
In Silico Strategies in Tuberculosis Drug Discovery

 , ,
, ,
Abstract
1. Introduction
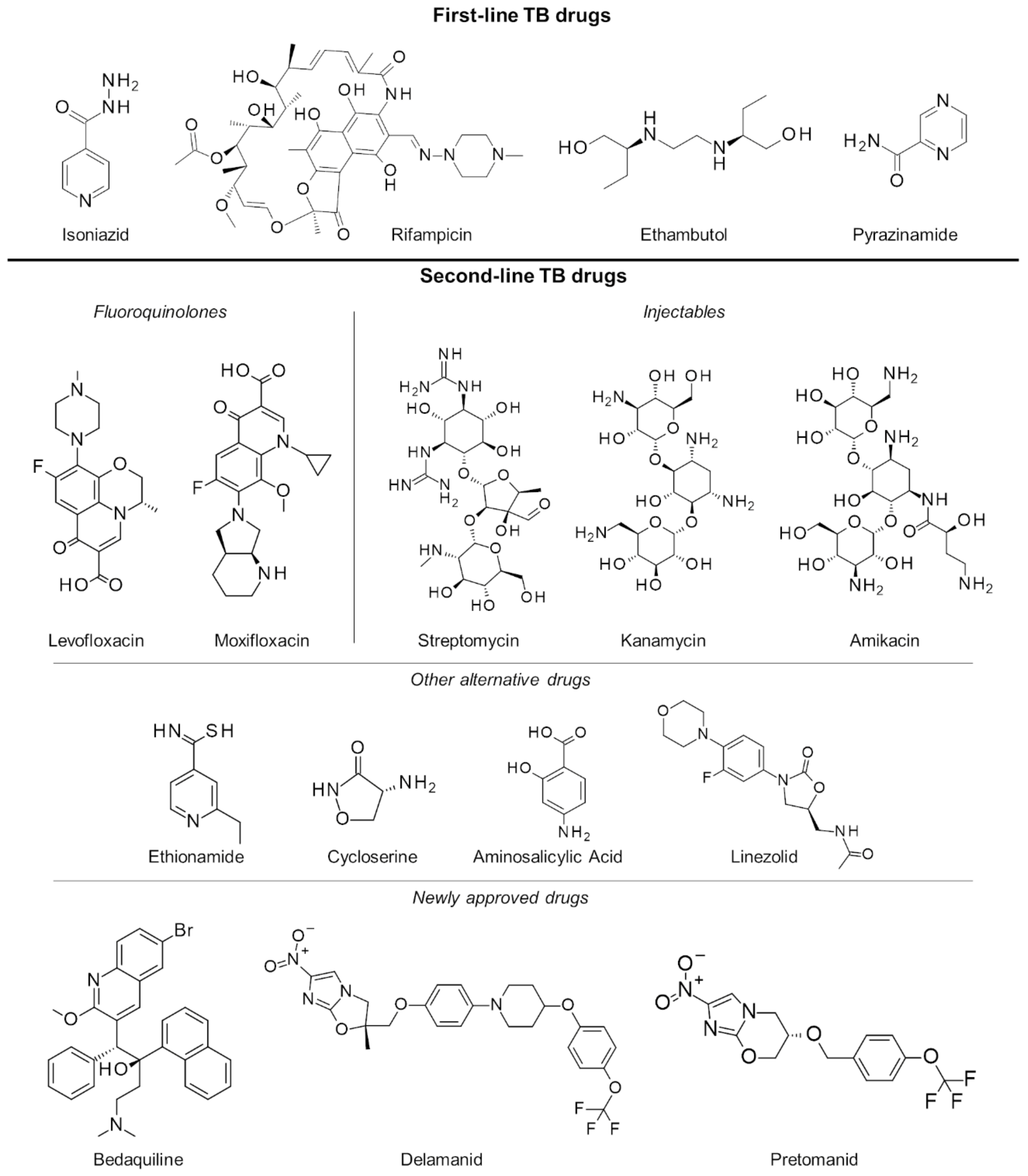
2. Current Tuberculosis Management
2.1. Latent Tuberculosis Infection
2.2. Active Drug-Sensitive Tuberculosis
2.3. Multiple and Extensively Drug-Resistant Tuberculosis
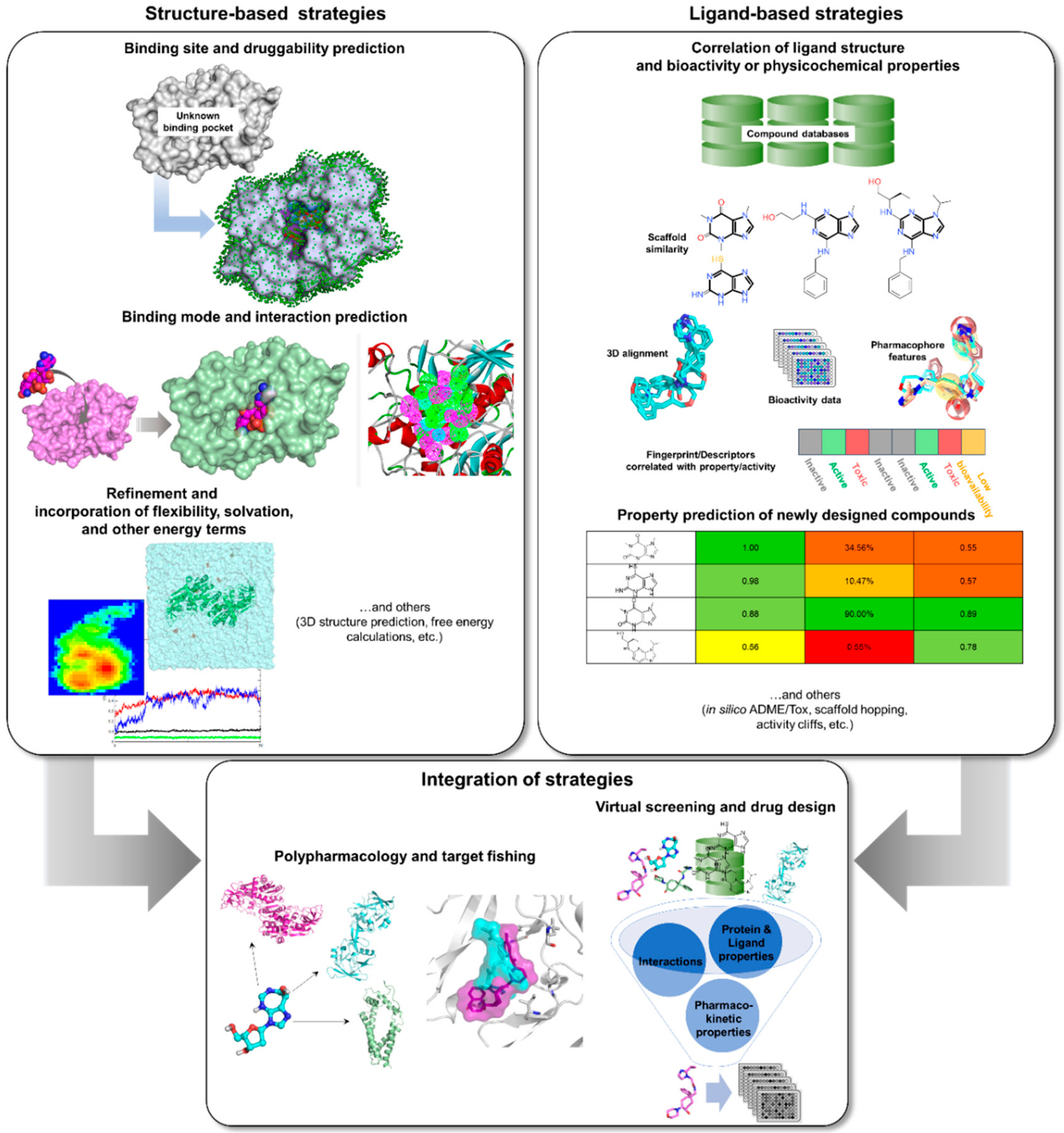
3. Rise of Computer-Aided Drug Design in TB Drug Discovery
3.1. Databases
3.2. Structure-Based Tools
3.2.1. Comparative Modeling, Binding Site Prediction, and Druggability
3.2.2. Pharmacophore Modeling and Molecular Docking
3.2.3. Molecular Dynamics
3.3. Ligand-Based Tools
3.3.1. Similarity-Based and Quantitative Structure-Activity/Property Relationship Methods
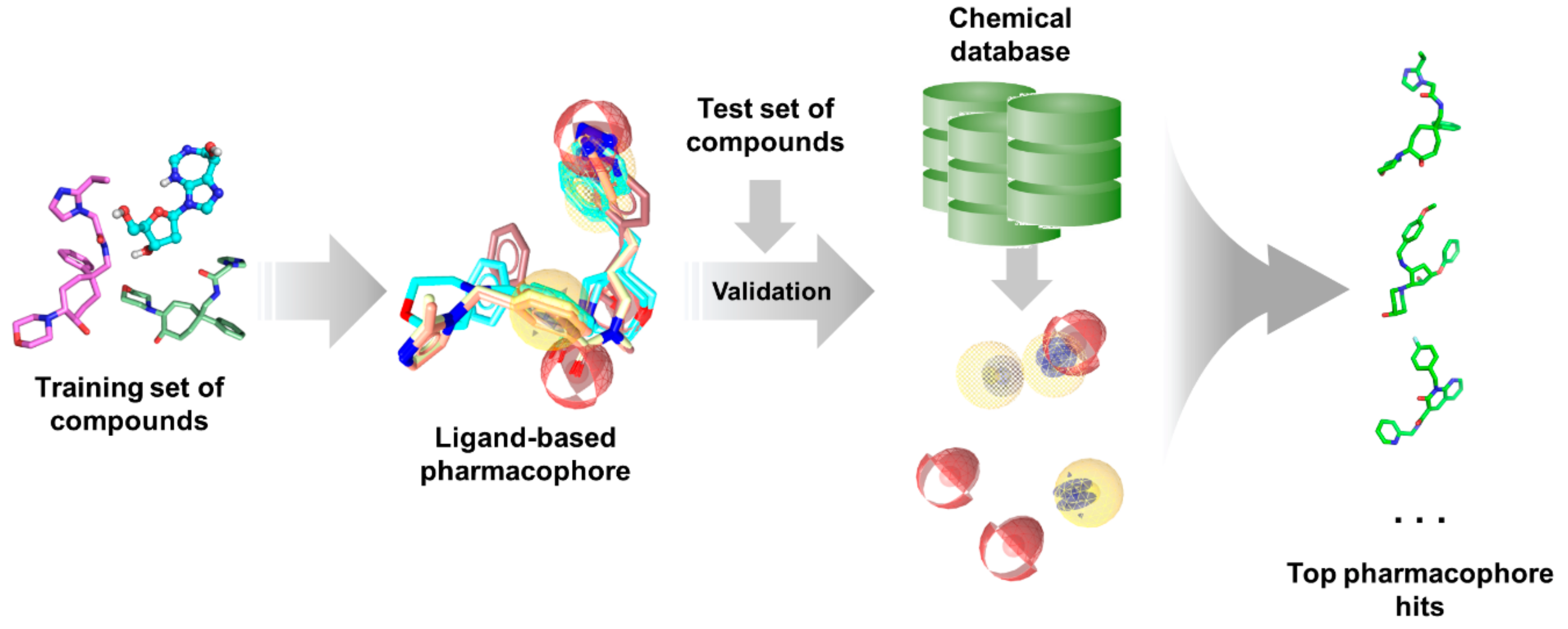
3.3.2. Ligand-Based Pharmacophore Modeling
3.3.3. Density Functional Theory
3.4. Integrated Tools
4. Edges and Pitfalls of In Silico Methods
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. [Google Scholar] [CrossRef] [PubMed]
- Havlir, D.V.; Getahun, H.; Sanne, I.; Nunn, P. Opportunities and challenges for HIV care in overlapping HIV and TB epidemics. JAMA 2008, 300, 423–430. [Google Scholar] [PubMed]
- Leung, C.C.; Yew, W.W.; Chan, C.K.; Chang, K.C.; Law, W.S.; Lee, S.N.; Tai, L.B.; Leung, E.C.; Au, R.K.; Huang, S.S.; et al. Smoking adversely affects treatment response, outcome and relapse in tuberculosis. Eur. Respir. J. 2015, 45, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, S.; Shield, K.D.; Roerecke, M.; Samokhvalov, A.V.; Lonnroth, K.; Rehm, J. Alcohol consumption as a risk factor for tuberculosis: Meta-analyses and burden of disease. Eur. Respir. J. 2017, 50, 1700216. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I. Diabetes and Tuberculosis. In Understanding the Host Immune Response against Mycobacterium tuberculosis Infection; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–21. [Google Scholar]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis infection. N. Engl. J. Med. 2015, 372, 2127–2135. [Google Scholar] [CrossRef]
- Miller, L.G.; Asch, S.M.; Yu, E.I.; Knowles, L.; Gelberg, L.; Davidson, P. A population-based survey of tuberculosis symptoms: How atypical are atypical presentations? Clin. Infect. Dis. 2000, 30, 293–299. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Tuberculosis Report 2019; World Health Organization (WHO): Geneva, Switzerland, 2019. [Google Scholar]
- World Health Organization (WHO). Guidelines on the Management of Latent Tuberculosis Infection; World Health Organization (WHO): Geneva, Switzerland, 2015. [Google Scholar]
- World Health Organization (WHO). Latent Tuberculosis infection: Updated and Consolidated Guidelines for Programmatic Management; World Health Organization (WHO): Geneva, Switzerland, 2018. [Google Scholar]
- Getahun, H.; Matteelli, A.; Abubakar, I.; Aziz, M.A.; Baddeley, A.; Barreira, D.; Den Boon, S.; Borroto Gutierrez, S.M.; Bruchfeld, J.; Burhan, E.; et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur. Respir. J. 2015, 46, 1563–1576. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Treatment of Tuberculosis: Guidelines; World Health Organization (WHO): Geneva, Switzerland, 2010. [Google Scholar]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195. [Google Scholar] [CrossRef]
- Volmink, J.; Garner, P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst. Rev. 2007, CD003343. [Google Scholar] [CrossRef]
- Horsburgh, C.R., Jr.; Barry, C.E., 3rd; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef]
- Saukkonen, J.J.; Cohn, D.L.; Jasmer, R.M.; Schenker, S.; Jereb, J.A.; Nolan, C.M.; Peloquin, C.A.; Gordin, F.M.; Nunes, D.; Strader, D.B.; et al. An official ATS statement: Hepatotoxicity of antituberculosis therapy. Am. J. Respir. Crit. Care Med. 2006, 174, 935–952. [Google Scholar] [CrossRef]
- Dheda, K.; Barry, C.E., 3rd; Maartens, G. Tuberculosis. Lancet 2016, 387, 1211–1226. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: From extensively drug-resistant to untreatable tuberculosis. Lancet. Respir. Med. 2014, 2, 321–338. [Google Scholar] [CrossRef]
- World Health Organization (WHO). WHO Treatment Guidelines for Drug-Resistant Tuberculosis 2016 Update; World Health Organization (WHO): Geneva, Switzerland, 2016. [Google Scholar]
- Walker, J.; Tadena, N. J&J Tuberculosis Drug Gets Fast-Track Clearance. Wall St. J. 2013. Available online: https://www.wsj.com/articles/SB10001424127887323320404578213421059138236 (accessed on 27 November 2019).
- Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 2013, 3, 1–2. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Deltyba Delamanid Summary of the European Public Assessment Report (EPAR) for Deltyba; EMA: Amsterdam, The Netherlands, 2014; pp. 1–3. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/deltyba (accessed on 15 November 2019).
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef]
- US FDA. FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis That Affects the Lungs; US FDA: Silver Spring, MD, USA, 2019.
- Baptista, R.; Fazakerley, D.M.; Beckmann, M.; Baillie, L.; Mur, L.A.J. Untargeted metabolomics reveals a new mode of action of pretomanid (PA-824). Sci. Rep. 2018, 8, 5084. [Google Scholar] [CrossRef]
- Thompson, A.M.; Bonnet, M.; Lee, H.H.; Franzblau, S.G.; Wan, B.; Wong, G.S.; Cooper, C.B.; Denny, W.A. Antitubercular Nitroimidazoles Revisited: Synthesis and Activity of the Authentic 3-Nitro Isomer of Pretomanid. ACS Med. Chem. Lett. 2017, 8, 1275–1280. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef]
- Reymond, J.-L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Choi, I.; Sarker, M.; Talcott, C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011, 19, 65–74. [Google Scholar] [CrossRef]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 1–37. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Schrödinger. Prime. Available online: https://www.schrodinger.com/prime (accessed on 26 October 2019).
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008, 9, 40. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins 2009, 77, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Soding, J. Fast and accurate automatic structure prediction with HHpred. Proteins 2009, 77, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. PocketQuery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, W387–W392. [Google Scholar] [CrossRef]
- Brady, G.P., Jr.; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Schrödinger. Glide. Available online: https://www.schrodinger.com/glide (accessed on 26 October 2019).
- Schrödinger. Induced Fit. Available online: https://www.schrodinger.com/induced-fit (accessed on 26 October 2019).
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Davis, I.W.; Baker, D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F., Jr. Docking with SwissDock. Methods Mol. Biol. 2019, 2053, 189–202. [Google Scholar]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and exact pharmacophore search. J. Chem. Inf. Model. 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Catalyst. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/pharmacophore-and-ligand-based-design.html (accessed on 26 October 2019).
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- Inte:Ligand. LigandScout. Available online: http://www.inteligand.com/ligandscout/ (accessed on 26 October 2019).
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Douguet, D. e-LEA3D: A computational-aided drug design web server. Nucleic Acids Res. 2010, 38, W615–W621. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Schrödinger. PHASE. Available online: https://www.schrodinger.com/phase (accessed on 26 October 2019).
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Miller, B.T.; Singh, R.P.; Klauda, J.B.; Hodoscek, M.; Brooks, B.R.; Woodcock, H.L., 3rd. CHARMMing: A new, flexible web portal for CHARMM. J. Chem. Inf. Model. 2008, 48, 1920–1929. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Desmond. Available online: https://www.schrodinger.com/desmond (accessed on 26 October 2019).
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef]
- Helguera, A.M.; Combes, R.D.; Gonzalez, M.P.; Cordeiro, M.N. Applications of 2D descriptors in drug design: A DRAGON tale. Curr. Top. Med. Chem. 2008, 8, 1628–1655. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Canvas. Available online: https://www.schrodinger.com/canvas (accessed on 26 October 2019).
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 26 October 2019).
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef]
- Dong, J.; Yao, Z.J.; Zhu, M.F.; Wang, N.N.; Lu, B.; Chen, A.F.; Lu, A.P.; Miao, H.; Zeng, W.B.; Cao, D.S. ChemSAR: An online pipelining platform for molecular SAR modeling. J. Cheminform. 2017, 9, 27. [Google Scholar] [CrossRef]
- BioSolveIT. SeeSAR version 9.2. Available online: https://www.biosolveit.de/SeeSAR/ (accessed on 26 October 2019).
- Schrödinger. QikProp. Available online: https://www.schrodinger.com/qikprop (accessed on 26 October 2019).
- SimulationsPlus. ADMET Predictor. Available online: https://www.simulations-plus.com/software/admetpredictor/ (accessed on 26 October 2019).
- ACD/Labs. Percepta Platform. Available online: https://www.acdlabs.com/products/percepta/ (accessed on 26 October 2019).
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. FAF-Drugs: Free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744. [Google Scholar] [CrossRef]
- Rasolohery, I.; Moroy, G.; Guyon, F. PatchSearch: A Fast Computational Method for Off-Target Detection. J. Chem. Inf. Model. 2017, 57, 769–777. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. DS TOPKAT. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 26 October 2019).
- Dassault Systèmes BIOVIA. DS ADMET. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-pipeline-pilot/component-collections/adme-tox.html (accessed on 26 October 2019).
- Poroikov, V.; Filimonov, D.; Lagunin, A.; Gloriozova, T.; Zakharov, A. PASS: Identification of probable targets and mechanisms of toxicity. SAR QSAR Environ. Res. 2007, 18, 101–110. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979. [Google Scholar] [CrossRef]
- Tcheremenskaia, O.; Benigni, R.; Nikolova, I.; Jeliazkova, N.; Escher, S.E.; Batke, M.; Baier, T.; Poroikov, V.; Lagunin, A.; Rautenberg, M.; et al. OpenTox predictive toxicology framework: Toxicological ontology and semantic media wiki-based OpenToxipedia. J. Biomed. Semant. 2012, 3, S7. [Google Scholar] [CrossRef] [PubMed]
- Smiesko, M.; Vedani, A. VirtualToxLab: Exploring the Toxic Potential of Rejuvenating Substances Found in Traditional Medicines. Methods Mol. Biol. 2016, 1425, 121–137. [Google Scholar]
- Vedani, A.; Dobler, M.; Smiesko, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A.; Smiesko, M.; Spreafico, M.; Peristera, O.; Dobler, M. VirtualToxLab—In silico prediction of the toxic (endocrine-disrupting) potential of drugs, chemicals and natural products. Two years and 2000 compounds of experience: A progress report. ALTEX 2009, 26, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A.; Dobler, M.; Spreafico, M.; Peristera, O.; Smiesko, M. VirtualToxLab—In silico prediction of the toxic potential of drugs and environmental chemicals: Evaluation status and internet access protocol. ALTEX 2007, 24, 153–161. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. Correction to “admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties”. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Rudik, A.; Bezhentsev, V.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. Metatox-Web application for generation of metabolic pathways and toxicity estimation. J. Bioinform. Comput. Biol. 2019, 17, 1940001. [Google Scholar] [CrossRef] [PubMed]
- Rudik, A.V.; Bezhentsev, V.M.; Dmitriev, A.V.; Druzhilovskiy, D.S.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. MetaTox: Web Application for Predicting Structure and Toxicity of Xenobiotics’ Metabolites. J. Chem. Inf. Model. 2017, 57, 638–642. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Rosenkrands, I.; King, A.; Weldingh, K.; Moniatte, M.; Moertz, E.; Andersen, P. Towards the proteome of Mycobacterium tuberculosis. Electrophoresis 2000, 21, 3740–3756. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Schaible, U.E.; Mollenkopf, H.J.; Zimny-Arndt, U.; Raupach, B.; Mattow, J.; Halada, P.; Lamer, S.; Hagens, K.; Kaufmann, S.H. Comparative proteome analysis of Mycobacterium tuberculosis and Mycobacterium bovis BCG strains: Towards functional genomics of microbial pathogens. Mol. Microbiol. 1999, 33, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE 2010, 5, e13938. [Google Scholar] [CrossRef]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2011, 2, e00100–e00111. [Google Scholar] [CrossRef]
- Rachman, H.; Strong, M.; Ulrichs, T.; Grode, L.; Schuchhardt, J.; Mollenkopf, H.; Kosmiadi, G.A.; Eisenberg, D.; Kaufmann, S.H. Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 2006, 74, 1233–1242. [Google Scholar] [CrossRef]
- Xu, D. Protein databases on the internet. Curr. Protoc. Protein Sci. 2012. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Williams, A.J. Public chemical compound databases. Curr. Opin. Drug Discov. Dev. 2008, 11, 393–404. [Google Scholar]
- UniProt, C. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011, 39, D214–D219. [Google Scholar]
- Reddy, T.B.; Riley, R.; Wymore, F.; Montgomery, P.; DeCaprio, D.; Engels, R.; Gellesch, M.; Hubble, J.; Jen, D.; Jin, H.; et al. TB database: An integrated platform for tuberculosis research. Nucleic Acids Res. 2009, 37, D499–D508. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Sisk, P.; Stolte, C.; Weiner, B.; Koehrsen, M.; Wymore, F.; Reddy, T.B.; Zucker, J.D.; Engels, R.; Gellesch, M.; et al. TB database 2010: Overview and update. Tuberculosis 2010, 90, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef]
- Rosenthal, A.; Gabrielian, A.; Engle, E.; Hurt, D.E.; Alexandru, S.; Crudu, V.; Sergueev, E.; Kirichenko, V.; Lapitskii, V.; Snezhko, E.; et al. The TB Portals: An Open-Access, Web-Based Platform for Global Drug-Resistant-Tuberculosis Data Sharing and Analysis. J. Clin. Microbiol. 2017, 55, 3267–3282. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Ryabukhin, S.; Bogolyubsky, A.V.; Mykytenko, D.M.; Chuprina, A.; Heilman, W.; Kostyuk, A.N.; Tolmachev, A. Enamine real database: Making chemical diversity real. Chem. Today 2007, 25, 58–59. [Google Scholar]
- Williams, A.J. ChemSpider: Integrating Structure-Based Resources Distributed across the Internet. In Enhancing Learning with Online Resources, Social Networking, and Digital Libraries; American Chemical Society: Washington, DC, USA, 2010; Volume 1060, pp. 23–39. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Voigt, J.H.; Bienfait, B.; Wang, S.; Nicklaus, M.C. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput Sci. 2001, 41, 702–712. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Fariselli, P.; Casadio, R. eSLDB: Eukaryotic subcellular localization database. Nucleic Acids Res. 2007, 35, D208–D212. [Google Scholar] [CrossRef]
- Sprenger, J.; Lynn Fink, J.; Karunaratne, S.; Hanson, K.; Hamilton, N.A.; Teasdale, R.D. LOCATE: A mammalian protein subcellular localization database. Nucleic Acids Res. 2008, 36, D230–D233. [Google Scholar] [CrossRef]
- Peabody, M.A.; Laird, M.R.; Vlasschaert, C.; Lo, R.; Brinkman, F.S. PSORTdb: Expanding the bacteria and archaea protein subcellular localization database to better reflect diversity in cell envelope structures. Nucleic Acids Res. 2016, 44, D663–D668. [Google Scholar] [CrossRef]
- Hendlich, M.; Bergner, A.; Gunther, J.; Klebe, G. Relibase: Design and development of a database for comprehensive analysis of protein-ligand interactions. J. Mol. Biol. 2003, 326, 607–620. [Google Scholar] [CrossRef]
- Chen, X.; Lin, Y.; Liu, M.; Gilson, M.K. The Binding Database: Data management and interface design. Bioinformatics 2002, 18, 130–139. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Franca, T.C. Homology modeling: An important tool for the drug discovery. J. Biomol. Struct. Dyn. 2015, 33, 1780–1793. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef]
- Madden, T. The BLAST Sequence Analysis Tool. In The NCBI Handbook [Internet], 2nd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. [Google Scholar]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. In Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 105–116. [Google Scholar]
- Lassmann, T.; Sonnhammer, E.L. Kalign—An accurate and fast multiple sequence alignment algorithm. BMC Bioinf. 2005, 6, 298. [Google Scholar] [CrossRef]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Hetenyi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Shi, Y.; Colombo, C.; Kuttiyatveetil, J.R.; Zalatar, N.; van Straaten, K.E.; Mohan, S.; Sanders, D.A.; Pinto, B.M. A Second, Druggable Binding Site in UDP-Galactopyranose Mutase from Mycobacterium tuberculosis? Chembiochem 2016, 17, 2264–2273. [Google Scholar] [CrossRef]
- Stahura, F.L.; Bajorath, J. Virtual screening methods that complement HTS. Comb. Chem. High Throughput Screen. 2004, 7, 259–269. [Google Scholar] [CrossRef]
- Steindl, T.M.; Schuster, D.; Wolber, G.; Laggner, C.; Langer, T. High-throughput structure-based pharmacophore modelling as a basis for successful parallel virtual screening. J. Comput. Aided Mol. Des. 2006, 20, 703–715. [Google Scholar] [CrossRef]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R.; Gillet, V.J.; Lewis, R.A.; Taylor, R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem. 2010, 53, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129. [Google Scholar] [CrossRef]
- Hein, M.; Zilian, D.; Sotriffer, C.A. Docking compared to 3D-pharmacophores: The scoring function challenge. Drug Discov. Today Technol. 2010, 7, e229–e236. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K. The scaffold hopping potential of pharmacophores. Drug Discov. Today Technol. 2010, 7, e263–e269. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef]
- Cummings, M.D.; DesJarlais, R.L.; Gibbs, A.C.; Mohan, V.; Jaeger, E.P. Comparison of automated docking programs as virtual screening tools. J. Med. Chem. 2005, 48, 962–976. [Google Scholar] [CrossRef]
- Annamala, M.K.; Inampudi, K.K.; Guruprasad, L. Docking of phosphonate and trehalose analog inhibitors into M. tuberculosis mycolyltransferase Ag85C: Comparison of the two scoring fitness functions GoldScore and ChemScore, in the GOLD software. Bioinformation 2007, 1, 339–350. [Google Scholar] [CrossRef][Green Version]
- Perola, E.; Walters, W.P.; Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. [Google Scholar] [CrossRef]
- Xu, W.; Lucke, A.J.; Fairlie, D.P. Comparing sixteen scoring functions for predicting biological activities of ligands for protein targets. J. Mol. Graph. Model. 2015, 57, 76–88. [Google Scholar] [CrossRef]
- Billones, J.B.; Carrillo, M.C.; Organo, V.G.; Sy, J.B.; Clavio, N.A.; Macalino, S.J.; Emnacen, I.A.; Lee, A.P.; Ko, P.K.; Concepcion, G.P. In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic acid synthase (Mtb BioA). Drug Des. Devel. Ther. 2017, 11, 563–574. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 26 October 2019).
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef]
- Ericksen, S.S.; Wu, H.; Zhang, H.; Michael, L.A.; Newton, M.A.; Hoffmann, F.M.; Wildman, S.A. Machine Learning Consensus Scoring Improves Performance Across Targets in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2017, 57, 1579–1590. [Google Scholar] [CrossRef]
- Li, D.D.; Meng, X.F.; Wang, Q.; Yu, P.; Zhao, L.G.; Zhang, Z.P.; Wang, Z.Z.; Xiao, W. Consensus scoring model for the molecular docking study of mTOR kinase inhibitor. J. Mol. Graph. Model. 2018, 79, 81–87. [Google Scholar] [CrossRef]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef]
- Clark, R.D.; Strizhev, A.; Leonard, J.M.; Blake, J.F.; Matthew, J.B. Consensus scoring for ligand/protein interactions. J. Mol. Graph. Model. 2002, 20, 281–295. [Google Scholar] [CrossRef]
- Harrison, A.J.; Yu, M.; Gardenborg, T.; Middleditch, M.; Ramsay, R.J.; Baker, E.N.; Lott, J.S. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J. Bacteriol. 2006, 188, 6081–6091. [Google Scholar] [CrossRef]
- Manos-Turvey, A.; Bulloch, E.M.; Rutledge, P.J.; Baker, E.N.; Lott, J.S.; Payne, R.J. Inhibition studies of Mycobacterium tuberculosis salicylate synthase (MbtI). ChemMedChem 2010, 5, 1067–1079. [Google Scholar] [CrossRef]
- Vasan, M.; Neres, J.; Williams, J.; Wilson, D.J.; Teitelbaum, A.M.; Remmel, R.P.; Aldrich, C.C. Inhibitors of the salicylate synthase (MbtI) from Mycobacterium tuberculosis discovered by high-throughput screening. ChemMedChem 2010, 5, 2079–2087. [Google Scholar] [CrossRef]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.R.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules 2018, 23, 1506. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In ANTS 2006: Ant Colony Optimization and Swarm Intelligence; Dorigo, M., Gambardella, L.M., Birattari, M., Martinoli, A., Poli, R., Stützle, T., Eds.; Springer: Heidelberg/Berlin, Germany, 2016; pp. 247–258. [Google Scholar]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Sotriffer, C.A. Accounting for induced-fit effects in docking: What is possible and what is not? Curr. Top. Med. Chem. 2011, 11, 179–191. [Google Scholar] [CrossRef]
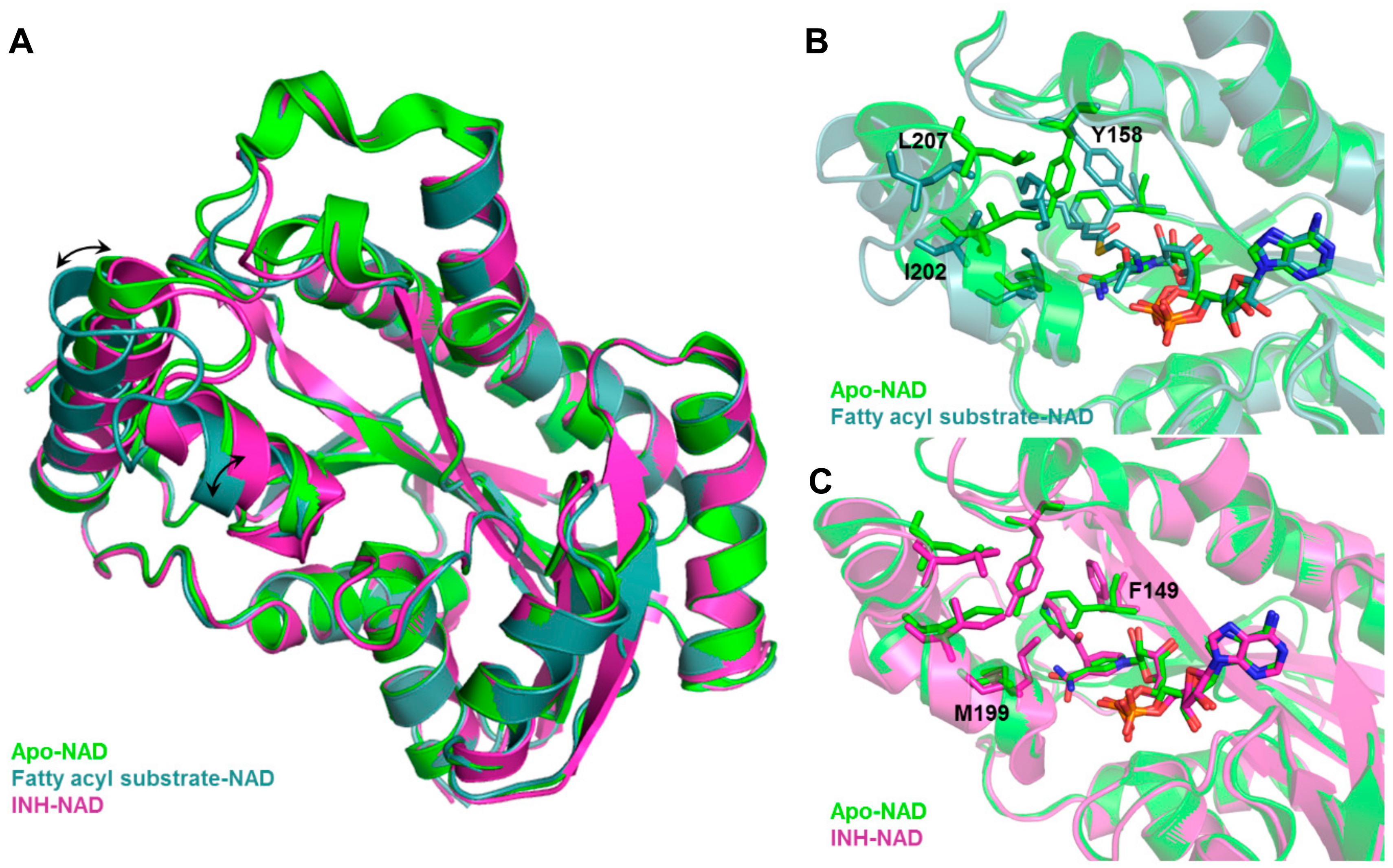
- Hartkoorn, R.C.; Sala, C.; Neres, J.; Pojer, F.; Magnet, S.; Mukherjee, R.; Uplekar, S.; Boy-Rottger, S.; Altmann, K.H.; Cole, S.T. Towards a new tuberculosis drug: Pyridomycin-nature’s isoniazid. EMBO Mol. Med. 2012, 4, 1032–1042. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilcheze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R., Jr.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef]
- Amaro, R.E.; Li, W.W. Emerging methods for ensemble-based virtual screening. Curr. Top. Med. Chem. 2010, 10, 3–13. [Google Scholar] [CrossRef]
- Brindha, S.; Sundaramurthi, J.C.; Velmurugan, D.; Vincent, S.; Gnanadoss, J.J. Docking-based virtual screening of known drugs against murE of Mycobacterium tuberculosis towards repurposing for TB. Bioinformation 2016, 12, 359–367. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Korb, O.; Howard, N.I.; Dias, M.V.B.; Blundell, T.L.; Abell, C. Discovery of Schaeffer’s Acid Analogues as Lead Structures of Mycobacterium tuberculosis Type II Dehydroquinase Using a Rational Drug Design Approach. ChemMedChem 2013, 8, 54–58. [Google Scholar] [CrossRef]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Bhabha, G.; Biel, J.T.; Fraser, J.S. Keep on moving: Discovering and perturbing the conformational dynamics of enzymes. Acc. Chem. Res. 2015, 48, 423–430. [Google Scholar] [CrossRef]
- Goh, C.S.; Milburn, D.; Gerstein, M. Conformational changes associated with protein-protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 104–109. [Google Scholar] [CrossRef]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar]
- Lee, Y.; Jeong, L.S.; Choi, S.; Hyeon, C. Link between allosteric signal transduction and functional dynamics in a multisubunit enzyme: S-adenosylhomocysteine hydrolase. J. Am. Chem. Soc. 2011, 133, 19807–19815. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- Larsson, P.; Hess, B.; Lindahl, E. Algorithm improvements for molecular dynamics simulations. WIREs Comput. Mol. Sci. 2011, 1, 93–108. [Google Scholar] [CrossRef]
- Orozco, M.; Orellana, L.; Hospital, A.; Naganathan, A.N.; Emperador, A.; Carrillo, O.; Gelpi, J.L. Coarse-grained representation of protein flexibility. Foundations, successes, and shortcomings. Adv. Protein Chem. Struct. Biol. 2011, 85, 183–215. [Google Scholar]
- Linge, J.P.; Williams, M.A.; Spronk, C.A.; Bonvin, A.M.; Nilges, M. Refinement of protein structures in explicit solvent. Proteins 2003, 50, 496–506. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of conformational change: Comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides†. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Daggett, V.; Levitt, M. Protein Unfolding Pathways Explored Through Molecular Dynamics Simulations. J. Mol. Biol. 1993, 232, 600–619. [Google Scholar] [CrossRef]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Papaleo, E. Integrating atomistic molecular dynamics simulations, experiments, and network analysis to study protein dynamics: Strength in unity. Front. Mol. Biosci. 2015, 2, 28. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Gomez-Gardenes, J.; Echenique, P.; Falo, F. Exploring the free energy landscape: From dynamics to networks and back. PLoS Comput. Biol. 2009, 5, e1000415. [Google Scholar] [CrossRef]
- Wahab, H.A.; Choong, Y.S.; Ibrahim, P.; Sadikun, A.; Scior, T. Elucidating isoniazid resistance using molecular modeling. J. Chem. Inf. Model. 2009, 49, 97–107. [Google Scholar] [CrossRef]
- Schroeder, E.K.; Basso, L.A.; Santos, D.S.; de Souza, O.N. Molecular dynamics simulation studies of the wild-type, I21V, and I16T mutants of isoniazid-resistant Mycobacterium tuberculosis enoyl reductase (InhA) in complex with NADH: Toward the understanding of NADH-InhA different affinities. Biophys. J. 2005, 89, 876–884. [Google Scholar] [CrossRef]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K.; Barros, C.A.L.; Neto, A. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J. Biomol. Struct. Dyn. 2019, 37, 1616–1627. [Google Scholar] [CrossRef]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets, M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259. [Google Scholar] [CrossRef]
- Nikolova, N.; Jaworska, J. Approaches to Measure Chemical Similarity—A Review. QSAR Comb. Sci. 2003, 22, 1006–1026. [Google Scholar] [CrossRef]
- Johnson, M.A.; Maggiora, G.M. American Chemical Society. In Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Bacilieri, M.; Moro, S. Ligand-based drug design methodologies in drug discovery process: An overview. Curr. Drug Discov. Technol. 2006, 3, 155–165. [Google Scholar] [CrossRef]
- Sukumar, N.; Das, S. Current trends in virtual high throughput screening using ligand-based and structure-based methods. Comb. Chem. High Throughput Screen. 2011, 14, 872–888. [Google Scholar] [CrossRef]
- Cereto-Massague, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallve, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Bajusz, D.; Racz, A.; Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- Faulon, J.-L.; Bender, A. Handbook of Chemoinformatics Algorithms; Chapman & Hall/CRC: Boca Raton, FL, USA, 2010; 440p. [Google Scholar]
- Haranczyk, M.; Holliday, J. Comparison of similarity coefficients for clustering and compound selection. J. Chem. Inf. Model. 2008, 48, 498–508. [Google Scholar] [CrossRef]
- Al Khalifa, A.; Haranczyk, M.; Holliday, J. Comparison of nonbinary similarity coefficients for similarity searching, clustering and compound selection. J. Chem. Inf. Model. 2009, 49, 1193–1201. [Google Scholar] [CrossRef]
- Ginn, C.M.R.; Willett, P.; Bradshaw, J. Combination of molecular similarity measures using data fusion. Perspect. Drug Discov. Des. 2000, 20, 1–16. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Maggiora, G.M.; Giulianotti, M.A.; Pinilla, C.; Houghten, R.A. A similarity-based data-fusion approach to the visual characterization and comparison of compound databases. Chem. Biol. Drug Des. 2007, 70, 393–412. [Google Scholar] [CrossRef]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Extending the activity cliff concept: Structural categorization of activity cliffs and systematic identification of different types of cliffs in the ChEMBL database. J. Chem. Inf. Model. 2012, 52, 1806–1811. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Advancing the activity cliff concept. F1000Res 2013, 2, 199. [Google Scholar] [CrossRef]
- Stumpfe, D.; de la Vega de Leon, A.; Dimova, D.; Bajorath, J. Advancing the activity cliff concept, part II. F1000Res 2014, 3, 75. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design—A review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Exploring QSAR. Environ. Sci. Technol. 1995, 29, 444A. [CrossRef]
- Bostrom, J.; Norrby, P.O.; Liljefors, T. Conformational energy penalties of protein-bound ligands. J. Comput. Aided Mol. Des. 1998, 12, 383–396. [Google Scholar] [CrossRef]
- Perola, E.; Charifson, P.S. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–2510. [Google Scholar] [CrossRef]
- Melo-Filho, C.C.; Braga, R.C.; Andrade, C.H. 3D-QSAR approaches in drug design: Perspectives to generate reliable CoMFA models. Curr. Comput. Aided Drug Des. 2014, 10, 148–159. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, II; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Bajpai, A.; Agarwal, N.; Gupta, S.P. A comparative 2D QSAR study on a series of hydroxamic acid-based histone deacetylase inhibitors vis-a-vis comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA). Indian J. Biochem. Biophys. 2014, 51, 244–252. [Google Scholar]
- Chhatbar, D.M.; Chaube, U.J.; Vyas, V.K.; Bhatt, H.G. CoMFA, CoMSIA, Topomer CoMFA, HQSAR, molecular docking and molecular dynamics simulations study of triazine morpholino derivatives as mTOR inhibitors for the treatment of breast cancer. Comput. Biol. Chem. 2019, 80, 351–363. [Google Scholar] [CrossRef]
- Singh, S.; Supuran, C.T. 3D-QSAR CoMFA studies on sulfonamide inhibitors of the Rv3588c beta-carbonic anhydrase from Mycobacterium tuberculosis and design of not yet synthesized new molecules. J. Enzym. Inhib. Med. Chem. 2014, 29, 449–455. [Google Scholar] [CrossRef]
- Punkvang, A.; Hannongbua, S.; Saparpakorn, P.; Pungpo, P. Insight into the structural requirements of aminopyrimidine derivatives for good potency against both purified enzyme and whole cells of M. tuberculosis: Combination of HQSAR, CoMSIA, and MD simulation studies. J. Biomol. Struct. Dyn. 2016, 34, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D. 3D pharmacophores as tools for activity profiling. Drug Discov. Today Technol. 2010, 7, e205–e211. [Google Scholar] [CrossRef] [PubMed]
- Tawari, N.R.; Degani, M.S. Predictive models for nucleoside bisubstrate analogs as inhibitors of siderophore biosynthesis in Mycobacterium tuberculosis: Pharmacophore mapping and chemometric QSAR study. Mol. Divers. 2011, 15, 435–444. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Sharma, S. Molecular Dynamics Simulation of Nanocomposites Using BIOVIA Materials Studio, Lammps and Gromacs, 1st ed.; Elsevier: Waltham, MA, USA, 2019. [Google Scholar]
- Fiolhais, C.; Nogueira, F.; Marques, M.A.L. A Primer in Density Functional Theory; Springer: Berlin, Germany; New York, NY, USA, 2003. [Google Scholar]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef]
- Rabi, S.; Patel, A.H.G.; Burger, S.K.; Verstraelen, T.; Ayers, P.W. Exploring the substrate selectivity of human sEH and M. tuberculosis EHB using QM/MM. Struct. Chem. 2017, 28, 1501–1511. [Google Scholar] [CrossRef]
- Ramalho, T.C.; Caetano, M.S.; Josa, D.; Luz, G.P.; Freitas, E.A.; da Cunha, E.F. Molecular modeling of Mycobacterium tuberculosis dUTpase: Docking and catalytic mechanism studies. J. Biomol. Struct. Dyn. 2011, 28, 907–917. [Google Scholar] [CrossRef]
- Oliveira, C.G.; da, S.M.P.I.; Souza, P.C.; Pavan, F.R.; Leite, C.Q.; Viana, R.B.; Batista, A.A.; Nascimento, O.R.; Deflon, V.M. Manganese(II) complexes with thiosemicarbazones as potential anti-Mycobacterium tuberculosis agents. J. Inorg. Biochem. 2014, 132, 21–29. [Google Scholar] [CrossRef]
- Chi, G.; Manos-Turvey, A.; O’Connor, P.D.; Johnston, J.M.; Evans, G.L.; Baker, E.N.; Payne, R.J.; Lott, J.S.; Bulloch, E.M. Implications of binding mode and active site flexibility for inhibitor potency against the salicylate synthase from Mycobacterium tuberculosis. Biochemistry 2012, 51, 4868–4879. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Indarto, A. Theoretical Modelling and Mechanistic Study of the Formation and Atmospheric Transformations of Polycyclic Aromatic Compounds and Carbonaceous Particles; Universal Publishers: Irvine, CA, USA, 2010. [Google Scholar]
- Hamada, I. van der Waals density functional made accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Berland, K.; Cooper, V.R.; Lee, K.; Schroder, E.; Thonhauser, T.; Hyldgaard, P.; Lundqvist, B.I. van der Waals forces in density functional theory: A review of the vdW-DF method. Rep. Prog. Phys. 2015, 78, 066501. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sanchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Polgar, T.; Keseru, G.M. Integration of virtual and high throughput screening in lead discovery settings. Comb. Chem. High Throughput Screen. 2011, 14, 889–897. [Google Scholar] [CrossRef]
- Tanrikulu, Y.; Kruger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef]
- Tan, L.; Geppert, H.; Sisay, M.T.; Gutschow, M.; Bajorath, J. Integrating structure- and ligand-based virtual screening: Comparison of individual, parallel, and fused molecular docking and similarity search calculations on multiple targets. ChemMedChem 2008, 3, 1566–1571. [Google Scholar] [CrossRef]
- Huang, S.Y.; Li, M.; Wang, J.; Pan, Y. HybridDock: A Hybrid Protein-Ligand Docking Protocol Integrating Protein- and Ligand-Based Approaches. J. Chem. Inf. Model. 2016, 56, 1078–1087. [Google Scholar] [CrossRef]
- Lam, P.C.; Abagyan, R.; Totrov, M. Ligand-biased ensemble receptor docking (LigBEnD): A hybrid ligand/receptor structure-based approach. J. Comput. Aided Mol. Des. 2018, 32, 187–198. [Google Scholar] [CrossRef]
- Mestres, J.; Knegtel, R.M.A. Similarity versus docking in 3D virtual screening. Perspect. Drug Discov. Des. 2000, 20, 191–207. [Google Scholar] [CrossRef]
- Kruger, D.M.; Evers, A. Comparison of structure- and ligand-based virtual screening protocols considering hit list complementarity and enrichment factors. ChemMedChem 2010, 5, 148–158. [Google Scholar] [CrossRef]
- Billones, J.B.; Carrillo, M.C.; Organo, V.G.; Macalino, S.J.; Sy, J.B.; Emnacen, I.A.; Clavio, N.A.; Concepcion, G.P. Toward antituberculosis drugs: In silico screening of synthetic compounds against Mycobacterium tuberculosisl,d-transpeptidase 2. Drug Des. Devel Ther. 2016, 10, 1147–1157. [Google Scholar] [CrossRef]
- Fakhar, Z.; Govender, T.; Maguire, G.E.M.; Lamichhane, G.; Walker, R.C.; Kruger, H.G.; Honarparvar, B. Differential flap dynamics in l,d-transpeptidase2 from Mycobacterium tuberculosis revealed by molecular dynamics. Mol. Biosyst. 2017, 13, 1223–1234. [Google Scholar] [CrossRef]
- Sandhu, P.; Akhter, Y. The drug binding sites and transport mechanism of the RND pumps from Mycobacterium tuberculosis: Insights from molecular dynamics simulations. Arch. Biochem. Biophys. 2016, 592, 38–49. [Google Scholar] [CrossRef]
- Shah, P.; Mistry, J.; Reche, P.A.; Gatherer, D.; Flower, D.R. In silico design of Mycobacterium tuberculosis epitope ensemble vaccines. Mol. Immunol. 2018, 97, 56–62. [Google Scholar] [CrossRef]
- Li, D.; Chi, B.; Wang, W.-W.; Gao, J.-M.; Wan, J. Exploring the possible binding mode of trisubstituted benzimidazoles analogues in silico for novel drug designtargeting Mtb FtsZ. Med. Chem. Res. 2017, 26, 153–169. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Eudes, R.; Miteva, M.A. Structure-based virtual ligand screening: Recent success stories. Comb. Chem. High Throughput Screen. 2009, 12, 1000–1016. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Clark, D.E. What has virtual screening ever done for drug discovery? Expert Opin. Drug Discov. 2008, 3, 841–851. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martinez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Coupez, B.; Lewis, R.A. Docking and scoring—Theoretically easy, practically impossible? Curr. Med. Chem. 2006, 13, 2995–3003. [Google Scholar]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Shaw, D.E. Picosecond to Millisecond Structural Dynamics in Human Ubiquitin. J. Phys. Chem. B 2016, 120, 8313–8320. [Google Scholar] [CrossRef] [PubMed]
- Noe, F. Beating the millisecond barrier in molecular dynamics simulations. Biophys. J. 2015, 108, 228–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shi, J.; Nobrega, R.P.; Schwantes, C.; Kathuria, S.V.; Bilsel, O.; Matthews, C.R.; Lane, T.J.; Pande, V.S. Atomistic structural ensemble refinement reveals non-native structure stabilizes a sub-millisecond folding intermediate of CheY. Sci. Rep. 2017, 7, 44116. [Google Scholar] [CrossRef]
- Fujita, T. Recent Success Stories Leading to Commercializable Bioactive Compounds with the Aid of Traditional QSAR Procedures. QSAR 1997, 16, 107–112. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, L.; Zhu, Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr. Comput. Aided Drug Des. 2010, 6, 37–49. [Google Scholar] [CrossRef]
- Sardari, S.; Dezfulian, M. Cheminformatics in anti-infective agents discovery. Mini Rev. Med. Chem. 2007, 7, 181–189. [Google Scholar] [CrossRef]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Software/ Webserver Name | Availability | Website |
|---|---|---|---|
| Comparative modeling | SWISS-MODEL [32] | Free webserver | https://swissmodel.expasy.org/ |
| Structural geometry confirmation | MODELLER [33] | Free standalone program for academic license or commercially available through BIOVIA | https://salilab.org/modeller/ https://www.3dsbiovia.com/ |
| Robetta [34] | Free webserver | http://new.robetta.org/ | |
| Prime [35] | Commercially available through Schrödinger | https://www.schrodinger.com/prime | |
| I-TASSER [36,37,38,39,40,41] | Free webserver or standalone program for academic license | https://zhanglab.ccmb.med.umich.edu/I-TASSER/ | |
| HHPred [42,43,44] | Free webserver | https://toolkit.tuebingen.mpg.de/tools/hhpred | |
| Structural geometry confirmation | PROCHECK [45] | Free webserver and source code | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| Druggability and binding site prediction Druggability and binding site prediction | ProSA [46] | Free webserver | https://prosa.services.came.sbg.ac.at/prosa.php |
| VERIFY3D [47] | Free webserver | https://servicesn.mbi.ucla.edu/Verify3D/ | |
| ERRAT [48] | Free webserver | https://servicesn.mbi.ucla.edu/ERRAT/ | |
| PockDrug [49] | Free webserver | http://pockdrug.rpbs.univ-paris-diderot.fr/cgi-bin/index.py?page=home | |
| DoGSiteScorer [50] | Free webserver | https://proteins.plus/ | |
| fpocket [51,52] | Free/open source platform | https://github.com/Discngine/fpocket | |
| CASTp [53,54,55] | Free webserver | http://sts.bioe.uic.edu/castp/calculation.html | |
| PocketQuery [56] | Free webserver | http://pocketquery.csb.pitt.edu/ | |
| PASS [57] | Free/open source platform | http://www.ccl.net/cca/software/UNIX/pass/overview.html | |
| SiteMap [58] | Commercially available through Schrödinger | https://www.schrodinger.com/sitemap | |
| Docking, pharmacophore, and virtual screening Docking, pharmacophore, and virtual screening | ConCavity [59] | Free webserver | https://compbio.cs.princeton.edu/concavity/ |
| PrankWeb [60] | Free webserver | http://prankweb.cz/ | |
| ProFunc [61] | Free webserver | http://www.ebi.ac.uk/thornton-srv/databases/ProFunc/ | |
| AutoDock [62] and AutoDock Vina [63] | Free standalone program | http://autodock.scripps.edu/ | |
| DOCK [64] | Free/open source platform | http://dock.compbio.ucsf.edu/ | |
| GOLD [65] | Commercially available through CCDC | https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ | |
| Glide [66] | Commercially available through Schrödinger | https://www.schrodinger.com/glide/ | |
| Induced Fit [67] | Commercially available through Schrödinger | https://www.schrodinger.com/induced-fit | |
| FlexX [68] | Commercially available through BioSolveIT | https://www.biosolveit.de/flexx/index.html | |
| RosettaLigand [69] | Free/open source platform for academic license | https://www.rosettacommons.org/software | |
| CDOCKER [70] | Commercially available through BIOVIA | https://www.3dsbiovia.com/ | |
| SwissDock [71,72] | Free webserver | http://www.swissdock.ch/docking | |
| Pharmer [73] | Free/open source platform | http://smoothdock.ccbb.pitt.edu/pharmer/ | |
| CATALYST [74] | Commercially available through BIOVIA | https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/pharmacophore-and-ligand-based-design.html | |
| PharmGist [75] | Free webserver | http://bioinfo3d.cs.tau.ac.il/pharma/php.php | |
| LigandScout [76] | Commercially available through Inte:Ligand | http://www.inteligand.com/ligandscout/ | |
| SwissSimilarity [77] | Free webserver | http://www.swisssimilarity.ch/ | |
| LEA3D [78] | Free webserver | https://chemoinfo.ipmc.cnrs.fr/LEA3D/index.html | |
| PyRx [79] | Free (no support) or commercially available | https://pyrx.sourceforge.io/ | |
| Phase [80] | Commercially available through Schrödinger | https://www.schrodinger.com/phase | |
| Molecular Dynamics | AMBER [81,82] | Commercially available | https://ambermd.org/ |
| CHARMM [83] | Free or commercially available through CHARMM or BIOVIA | http://charmm.chemistry.harvard.edu/ https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/simulations.html | |
| CHARMMing [84] | Free webserver | https://www.charmming.org/charmming | |
| GROMACS [85,86] | Free/open source platform | http://www.gromacs.org/ | |
| NAMD [87] | Free/open source platform | https://www.ks.uiuc.edu/Research/namd/ | |
| Desmond [88] | Commercially available through Schrödinger | https://www.schrodinger.com/desmond | |
| SwissParam [89] | Free webserver | http://www.swissparam.ch/ | |
| CHARMM-GUI [90] | Free webserver | http://www.charmm-gui.org/ | |
| ParamChem CGenFF [91,92,93] | Free webserver | https://cgenff.umaryland.edu/ | |
| VMD [94] | Free/open source platform | https://www.ks.uiuc.edu/Research/vmd/ | |
| Molecular Descriptors, Fingerprints, and Quantitative Structure-Activity Relationship | Dragon [95] | Commercially available through Talete | http://www.talete.mi.it/products/dragon_description.htm |
| E-Dragon [96] | Free webserver | http://146.107.217.178/lab/edragon/start.html | |
| Canvas [97] | Commercially available through Schrödinger | https://www.schrodinger.com/canvas | |
| RDKit [98] | Free/open source platform | https://www.rdkit.org/docs/source/rdkit.ML.Descriptors.MoleculeDescriptors.html | |
| PyDescriptor [99] | Free/open source platform | https://ochem.eu/home/show.do | |
| Mordred [100] | Free/open source platform | https://github.com/mordred-descriptor/mordred | |
| Open3DQSAR [101] | Free/open source platform | http://open3dqsar.sourceforge.net/?Home | |
| ChemSAR [102] | Free webserver | http://chemsar.scbdd.com/ | |
| SeeSAR [103] | Commercially available through BioSolveIT | https://www.biosolveit.de/SeeSAR/ | |
| Pharmacokinetic properties | QikProp [104] | Commercially available through Schrödinger | https://www.schrodinger.com/qikprop |
| ADMET Predictor [105] | Commercially available through SimulationsPlus, Inc. | https://www.simulations-plus.com/software/overview/ | |
| ACD Percepta [106] | Commercially available through ACD/Labs | https://www.acdlabs.com/products/percepta/index.php | |
| FAF-Drugs4 [107] | Free webserver | http://fafdrugs4.mti.univ-paris-diderot.fr/ | |
| PatchSearch [108] | Free webserver | http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PatchSearch | |
| TOPKAT [109] and ADMET [110] | Commercially available through BIOVIA | https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html | |
| PASS Online [111] | Free webserver or commercially available standalone program | http://pharmaexpert.ru/Passonline/index.php | |
| SwissADME [112] | Free webserver | http://www.swissadme.ch/ | |
| MetaSite [113] | Commercially available through Molecular Discovery | https://www.moldiscovery.com/software/metasite/ | |
| ToxPredict [114] | Free webserver | https://apps.ideaconsult.net/ToxPredict# | |
| VirtualToxLab [115,116,117,118] | Free standalone software | http://www.biograf.ch/index.php?id=home | |
| admetSAR [119,120,121] | Free webserver | http://lmmd.ecust.edu.cn/admetsar1/home/ | |
| MetaTox [122,123] | Free webserver | http://way2drug.com/mg2/ |
| Database | Size (Approximate) | Website |
|---|---|---|
| GDB-17 [140] | 166 billion | http://gdb.unibe.ch/ |
| Enamine REAL [141] | 700 million | https://enamine.net/ |
| PubChem [131] | 97 million | https://pubchem.ncbi.nlm.nih.gov/ |
| ChemSpider [142] | 77 million | http://www.chemspider.com/ |
| ZINC [143] | 230 million | http://zinc.docking.org/ |
| ChEMBL [144] | 1.9 million | https://www.ebi.ac.uk/chembl/ |
| NCI [145] | 460,000 | https://cactus.nci.nih.gov/download/roadmap/ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macalino, S.J.Y.; Billones, J.B.; Organo, V.G.; Carrillo, M.C.O. In Silico Strategies in Tuberculosis Drug Discovery. Molecules 2020, 25, 665. https://doi.org/10.3390/molecules25030665
Macalino SJY, Billones JB, Organo VG, Carrillo MCO. In Silico Strategies in Tuberculosis Drug Discovery. Molecules. 2020; 25(3):665. https://doi.org/10.3390/molecules25030665
Chicago/Turabian StyleMacalino, Stephani Joy Y., Junie B. Billones, Voltaire G. Organo, and Maria Constancia O. Carrillo. 2020. "In Silico Strategies in Tuberculosis Drug Discovery" Molecules 25, no. 3: 665. https://doi.org/10.3390/molecules25030665
APA StyleMacalino, S. J. Y., Billones, J. B., Organo, V. G., & Carrillo, M. C. O. (2020). In Silico Strategies in Tuberculosis Drug Discovery. Molecules, 25(3), 665. https://doi.org/10.3390/molecules25030665