: Possible analytical applications. Acta Biochim. Polon. 2019, 66, 2885. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.J.T. Purine nucleoside phosphorylase-kinetic mechanism of the enzyme from calf spleen. J. Biol. Chem. 1992, 267, 7342–7351. [Google Scholar] [PubMed]
- Wierzchowski, J.; Bzowska, A.; Stępniak, K.; Shugar, D. Interactions of Calf Spleen Purine Nucleoside Phosphorylase with 8-Azaguanine, and a Bisubstrate Analogue Inhibitor: Implications for the Reaction Mechanism. Z Nat. C J Biosci. 2004, 59, 713–725. [Google Scholar] [CrossRef]
- Wierzchowski, J.; Stępniak, K.; Bzowska, A.; Shugar, D. Spectroscopic and kinetic studies of interactions of calf spleen purine nucleoside phosphorylase with 8-azaguanine and its 9-(2-phosphonylmethoxyethyl) derivative. Nucleosides Nucleotides Nucleic Acids 2005, 24, 459–464. [Google Scholar] [CrossRef]
- Bzowska, A. Calf spleen purine nucleoside phosphorylase: Complex kinetic mechanism, hydrolysis of 7-methylguanosine, and oligomeric state in solution. Biochim. Biopys. Acta 2002, 1596, 293–317. [Google Scholar] [CrossRef]
- Krentitsky, T.A.; Tuttle, J.V. Correlation of substrate stabilization patterns with proposed mechanisms for three nucleoside phosphorylases. Biochim. Biophys. Acta 1982, 703, 247–249. [Google Scholar] [CrossRef]
- Wierzchowski, J.; Mędza, G.; Szabelski, M.; Stachelska-Wierzchowska, A. Properties of 2,6-diamino-8-azapurine, a highly fluorescent purine analog and its N-alkyl derivatives: Tautomerism and excited-state proton transfer reactions. J. Photochem. Photobiol. A 2013, 265, 49–57. [Google Scholar] [CrossRef]
- Koos, M.R.M.; Kummerlöwe, G.; Kaltschnee, L.; Thiele, C.M.; Luy, B. CLIP-COSY: A Clean In-Phase Experiment for the Rapid Acquisition of COSY-type Correlations. Angew. Chem. Internat. Ed. 2016, 55, 7655–7659. [Google Scholar] [CrossRef]
- Davis, A.L.; Keeler, J.; Laue, E.D.; Moskau, D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 1992, 98, 207–216. [Google Scholar] [CrossRef]
- Saurí, J.; Parella, T.; Espinosa, J.F. CLIP-HSQMBC: Easy measurement of small proton–carbon coupling constants in organic molecules. Org. Biomol. Chem. 2013, 11, 4473. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Pure Appl. Chem. 2008, 80, 59–84. [Google Scholar] [CrossRef]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2000. [Google Scholar]
- Mikleuševic, G.; Štefanic, Z.; Narczyk, M.; Wielgus-Kutrowska, B.; Bzowska, A.; Luic, M. Validation of the catalytic mechanism of Escherichia coli purine nucleoside phosphorylase by structural and kinetic studies. Biochimie 2011, 93, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Breer, K.; Girstun, A.; Wielgus-Kutrowska, B.; Staron, K.; Bzowska, A. Overexpression, purification and characterization of functional calf purine nucleoside phosphorylase (PNP). Protein Expr. Purif. 2008, 61, 122–130. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1,N2-etheno-2-aminopurine (1), N2,3-etheno-2-aminopurine (2) are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pKa | form (pH) | UV Absorption λmax (nm) εmax | Fluorescence λmax (nm) φ | τ [ns] |
|---|---|---|---|---|---|
| 1,N2-etheno-2-aminopurine (1) | 5.6; 8.2 | n(7) | 348 2560 | 472 0.18 | 6.9; 10.3 |
| c(3) | 285 5050 | 467 ~0.04 | Nd | ||
| a(11) | 367 3500 | 463 ~0.40 | Nd | ||
| N2-β-d-Ribofuranoside (4) | 6.3 | n(8) | 338 4260 | 406 0.73 | Nd |
| c(3) | 326 Nd | 406 0.73 | Nd | ||
| N9-β-d-Ribofuranoside (3) | - | n(8) | 295 5700 1 | 463 0.14 | Nd |
| c(3) | 318 6120 | 465 0.06 | Nd | ||
| N2,3-etheno-2-aminopurine (2) | 5.0; ~9.5 | n(7) | 315 ~5600 | 406 0.73 | 3.8; 8.5 |
| c(2.5) | 306 ~7700 | 455 ~0.30 | Nd | ||
| a(12) | 320 ~6700 | 390 ~0.40 | Nd | ||
| N2-β-d-Ribofuranoside (5) | 6.15 | n(8) | 315 ~10,700 | 357 0.29 | 2.15 |
| c(3) | 310 ~9400 | 373 ~0.15 | Nd | ||
| 1,N6-ethenotubercidine (6) | 5.2 | n(7) | 288 5800 2 | 415 0.53 2 | Nd |
| c(3) | 281 8700 | 415 Nd | Nd |
| Nucleus | 1,N2-ε2AP (1) | 1,N2-ε2AP- N9-ribofuranoside (3) | 1,N2-ε2AP- N2-ribofuranoside (4) | N2,3-ε2AP (2) | N2,3-ε2AP- N2-ribofuranoside (5) |
|---|---|---|---|---|---|
| H6 | 9.445 | 9.541 | 9.246 | 8.879 | 8.992 |
| H8 | 8.513 | 8.768 | 8.403 | 8.408 | 8.067 |
| H10 1 | 7.624 | 7.661 | 7.987 | 8.115 | 8.356 |
| H11 1 | 7.895 | 7.931 | 8.092 | 7.719 | 8.351 |
| H1′ | -- | 5.969 | 6.212 | -- | 6.286 |
| H2′ | -- | 4.617 | 4.418 | -- | 4.415 |
| OH2′ | -- | 5.531 | 5.592 | -- | 5.612 |
| H3′ | -- | 4.189 | 4.152 | -- | 4.177 |
| OH3′ | -- | 5.217 | 5.343 | -- | 5.249 |
| H4′ | -- | 3.962 | 3.985 | -- | 4.010 |
| H5′/H5′′ | -- | 3.581/3.690 | 3.601/3.694 | -- | 3.630/3.733 |
| OH5′ | -- | 5.090 | 5.162 | -- | 5.210 |
| C2 | 147.470 | 146.868 | 141.394 | 147.740 | 139.817 |
| C4 | 152.793 | 150.122 | 165.789 | 143.330 | 147.377 |
| C5 | 126.637 | 127.994 | 134.105 | 116.873 | 132.921 |
| C8 | 149.972 | 149.029 | 167.161 | 142.398 | 155.724 |
| C10 1 | 133.727 | 134.305 | 111.629 | 107.780 | 108.777 |
| C11 1 | 110.641 | 111.080 | 118.837 | 133.172 | 119.946 |
| C6 | 125.239 | 127.102 | 123.368 | 138.144 | 144.668 |
| C1′ | -- | 87.495 | 88.07 | -- | 89.037 |
| C2′ | -- | 73.529 | 74.439 | -- | 74.969 |
| C3′ | -- | 70.568 | 70.471 | -- | 70.151 |
| C4′ | -- | 85.706 | 85.761 | -- | 85.798 |
| C5′ | -- | 61.603 | 61.443 | -- | 61.059 |
| Substrate | Enzyme (PNP Source) | Km (μM) | Vmax (Relative) 1 | Ribosylation Site |
|---|---|---|---|---|
| 1,N2-etheno-2-aminopurine (1) | calf-wt | nr | < 0.1 | - |
| “ | calf—N243D | nd | traces | nd |
| “ | E. coli-wt | <10 | ~1 | mostly N2 |
| N2,3-etheno-2-aminopurine (2) | calf-wt | ~8 | ~5 | predominantly N2 |
| “ | calf—N243D | 110 | 29 | predominantly N2 |
| “ | E. coli-wt | 11 | 20 | predominantly N2 |
| “ | E. coli—D204N | 12 | 30 | predominantly N2 |
| “ | E. coli—S90A | ~20 | 23 | predominantly N2 |
| Substrate | Enzyme 2 | Km (μM) | Vmax (relative) 1 |
|---|---|---|---|
| 1,N2-etheno-2-aminopurine-N9-β-d-riboside (3) | E. coli PNP-wt | nr | <1 |
| - | calf PNP-wt | nr | <0.1 |
| 1,N2-etheno-2-aminopurine-N2-β-d-riboside (4) | E. coli PNP-wt | 47 | 4 |
| - | calf PNP-wt | nr | <0.1 |
| N2,3-etheno-2-aminopurine-N2-β-d-riboside (5) | E. coli PNP-wt | ~ 20 | 115 |
| - | calf PNP-wt | 4.6 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachelska-Wierzchowska, A.; Wierzchowski, J.; Górka, M.; Bzowska, A.; Stolarski, R.; Wielgus-Kutrowska, B. Tricyclic Nucleobase Analogs and Their Ribosides as Substrates and Inhibitors of Purine-Nucleoside Phosphorylases III. Aminopurine Derivatives. Molecules 2020, 25, 681. https://doi.org/10.3390/molecules25030681
Stachelska-Wierzchowska A, Wierzchowski J, Górka M, Bzowska A, Stolarski R, Wielgus-Kutrowska B. Tricyclic Nucleobase Analogs and Their Ribosides as Substrates and Inhibitors of Purine-Nucleoside Phosphorylases III. Aminopurine Derivatives. Molecules. 2020; 25(3):681. https://doi.org/10.3390/molecules25030681
Chicago/Turabian StyleStachelska-Wierzchowska, Alicja, Jacek Wierzchowski, Michał Górka, Agnieszka Bzowska, Ryszard Stolarski, and Beata Wielgus-Kutrowska. 2020. "Tricyclic Nucleobase Analogs and Their Ribosides as Substrates and Inhibitors of Purine-Nucleoside Phosphorylases III. Aminopurine Derivatives" Molecules 25, no. 3: 681. https://doi.org/10.3390/molecules25030681
APA StyleStachelska-Wierzchowska, A., Wierzchowski, J., Górka, M., Bzowska, A., Stolarski, R., & Wielgus-Kutrowska, B. (2020). Tricyclic Nucleobase Analogs and Their Ribosides as Substrates and Inhibitors of Purine-Nucleoside Phosphorylases III. Aminopurine Derivatives. Molecules, 25(3), 681. https://doi.org/10.3390/molecules25030681