
Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease



Abstract
:1. Introduction
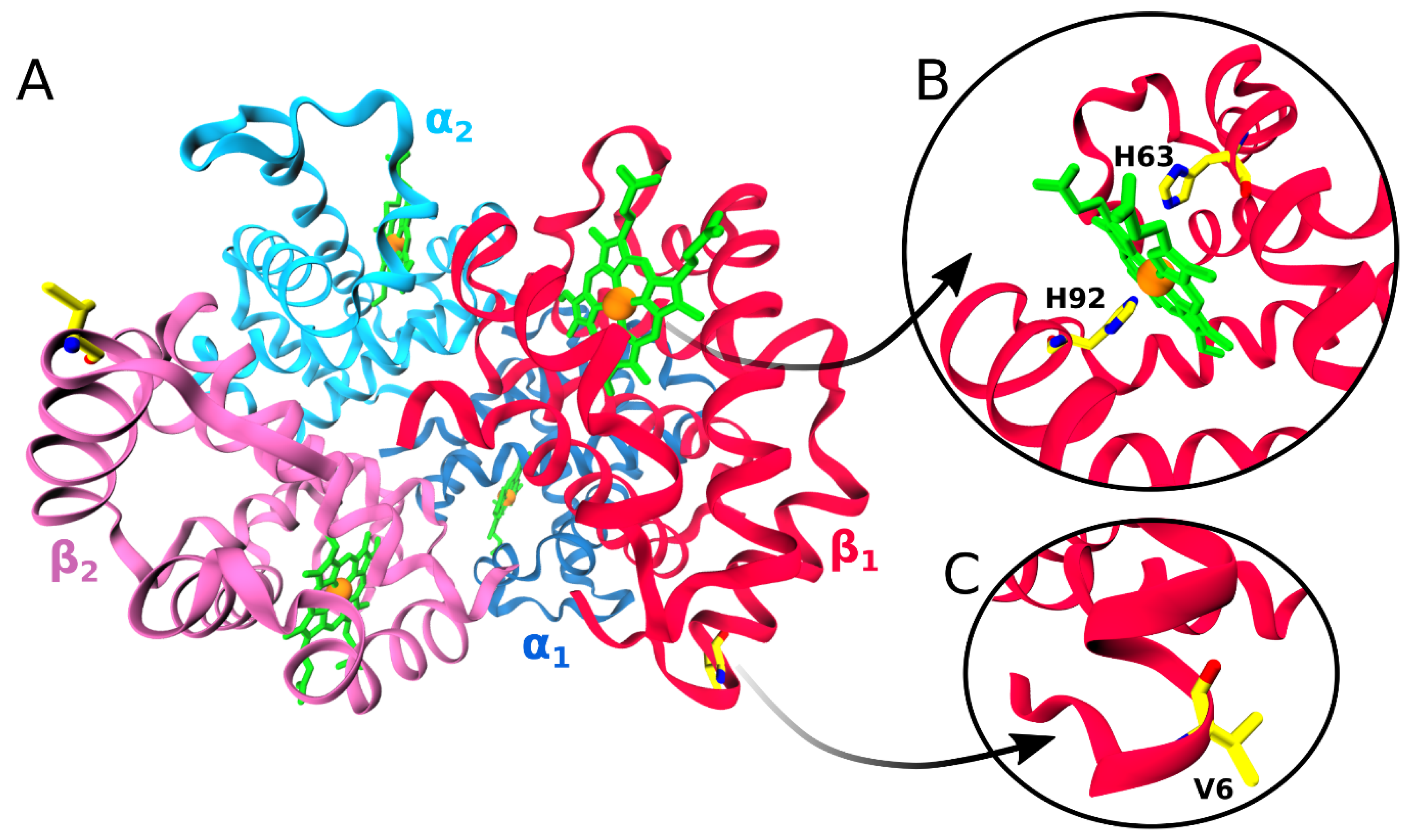
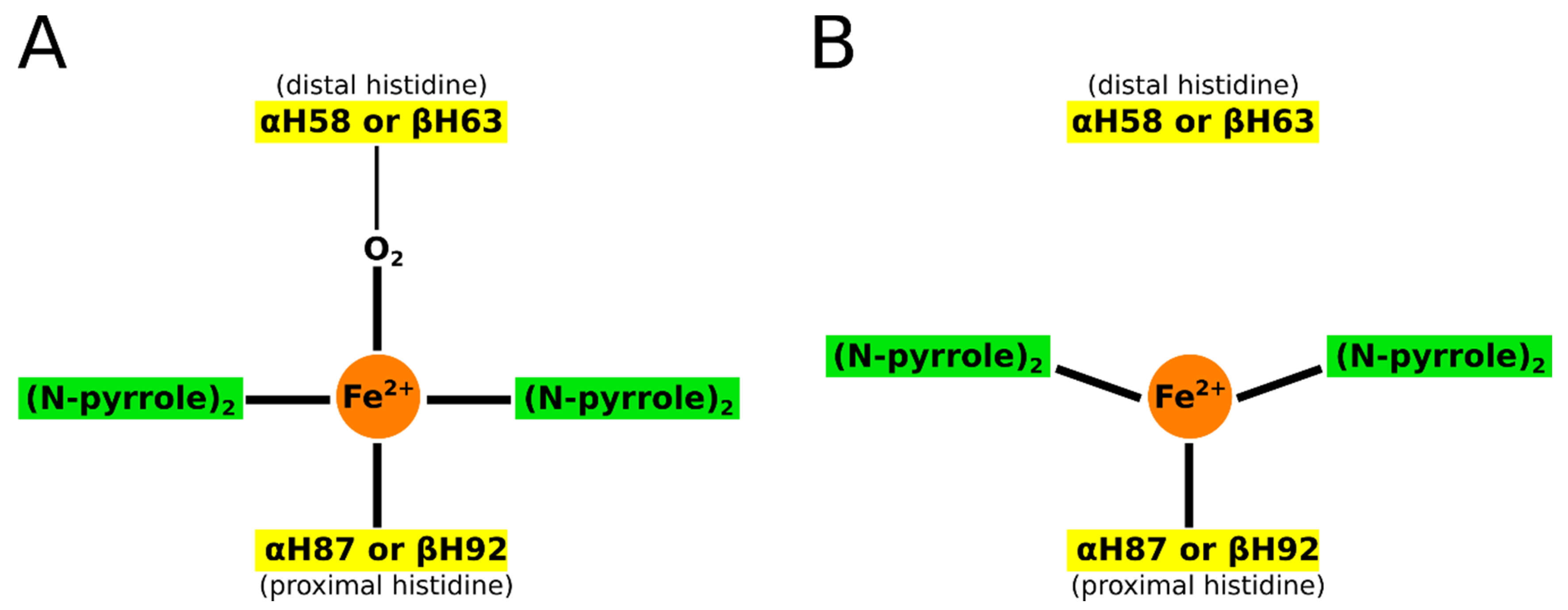
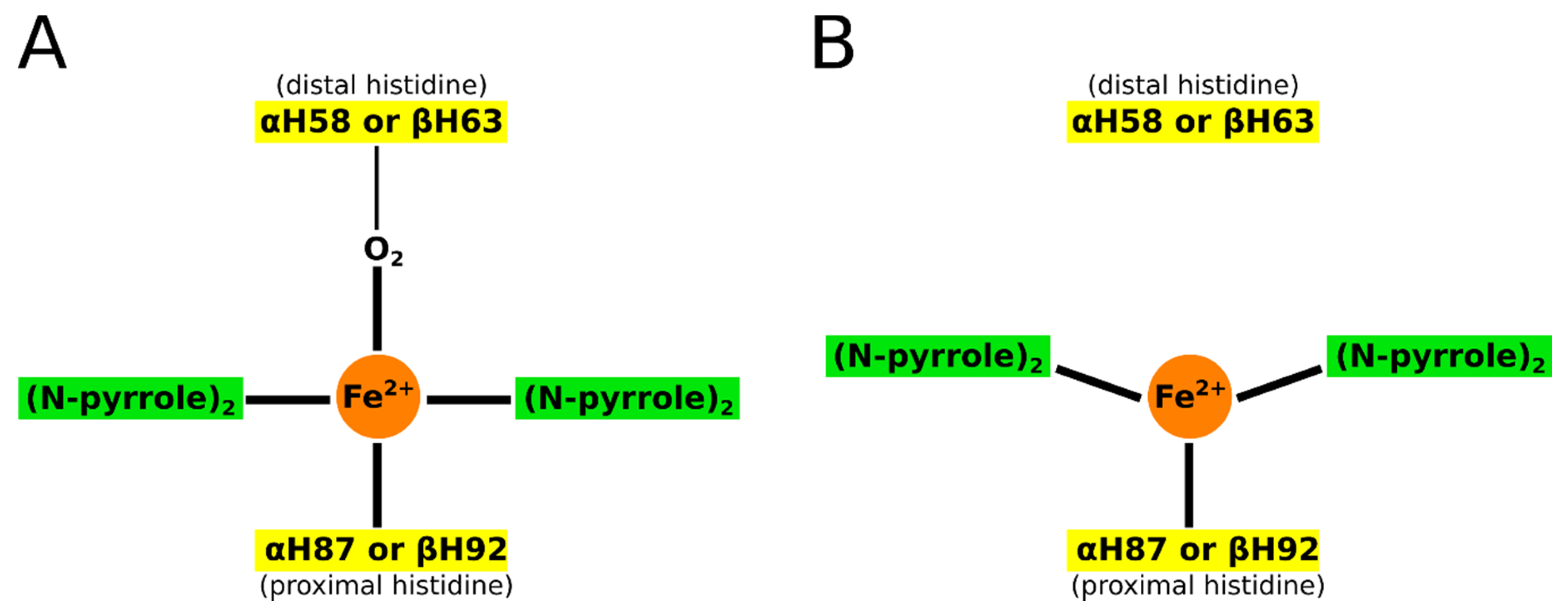
2. Hemoglobins: Structure, Function, and Aggregation
3. HbS as a Target for Drug Design
3.1. HbS Aggregation Is An Inefficient Process
3.2. Antisickling Effect and HbS Conformation
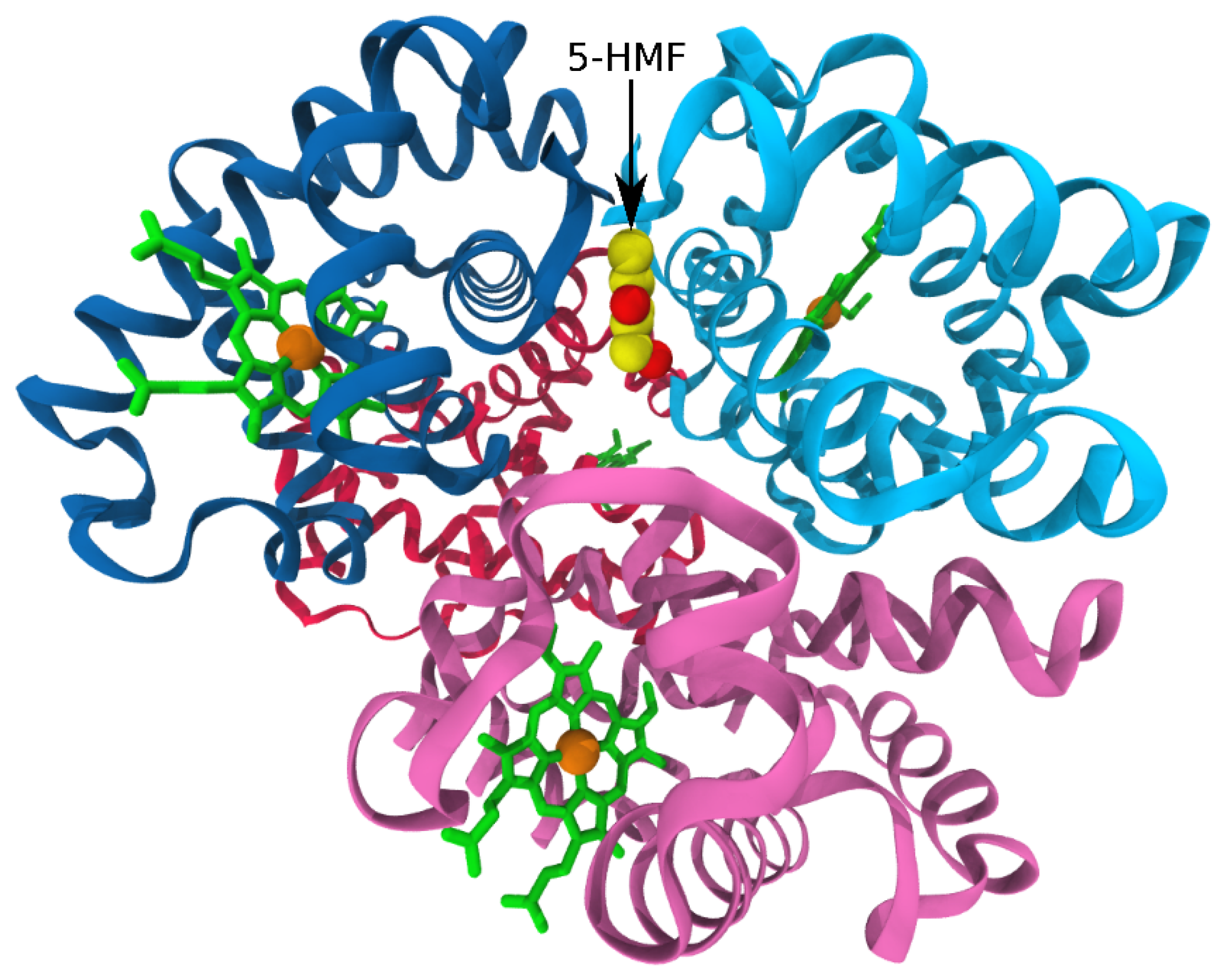
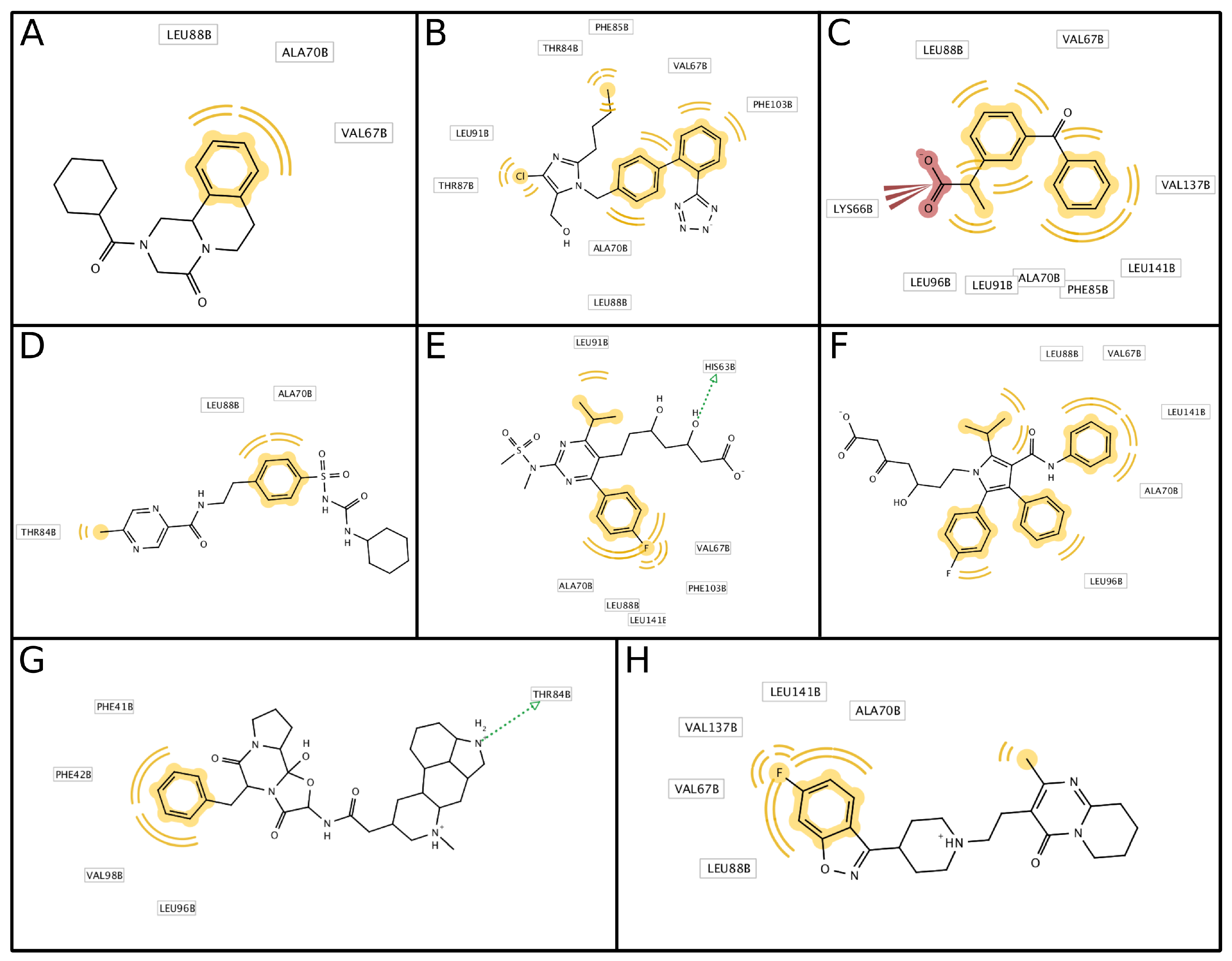
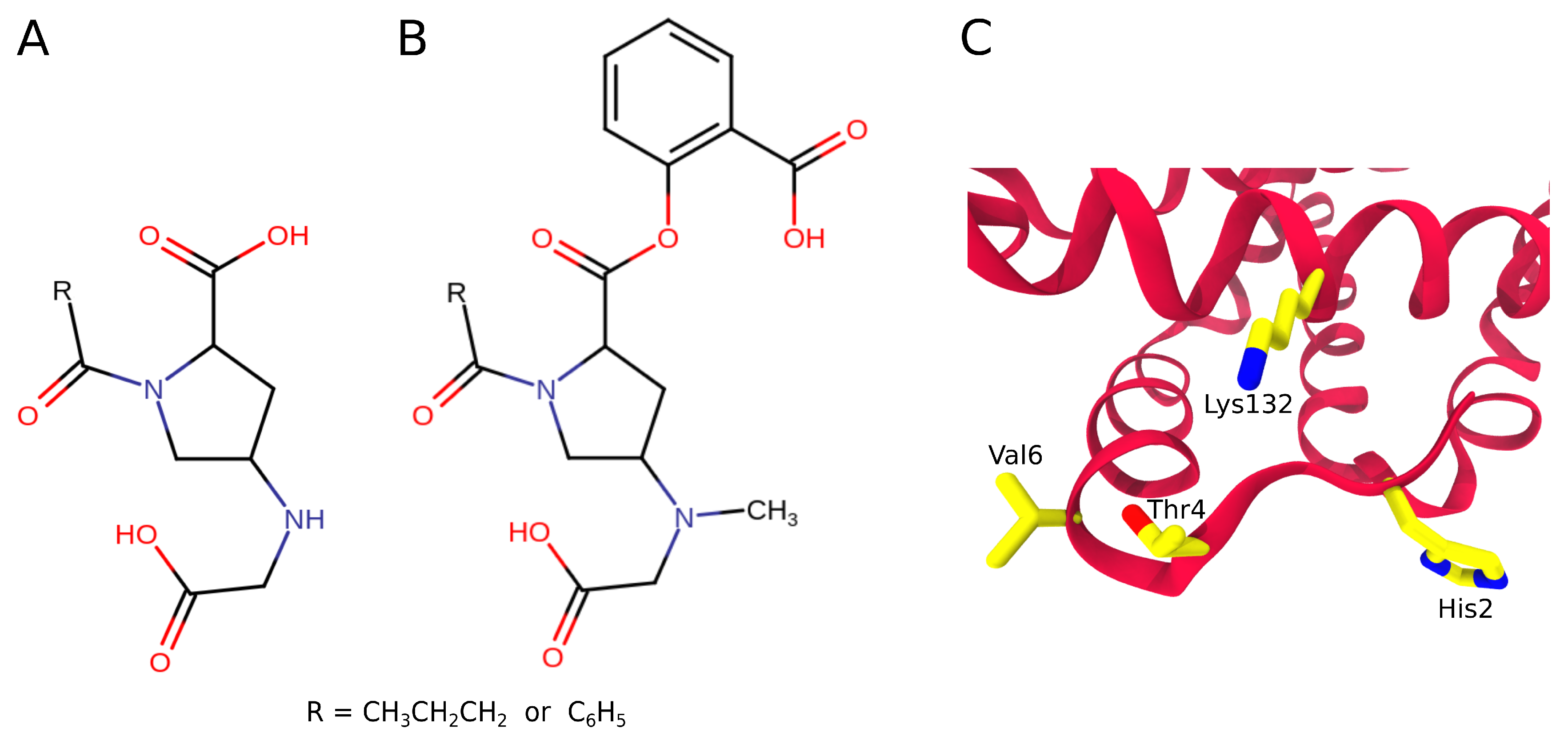
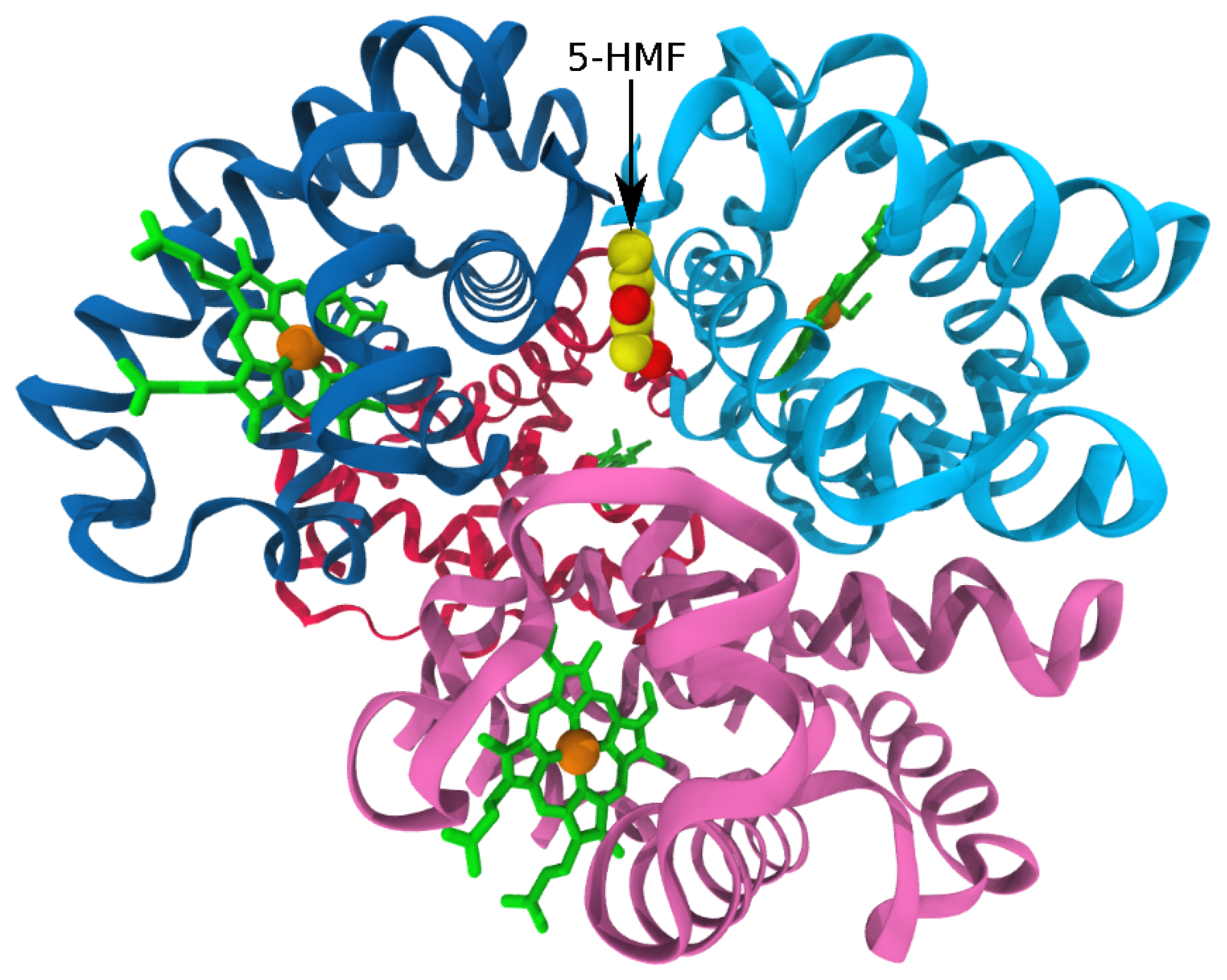
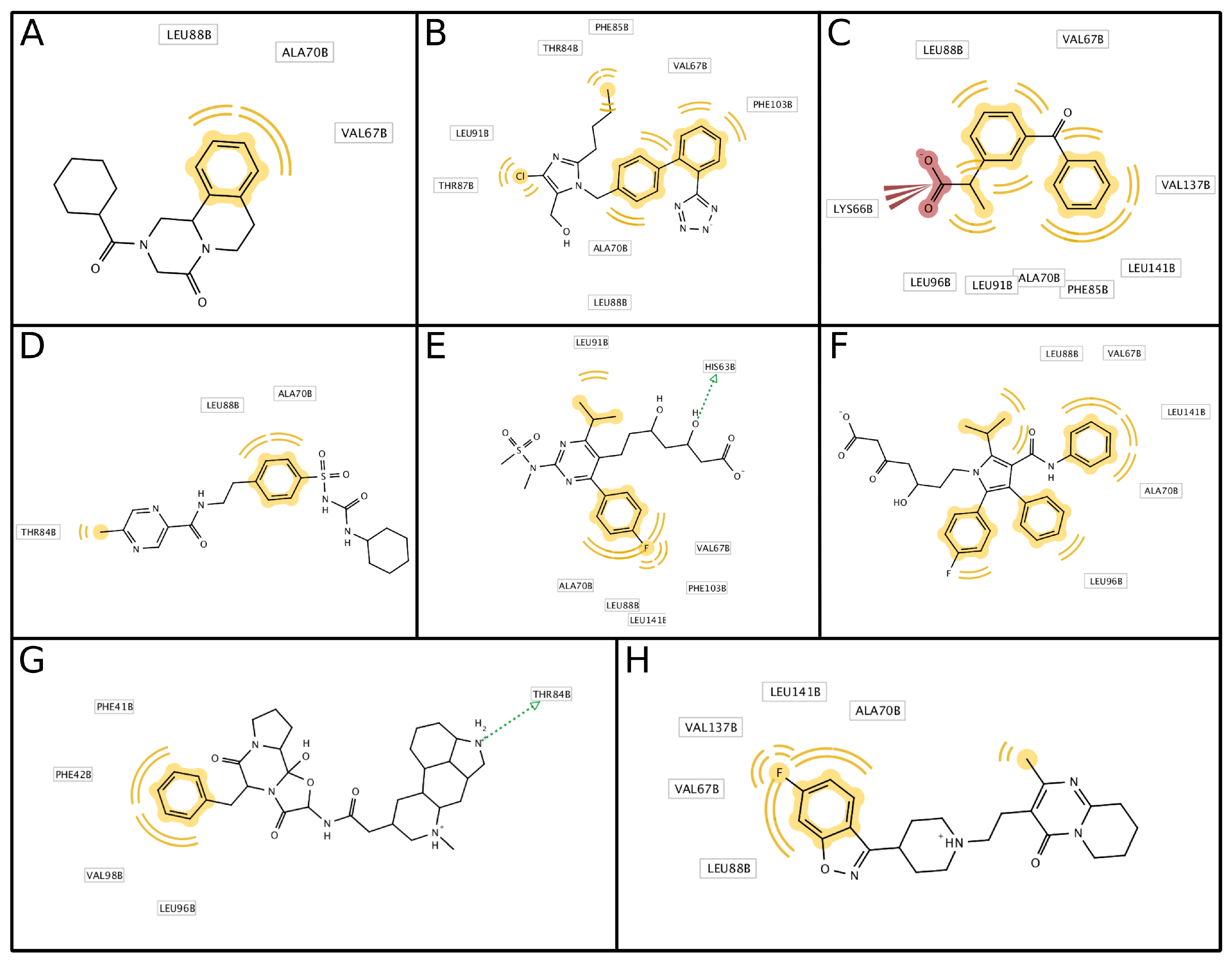
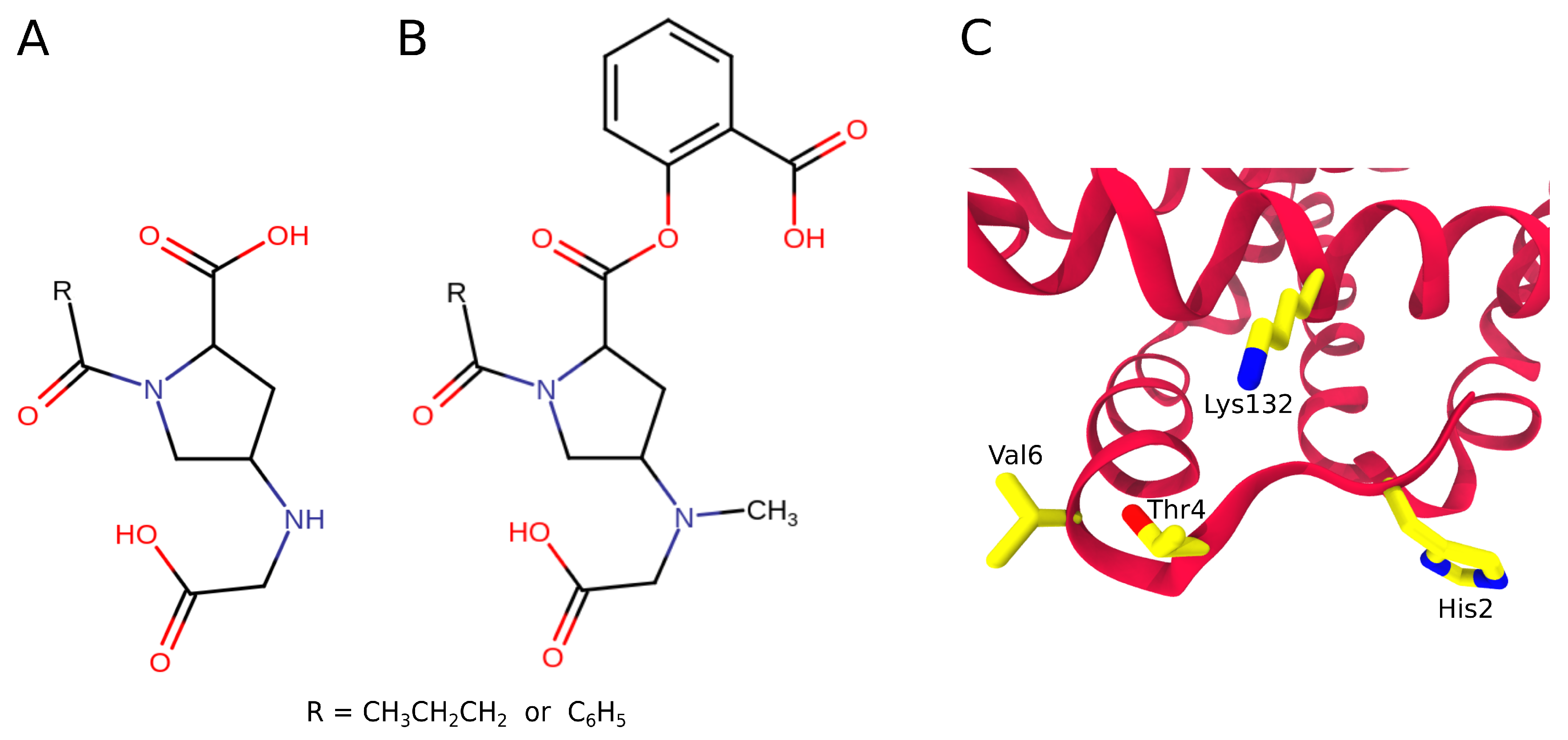
3.3. Antisickling Agents from In Silico Screening
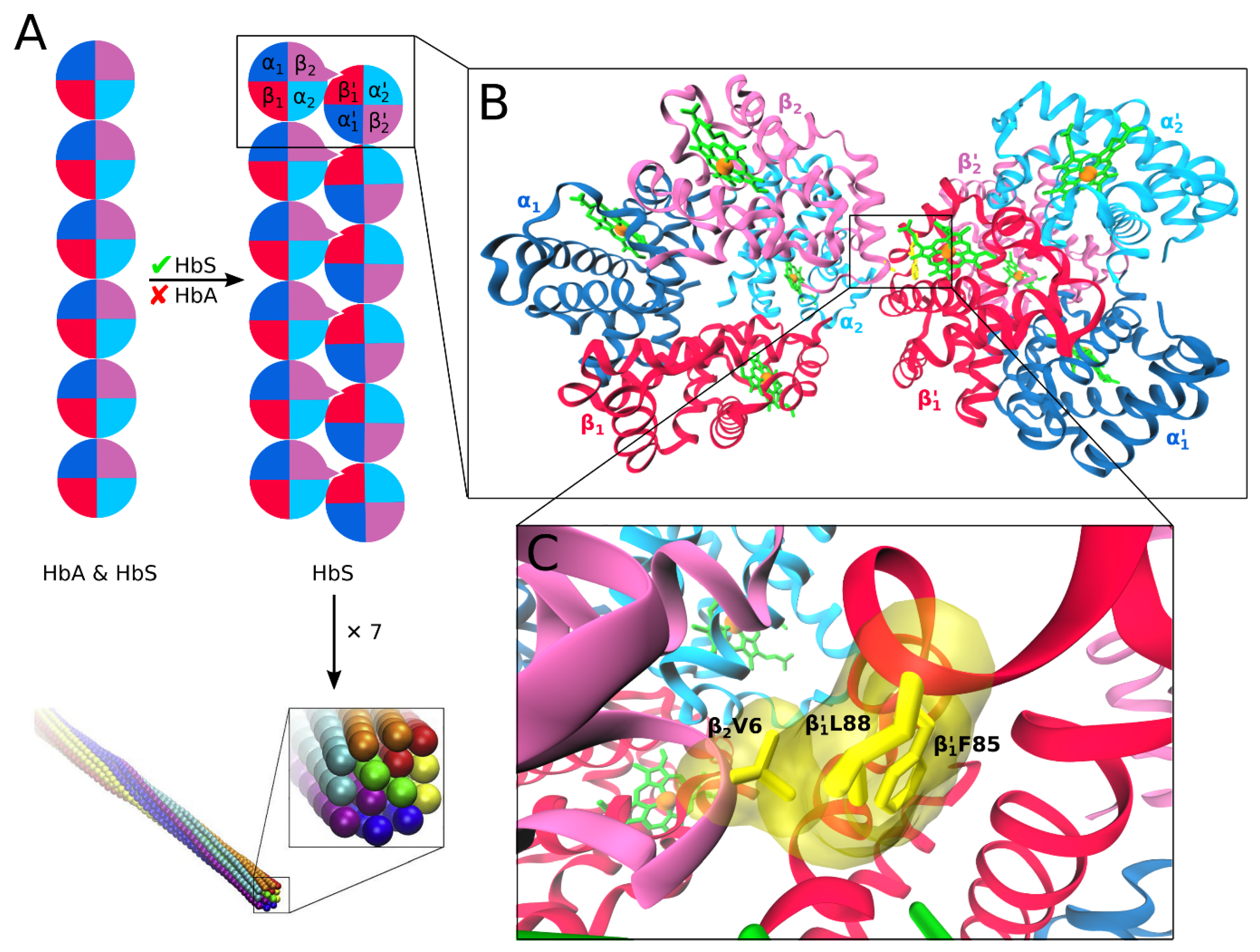
3.4. Interprotein Contacts During HbS Aggregation
4. Amino Acid-Derived Antisickling Compounds
4.1. Peptide Length and HbS Polymerization Inhibition
4.2. Peptide Hydrophobicity and Hydrophilicity
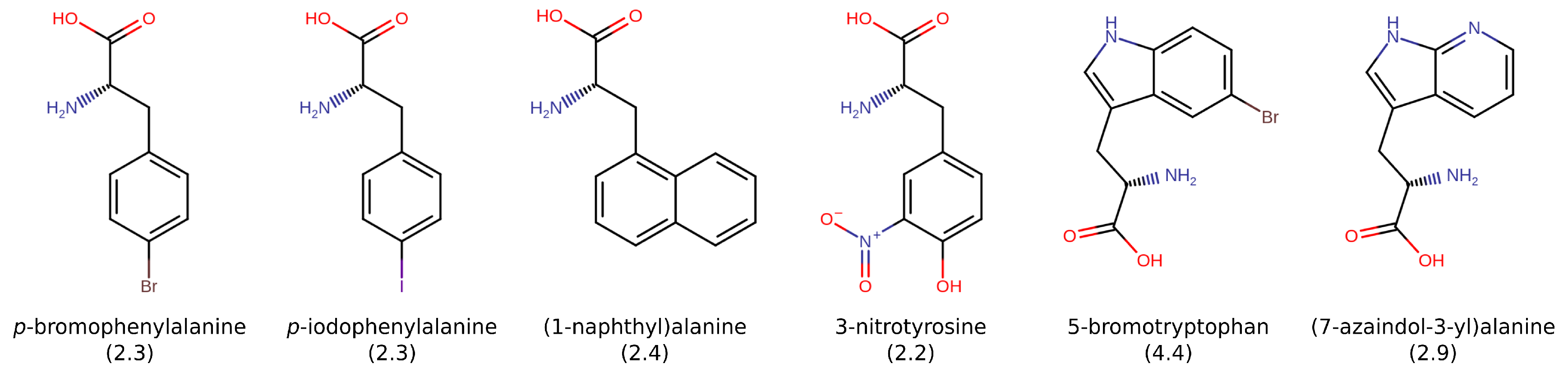
4.3. Effects of Amino Acids and Specific Chemical Properties
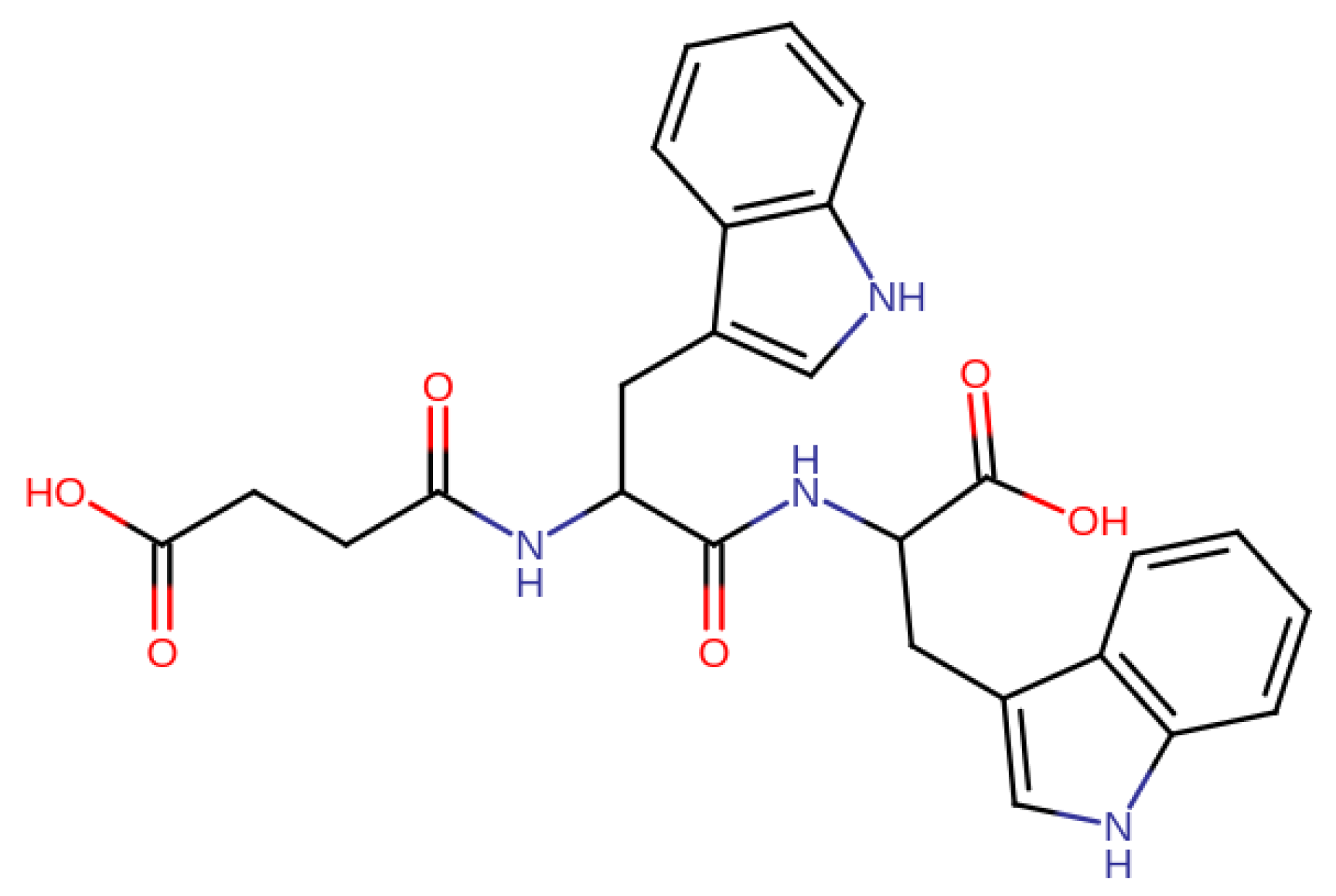
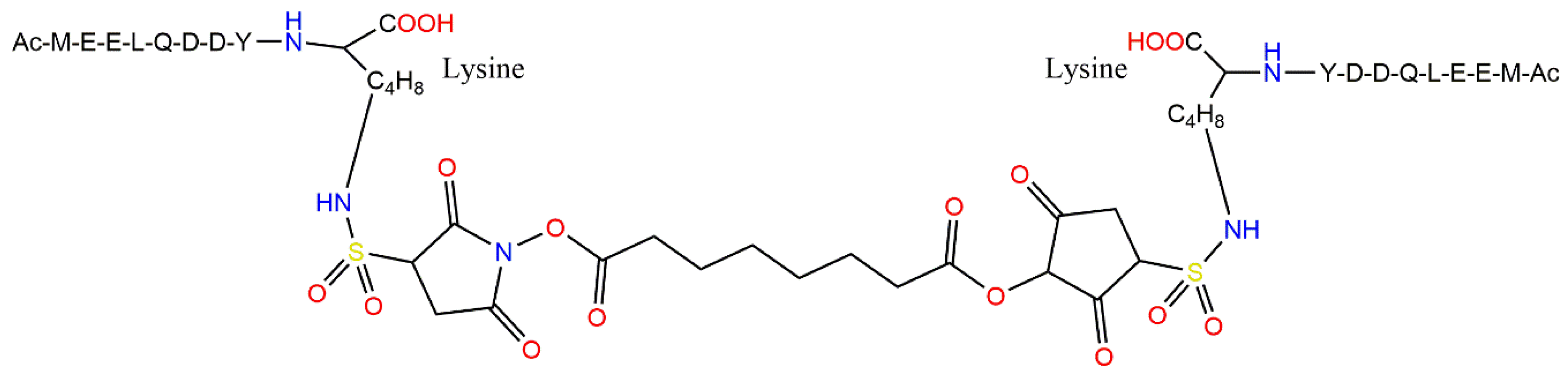
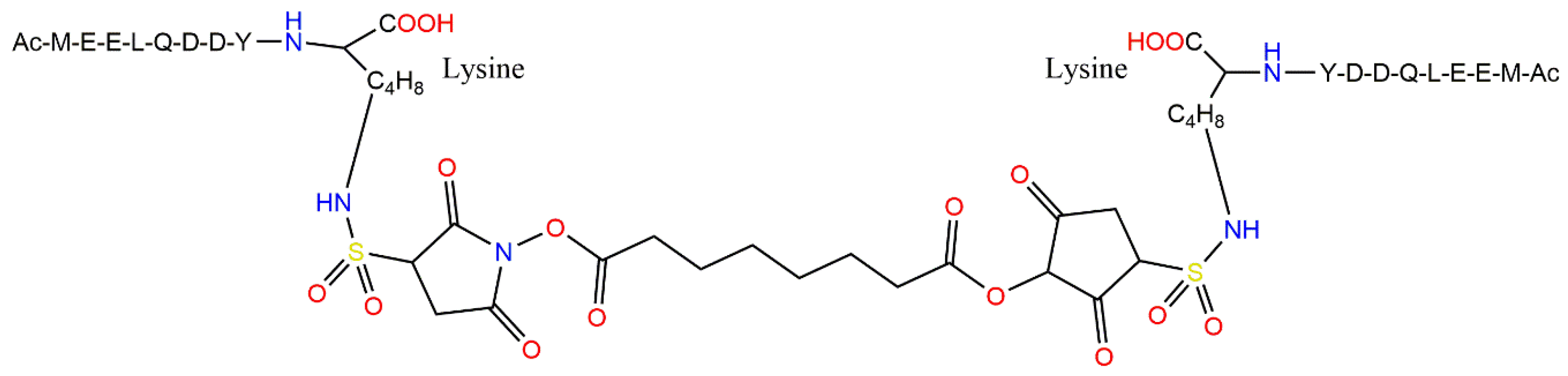
4.4. Highly Potent Peptide Inhibitors
5. Benefits and Challenges Associated with Peptide-Based Drugs
6. Conclusions
Funding
Conflicts of Interest
References
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ingram, V.M. Abnormal human haemoglobins: I. The comparison of normal human and sickle-cell haemoglobins by fingerprinting. Biochim. Biophys. Acta 1958, 28, 539–545. [Google Scholar] [CrossRef]
- Poillon, W.N.; Kim, B.C.; Labotka, R.J.; Hicks, C.U.; Kark, J.A. Antisickling effects of 2,3-diphosphoglycerate depletion. Blood 1995, 85, 3289–3296. [Google Scholar] [CrossRef] [PubMed]
- Poillon, W.N.; Kim, B.C. 2,3-Diphosphoglycerate and intracellular pH as interdependent determinants of the physiologic solubility of deoxyhemoglobin S. Blood 1990, 76, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Li, X.; Dao, M.; Lykotrafitis, G.; Karniadakis, G.E. Biomechanics and biorheology of red blood cells in sickle cell anemia. J. Biomech. 2017, 50, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Conner, B.J.; Reyes, A.A.; Morin, C.; Itakura, K.; Teplitz, R.L.; Wallace, R.B. Detection of sickle cell beta S-globin allele by hybridization with synthetic oligonucleotides. Proc. Natl. Acad. Sci. USA 1983, 80, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Hahn, E.V.; Gillespie, E.B. Sickle cell anemia. Report of a case greatly improved by splenectomy. Experimental study of sickle cell formation. Arch. Intern. Med. 1927, 39, 233–254. [Google Scholar] [CrossRef]
- Brugnara, C.; de Franceschi, L.; Alper, S.L. Inhibition of Ca2+-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Investig. 1993, 92, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Mohandas, N.; Evans, E. Adherence of sickle erythrocytes to vascular endothelial cells: Requirement for both cell membrane changes and plasma factors. Blood 1984, 64, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Chien, S.; Usami, S.; Bertles, J.F. Abnormal rheology of oxygenated blood in sickle cell anemia. J. Clin. Investig. 1970, 49, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.V. Sickle Cell Disease: Advances in Treatment. Ochsner J. 2018, 18, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, M.H.; Barton, F.; Castro, O.; Pegelow, C.H.; Ballas, S.K.; Kutlar, A.; Orringer, E.; Bellevue, R.; Olivieri, N.; Eckman, J.; et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Strouse, J.J.; Lanzkron, S.; Beach, M.C.; Haywood, C.; Park, H.; Witkop, C.; Wilson, R.F.; Bass, E.B.; Segal, J.B. Hydroxyurea for sickle cell disease: A systematic review for efficacy and toxicity in children. Pediatrics 2008, 122, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Inusa, B.P.D.; Atoyebi Wale, A.A.; Idhate, T.; Dogara, L.; Ijei, I.; Qin, Y.; Anie, K.; Lawson, J.O.; Hsu, L. Low-dose hydroxycarbamide therapy may offer similar benefit as maximum tolerated dose for children and young adults with sickle cell disease in low-middle-income Settings. F1000Res 2018, 7. F1000 Faculty Rev-1407. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Kaya, B.; Pancham, S.; Keenan, R.; Anderson, J.; Akanni, M.; Howard, J.; British Society for Haematology. Guidelines for the use of hydroxycarbamide in children and adults with sickle cell disease: A British Society for Haematology Guideline. Br. J. Haematol. 2018, 181, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Awwalu, S.; Okpetu, L.; Waziri, A.D. Effect of hydroxyurea on clinical and laboratory parameters of sickle cell anaemia patients in North–West Nigeria. Egypt. J. Haematol. 2017, 42, 70. [Google Scholar] [CrossRef]
- Wong, T.E.; Brandow, A.M.; Lim, W.; Lottenberg, R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014, 124, 3850–3857. [Google Scholar] [CrossRef] [Green Version]
- Murad, M.H.; Hazem, A.; Prokop, L. Hydroxyurea for Sickle Cell Disease: A Systematic Review of Benefits, Harms, and Barriers of Utilization, 2012 Prepared for the National Heart, Lung, and Blood Institute (NHLBI) Prepared by the Knowledge and Encounter Research Unit. Mayo Clin. 2012, 2012, 1–116. [Google Scholar]
- Ware, R.E. Optimizing hydroxyurea therapy for sickle cell anemia. ASH Educ. Program Book 2015, 2015, 436–443. [Google Scholar]
- Agrawal, R.K.; Patel, R.K.; Shah, V.; Nainiwal, L.; Trivedi, B. Hydroxyurea in Sickle Cell Disease: Drug Review. Indian J. Hematol. Blood Transfus. 2014, 30, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; Jerrell, J.M.; Stallworth, J.R. Clinical complications in severe pediatric sickle cell disease and the impact of hydroxyurea. Pediatr. Blood Cancer 2011, 56, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, O.; Yovetich, N.A.; Scott, J.P.; Owen, W.; Miller, S.T.; Schultz, W.; Lockhart, A.; Aygun, B.; Flanagan, J.; Bonner, M.; et al. Investigators of the Stroke with Transfusions Changing to Hydroxyurea Clinical Trial (SwiTCH). Pain and other non-neurological adverse events in children with sickle cell anemia and previous stroke who received hydroxyurea and phlebotomy or chronic transfusions and chelation: Results from the SWiTCH clinical trial. Am. J. Hematol. 2013, 88, 932–938. [Google Scholar] [PubMed] [Green Version]
- Nzouakou, R.; Bachir, D.; Lavaud, A.; Habibi, A.; Lee, K.; Lionnet, F.; Hulin, A.; Jouault, H.; Préhu, C.; Roudot-Thoraval, F.; et al. Clinical follow-up of hydroxyurea-treated adults with sickle cell disease. Acta Haematol. 2011, 125, 145–152. [Google Scholar] [CrossRef]
- Wang, W.C.; Ware, R.E.; Miller, S.T.; Iyer, R.V.; Casella, J.F.; Minniti, C.P.; Rana, S.; Thornburg, C.D.; Rogers, Z.R.; Kalpatthi, R.V.; et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet 2011, 377, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, C.D.; Files, B.A.; Luo, Z.; Miller, S.T.; Kalpatthi, R.; Iyer, R.; Seaman, P.; Lebensburger, J.; Alvarez, O.; Thompson, B.; et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012, 120, 4304–4310. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, A.; Cho, G.; Howard, J.; Layton, M.; Afif, M.; Hughes, R.G.; Philpott, N.J.; Patankar, S.; Davies, S.C. North West London Haemoglobinopathy Registry Group. Feasibility and benefit of hydroxycarbamide as a long-term treatment for sickle cell disease patients: Results from the North West London Sickle Cell Disease Registry. Am. J. Hematol. 2011, 86, 958–961. [Google Scholar] [CrossRef]
- Lobo, C.L.; Pinto, J.F.; Nascimento, E.M.; Moura, P.G.; Cardoso, G.P.; Hankins, J.S. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br. J. Haematol. 2013, 161, 852–860. [Google Scholar] [CrossRef]
- Sharef, S.W.; Al-Hajri, M.; Beshlawi, I.; Al-Shahrabally, A.; Elshinawy, M.; Zachariah, M.; Mevada, S.T.; Bashir, W.; Rawas, A.; Taqi, A.; et al. Optimizing Hydroxyurea use in children with sickle cell disease: Low dose regimen is effective. Eur. J. Haematol. 2013, 90, 519–524. [Google Scholar] [CrossRef]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef]
- Hansen, I.O.; Sørensen, A.L.; Hasselbalch, H.C. Second malignancies in hydroxyurea and interferon-treated Philadelphia-negative myeloproliferative neoplasms. Eur. J. Haematol. 2017, 98, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; McCarthy, W.F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am. J. Hematol. 2010, 85, 403–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGann, P.T.; Flanagan, J.M.; Howard, T.A.; Dertinger, S.D.; He, J.; Kulharya, A.S.; Thompson, B.W.; Ware, R.E.; BABY HUG Investigators. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: Results from the BABY-HUG Phase III Clinical Trial. Pediatr. Blood Cancer 2012, 59, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A phase 3 trial of l-glutamine in sickle cell disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Rumen, N.M. Inhibition of sickling in erythrocytes by amino acids. Blood 1975, 45, 45–48. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, R.D.; Gleason, E.M.; Schatzman, G.L.; Cawein, M.J. An In Vitro method for screening compounds for the effect on the rate of sickling of erythrocytes. J. Int. Med. Res. 1976, 4, 375–381. [Google Scholar] [CrossRef]
- Quinn, C.T. L-Glutamine for sickle cell anemia: More questions than answers. Blood 2018, 132, 689–693. [Google Scholar] [CrossRef]
- Zerez, C.R.; Lachant, N.A.; Lee, S.J.; Tanaka, K.R. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood 1988, 71, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Niihara, Y.; Zerez, C.R.; Akiyama, D.S.; Tanaka, K.R. Increased red cell glutamine availability in sickle cell anemia: Demonstration of increased active transport, affinity, and increased glutamate level in intact red cells. J. Lab. Clin. Med. 1997, 130, 83–90. [Google Scholar] [CrossRef]
- Kiessling, K.; Roberts, N.; Gibson, J.S.; Ellory, J.C. A comparison in normal individuals and sickle cell patients of reduced glutathione precursors and their transport between plasma and red cells. Hematol. J. 2000, 1, 243–249. [Google Scholar] [CrossRef]
- Morris, C.R.; Suh, J.H.; Hagar, W.; Larkin, S.; Bland, D.A.; Steinberg, M.H.; Vichinsky, E.P.; Shigenaga, M.; Ames, B.; Kuypers, F.A.; et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 2008, 111, 402–410. [Google Scholar] [CrossRef]
- Bhatia, M.; Sheth, S. Hematopoietic stem cell transplantation in sickle cell disease: Patient selection and special considerations. J. Blood Med. 2015, 6, 229–238. [Google Scholar] [PubMed] [Green Version]
- Mentzer, W.C.; Heller, S.; Pearle, P.R.; Hackney, E.; Vichinsky, E. Availability of related donors for bone marrow transplantation in sickle cell anemia. Am. J. Pediatr. Hematol. Oncol. 1994, 16, 27–29. [Google Scholar] [PubMed]
- Dallas, M.H.; Triplett, B.; Shook, D.R.; Hartford, C.; Srinivasan, A.; Laver, J.; Ware, R.; Leung, W. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2013, 19, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Bernaudin, F.; Socie, G.; Kuentz, M.; Chevret, S.; Duval, M.; Bertrand, Y.; Vannier, J.P.; Yakouben, K.; Thuret, I.; Bordigoni, P.; et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007, 110, 2749–2756. [Google Scholar] [CrossRef]
- Vermylen, C.; Cornu, G.; Ferster, A.; Brichard, B.; Ninane, J.; Ferrant, A.; Zenebergh, A.; Maes, P.; Dhooge, C.; Benoit, Y.; et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998, 22, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simões, B.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Eurocord, the Pediatric Working Party of the European Society for Blood and Marrow Transplantation, and the Center for International Blood and Marrow Transplant Research. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar]
- Lopez, A.D.; Williams, T.N.; Levin, A.; Tonelli, M.; Singh, J.A.; Burney, P.G.; Rehm, J.; Volkow, N.D.; Koob, G.; Ferri, C.P. Remembering the forgotten non-communicable diseases. BMC Med. 2014, 12, 200. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.N. Sickle Cell Disease in sub-Saharan Africa. Hematol. Oncol. Clin. N. Am. 2016, 30, 343–358. [Google Scholar] [CrossRef]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global burden of sickle cell anaemia in children under five, 2010-2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [Green Version]
- Marilyn, J.; Malik, T.P.; Vercellotti, G.M. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nat. Rev. Drug Discov. 2019, 18, 139–158. [Google Scholar]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Mehanna, A.S.; Wireko, F.C.; Whitney, J.; Thomas, R.P.; Orringer, E.P. Vanillin, a Potential Agent for the Treatment of Sickle Cell Anemia. Blood 1991, 77, 1334–1341. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, A.; Lui, F.E.; Wassaf, D.; Yefidoff-Freedman, R.; Casalena, D.; Palmer, M.A.; Meadows, J.; Mozzarelli, A.; Ronda, L.; Abdulmalik, O.; et al. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS Chem. Biol. 2014, 9, 2318–2325. [Google Scholar] [CrossRef]
- Stocker, J.W.; De Franceschi, L.; McNaughton-Smith, G.A.; Corrocher, R.; Beuzard, Y.; Brugnara, C. ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood 2003, 101, 2412–2418. [Google Scholar] [CrossRef]
- Ataga, K.I.; Reid, M.; Ballas, S.K.; Yasin, Z.; Bigelow, C.; James, L.S.; Smith, W.R.; Galacteros, F.; Kutlar, A.; Hull, J.H.; et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: A phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br. J. Haematol. 2011, 153, 92–104. [Google Scholar]
- McArthur, J.G.; Svenstrup, N.; Chen, C.; Fricot, A.; Carvalho, C.; Nguyen, J.; Nguyen, P.; Prachikova, A.; Abdulla, F.; Vercellotti, G.M.; et al. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica 2019. [Google Scholar] [CrossRef] [Green Version]
- Iyamu, E.W.; Turner, E.A.; Asakura, T. In vitro effects of NIPRISAN (Nix-0699): A naturally occurring, potent antisickling agent. Br. J. Haematol. 2002, 118, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Swift, R.; Abdulmalik, O.; Chen, Q.K.; Asakura, T.; Gustafson, K.; Simon, J.E.; Zaman, V.; Quiusky, K.A.; Hassell, K.L.; Shapira, I.; et al. SCD-101: A new anti-sickling drug reduces pain and fatigue and improves red blood cell shape in peripheral blood of patients with sickle cell disease. Blood 2016, 128, 121. [Google Scholar] [CrossRef]
- Eaton, W.A.; Bunn, H.F. Treating sickle cell disease by targeting HbS polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, T.; Wood, F.; Strader, M.B.; Alayash, A.I. Antisickling Drugs Targeting βCys93 Reduce Iron Oxidation and Oxidative Changes in Sickle Cell Hemoglobin. Front. Physiol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatge, M.S.; Ahmed, M.H.; Omar, A.S.; Pagare, P.P.; Rosef, S.; Kellogg, G.E.; Abdulmalik, O.; Safo, M.K. Crystal structure of carbonmonoxy sickle hemoglobin in R-state conformation. J. Struct. Biol. 2016, 194, 446–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitagliano, L.; Mazzarella, L.; Merlino, A.; Vergara, A. Fine sampling of the R→T quaternary-structure transition of a tetrameric hemoglobin. Chem. Eur. J. 2017, 23, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, A.S. Sickle cell anemia and antisickling agents then and now. Curr. Med. Chem. 2001, 8, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Asakura, T. Gelation of deoxyhemoglobin A in concentrated phosphate buffer. Exhibition of delay time prior to aggregation and crystallization of deoxyhemoglobin A. J. Biol. Chem. 1979, 254, 12273–12276. [Google Scholar]
- Adachi, K.; Matarasso, S.L.; Asakura, T. Nucleation-controlled aggregation of deoxyhemoglobin S. Effect of organic phosphates on the kinetics of aggregation of deoxyhemoglobin S in concentrated phosphate buffer. Biochim. Biophys. Acta 1980, 624, 372–377. [Google Scholar] [CrossRef]
- Adachi, K.; Asakura, T. Polymerization of deoxyhemoglobin CHarlem (beta 6 Glu replaced by Val, beta 73 Asp replaced by Asn). The effect of beta 73 asparagine on the gelation and crystallization of hemoglobin. J. Mol. Biol. 1980, 144, 467–480. [Google Scholar] [CrossRef]
- Adachi, K.; Asakura, T. Hemoglobin gelation. Tex. Rep. Biol. Med. 1980, 40, 251–260. [Google Scholar]
- Adachi, K.; Asakura, T. Gelation and crystallization of sickle (Hb S and Hb C Harlem) and non-sickle hemoglobins (Hb A and Hb C) in concentrated phosphate buffer. Prog. Clin. Biol. Res. 1981, 55, 123–144. [Google Scholar]
- Adachi, K.; Asakura, T. Aggregation and crystallization of hemoglobins A, S, and C. Probable formation of different nuclei for gelation and crystallization. J. Biol. Chem. 1981, 256, 1824–1830. [Google Scholar] [PubMed]
- Asakura, T.; Ohnishi, S.T.; Adachi, K.; Ozguc, M.; Hashimoto, K.; Devlin, M.T.; Schwartz, E. Effect of piracetam on sickle erythrocytes and sickle hemoglobin. Biochim. Biophys. Acta 1981, 668, 397–405. [Google Scholar] [CrossRef]
- Adachi, K.; Asakura, T. Kinetics of the polymerization of hemoglobin in high and low phosphate buffers. Blood Cells 1982, 8, 213–224. [Google Scholar] [PubMed]
- Delalic, Z.; Takashima, S.; Adachi, K.; Asakura, T. Dielectric constant of sickle cell hemoglobin. Dielectric properties of sickle cell hemoglobin in solution and gel. J. Mol. Biol. 1983, 168, 659–671. [Google Scholar] [CrossRef]
- Adachi, K.; Kim, J.; Travitz, R.; Harano, T.; Asakura, T. Effect of amino acid at the beta 6 position on surface hydrophobicity, stability, solubility, and the kinetics of polymerization of hemoglobin. Comparisons among Hb A (Glu beta 6), Hb C (Lys beta 6), Hb Machida (Gln beta 6), and Hb S (Val beta 6). J. Biol. Chem. 1987, 262, 12920–12925. [Google Scholar]
- Bridges, K.R.; Barabino, G.D.; Brugnara, C.; Cho, M.R.; Christoph, G.W.; Dover, G.; Ewenstein, B.M.; Golan, D.E.; Guttmann, C.R.; Hofrichter, J.; et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996, 88, 4701–4710. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Ballas, S.K.; Hantgan, R.R.; Kim-Shapiro, D.B. Aggregation of normal and sickle hemoglobin in high concentration phosphate buffer. Biophys. J. 2004, 87, 4113–4121. [Google Scholar] [CrossRef] [Green Version]
- Chikezie, P.C. Sodium metabisulfite-induced polymerization of sickle cell hemoglobin incubated in the extracts of three medicinal plants (Anacardium occidentale, Psidium guajava, and Terminalia catappa). Pharmacogn. Mag. 2011, 7, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Perutz, M.F.; Fermi, G.; Abraham, D.J.; Poyart, C.; Bursaux, E. Hemoglobin as a Receptor of Drugs and Peptides: X-ray Studies of the Stereochemistry of Binding. J. Am. Chem. Soc. 1986, 108, 1064–1078. [Google Scholar] [CrossRef]
- Abraham, D.J. Correlation of partition coefficients with antisickling activity of simple alcohols, amides and ureas. Blood Cells 1982, 8, 345–355. [Google Scholar]
- Abraham, E.C.; Stallings, M.; Abraham, A.; Garbutt, G.J. Modification of sickle hemoglobin by acetaldehyde and its effect on oxygenation, gelation and sickling. Biochim. Biophys. Acta 1982, 705, 76–81. [Google Scholar] [CrossRef]
- Abraham, D.J.; Perutz, M.F.; Phillips, S.E. Physiological and x-ray studies of potential antisickling agents. Proc. Natl. Acad. Sci. USA 1983, 80, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Li, X.; Vekilov, P.G.; Karniadakis, G.E. Probing the Twisted Structure of Sickle Hemoglobin Fibers via Particle Simulations. Biophys. J. 2016, 110, 2085–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, D.J.; Adachi, K.; Royer, W.E., Jr. The high resolution crystal structure of deoxyhemoglobin S. J. Mol. Biol. 1997, 272, 398–407. [Google Scholar] [CrossRef]
- Oder, E.; Safo, M.K.; Abdulmalik, O.; Kato, G.J. New developments in anti-sickling agents: Can drugs directly prevent the polymerization of sickle haemoglobin in vivo? Br. J. Haematol. 2016, 175, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, Z.; Li, M.; McKendry, R.; Horton, M.; Caruana, D.J. Investigation of Sickle-Cell Haemoglobin Polymerisation under Electrochemical Control. ChemPhysChem 2013, 14, 2143–2148. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J. Mol. Biol. 1985, 183, 611–631. [Google Scholar] [CrossRef]
- Eaton, W.A.; Hofrichter, J. Sickle cell hemoglobin polymerization. Adv. Prot. Chem. 1990, 40, 63–279. [Google Scholar]
- Lu, L.; Li, Z.; Li, H.; Li, X.; Vekilov, P.G.; Karniadaki, G.E. Quantitative prediction of erythrocyte sickling for the development of advanced sickle cell therapies. Sci. Adv. 2019, 5, eaax3905. [Google Scholar] [CrossRef] [Green Version]
- Castle, B.T.; Odde, D.J.; Wood, D.K. Rapid and inefficient kinetics of sickle hemoglobin fiber growth. Sci. Adv. 2019, 5, eaau1086. [Google Scholar] [CrossRef] [Green Version]
- Oosawa, F.; Asakura, S. Thermodynamics of the Polymerization of Protein; Academic Press: London, UK; New York, NY, USA, 1975. [Google Scholar]
- Gardner, M.K.; Charlebois, B.D.; Jánosi, I.M.; Howard, J.; Hunt, A.J.; Odde, D.J. Rapid microtubule self-assembly kinetics. Cell 2011, 146, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ferrone, F.A. Dissecting the Energies that Stabilize Sickle Hemoglobin Polymers. Biophys. J. 2013, 105, 2149–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Ferrone, F.A. Homogeneous nucleation in sickle hemoglobin: Stochastic measurements with a parallel method. Biophys. J. 1997, 72, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Otto, J.M.; Plumb, J.O.M.; Clissold, E.; Kumar, S.B.; Wakeham, D.J.; Schmidt, W.; Grocott, M.P.W.; Richards, T.; Montgomery, H.E. Hemoglobin concentration, total hemoglobin mass and plasma volume in patients: Implications for anemia. Haematologica 2017, 102, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Akinbami, A.; Dosunmu, A.; Adediran, A.; Oshinaike, O.; Adebola, P.; Arogundade, O. Haematological values in homozygous sickle cell disease in steady state and haemoglobin phenotypes AA controls in Lagos, Nigeria. BMC Res. Notes 2012, 5, 396. [Google Scholar] [CrossRef] [Green Version]
- Abere, C.J.; Okoye, C.J.; Agoreyo, F.O.; Eze, G.I.; Jesuorobo, R.I.; Egharevba, C.O.; Aimator, P.O. Antisickling and toxicological evaluation of the leaves of Scoparia dulcis Linn (Scrophulariaceae). BMC Complement. Altern. Med. 2015, 15, 414. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.G.; Pagare, P.P.; Ghatge, M.S.; Safo, R.P.; Gazi, A.; Chen, Q.; David, T.; Alabbas, A.B.; Musayev, F.N.; Venitz, J.; et al. Design, Synthesis, and Biological Evaluation of Ester and Ether Derivatives of Antisickling Agent 5-HMF for the Treatment of Sickle Cell Disease. Mol. Pharm. 2017, 14, 3499–3511. [Google Scholar] [CrossRef] [Green Version]
- Abdulmalik, O.; Ghatge, M.S.; Musayev, F.N.; Parikh, A.; Chen, Q.; Yang, J.; Nnamani, I.; Danso-Danquah, R.; Eseonu, D.N.; Asakura, T.; et al. Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin. Acta Crystallogr. Sect. D 2011, 67, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Dash, B.P.; Archana, Y.; Satapathy, N.; Naik, S.K. Search for antisickling agents from plants. Pharmacogn. Rev. 2013, 7, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Henry, E.R.; Hofrichter, J.; Smith, J.F.; Cellmer, T.; Dunkelberger, E.B.; Metaferia, B.B.; Jones-Straehle, S.; Boutom, S.; Christoph, G.W.; et al. Kinetic assay shows that increasing red cell volume could be a treatment for sickle cell disease. Proc. Natl. Acad. Sci. USA 2017, 114, E689–E696. [Google Scholar] [CrossRef] [Green Version]
- Padmos, M.A.; Sackey, K.; Roberts, G.T.; Kulozik, A.; Bail, S.; Morris, J.S.; Serjeant, B.E.; Serjeant, G.R. Two different forms of homozygous sickle cell disease occur in Saudi Arabia. Br. J. Haematol. 1991, 79, 93–98. [Google Scholar] [CrossRef] [PubMed]
- El-Hazmi, M.A.F. Heterogeneity and Variation of Clinical and Haematological Expression of Haemoglobin S in Saudi Arabs. Acta Haematol. 1992, 88, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.; Jasuja, R.; Kwong, S.; Briehl, R.W.; Ferrone, F.A. Nonideality and the nucleation of sickle hemoglobin. Biophys. J. 2000, 79, 1016–1022. [Google Scholar] [CrossRef] [Green Version]
- Safo, M.K.; Ko, T.P.; Schreiter, E.R.; Russell, J.E. Structural basis for the antipolymer activity of Hb ζ2βs2 trapped in a tense conformation. J. Mol. Struct. 2015, 1099, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 2017, 8, 321–326. [Google Scholar] [CrossRef]
- Pagare, P.P.; Ghatge, M.S.; Musayev, F.N.; Deshpande, T.M.; Chen, Q.; Braxton, C.; Kim, S.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; et al. Rational design of pyridyl derivatives of vanillin for the treatment of sickle cell disease. Bioorg. Med. Chem. 2018, 26, 2530–2538. [Google Scholar] [CrossRef]
- Oksenberg, D.; Dufu, K.; Patel, M.P.; Chuang, C.; Li, Z.; Xu, Q.; Silva-Garcia, A.; Zhou, C.; Hutchaleelaha, A.; Patskovska, L.; et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br. J. Haematol. 2016, 175, 141–153. [Google Scholar] [CrossRef]
- Dufu, K.; Patel, M.; Oksenberg, D.; Cabrales, P. GBT440 improves red blood cell deformability and reduces viscosity of sickle cell blood under deoxygenated conditions. Clin. Hemorheol. Microcirc. 2018, 70, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Nnamani, I.N.; Joshi, G.S.; Danso-Danquah, R.; Abdulmalik, O.; Asakura, T.; Abraham, D.J.; Safo, M.K. Pyridyl derivatives of benzaldehyde as potential antisickling agents. Chem Biodivers. 2008, 5, 1762–1769. [Google Scholar] [CrossRef]
- Olubiyi, O.O.; Olagunju, M.O.; Oni, J.O.; Olubiyi, A.O. Structural basis of antisickling effects of selected FDA approved drugs: A drug repurposing study. Curr. Comput. Aided Drug Des. 2018, 14, 106–116. [Google Scholar] [CrossRef]
- Yee, M.E.; Lane, P.A.; Archer, D.R.; Joiner, C.H.; Eckman, J.R.; Guasch, A. Losartan therapy decreases albuminuria with stable glomerular filtration and permselectivity in sickle cell anemia. Blood Cells Mol. Dis. 2018, 69, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T.; Saraf, S.L.; Gordeuk, V.R.; Fitzhugh, C.D.; Creary, S.E.; Bodas, P.; George, A.; Raj, A.B.; Nero, A.C.; Terrell, C.E.; et al. Losartan for the nephropathy of sickle cell anemia: A phase-2, multicenter trial. Am. J. Hematol. 2017, 92, E520–E528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manno, M.; San Biagio, P.L.; Palma, M.U. The role of pH on instability and aggregation of sickle hemoglobin solutions. Proteins 2004, 55, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.A. Polymerization and sickle cell disease: A molecular view. Microcirculation 2004, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Benesch, R.E.; Kwong, S.; Benesch, R. Structural basis of antisickling effects of selected FDA approved drugs: A drug repurposing study. Nature 1982, 299, 231. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.L.; Johnson, J.; Bookchin, R.M.; Garel, M.C.; Rosa, J.; Schiliro, G.; Wajcman, H.; Labie, D.; Moo-Penn, W.; Castro, O. β-Chain contact sites in the haemoglobin S polymer. Nature 1980, 283, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Rhoda, M.-D.; Martin, J.; Blouquit, Y.; Garel, M.-C.; Edelstein, S.J.; Rosa, J. Sickle cell hemoglobin fiber formation strongly inhibited by the Stanleyville II mutation (alpha 78 Asn leads to Lys). Biochem. Biophys. Res. Commun. 1983, 111, 8–13. [Google Scholar] [CrossRef]
- Benesch, R.E.; Kwong, S.; Edalji, R.; Benesch, R. α Chain mutations with opposite effects on the gelation of hemoglobin S. J. Biol. Chem. 1979, 254, 8169–8172. [Google Scholar]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Gorecki, M.; Votano, J.R.; Rich, A. Peptide inhibitors of sickle hemoglobin aggregation: Effect of hydrophobicity. Biochemistry 1980, 19, 1564–1568. [Google Scholar] [CrossRef]
- Votano, J.R.; Gorecki, M.; Rich, A. Sickle hemoglobin aggregation: A new class of inhibitors. Science 1977, 196, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Olubiyi, O.O.; Frenzel, D.; Bartnik, D.; Gluck, J.M.; Brener, O.; Nagel-Steger, L.; Funke, S.A.; Willbold, D.; Strodel, B. Amyloid aggregation inhibitory mechanism of arginine-rich D-Peptides. Curr. Med. Chem. 2014, 21, 1448–1457. [Google Scholar] [CrossRef]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1-42 by mirror image phage display. ChemBioChem 2003, 4, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Willbold, D. Peptides for therapy and diagnosis of Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.S.; Xu, H.; Flewelen, T.C.; Holzhauer, S.L.; Retherford, D.; Jones, D.W.; Frei, A.C.; Pritchard, K.A., Jr.; Hillery, C.A.; Hogg, N.; et al. A novel hemoglobin-binding peptide reduces cell-free hemoglobin in murine hemolytic anemia. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H328–H336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, S.; Yang, J.T. Oligopeptides as potential antiaggregation agents for deoxyhemoglobin S. Proc. Natl. Acad. Sci. USA 1977, 74, 5431–5434. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, C.T.; Schechter, A.N. Inhibition of sickle hemoglobin gelation by amino acids and related compounds. Biochemistry 1978, 17, 5455–5459. [Google Scholar] [CrossRef]
- Schechter, A.N. Stereospecific inhibitors of the gelation of sickle hemoglobin. Hemoglobin 1980, 4, 335–345. [Google Scholar] [CrossRef]
- Adachi, K.; Ding, M.; Wehrli, S.; Reddy, K.S.; Surrey, S.; Horiuchi, K. Effects of different beta73 amino acids on formation of 14-stranded fibers of Hb S versus double-stranded crystals of Hb C-Harlem. Biochemistry 2003, 42, 4476–4484. [Google Scholar] [CrossRef]
- Akbar, M.G.K.; Tamura, Y.; Ding, M.; Ding, H.; Rosenblatt, M.M.; Reddy, K.S.; Surrey, S.; Adachi, K. Inhibition of Hb S Polymerization in vitro by a novel 15-mer EF helix β73 His-containing peptide. Biochemistry 2006, 45, 8358–8367. [Google Scholar] [CrossRef] [Green Version]
- Ross, P.D.; Subramanian, S. Inhibition of sickle cell hemoglobin gelation by some aromatic compounds. Biochem. Biophys. Res. Commun. 1977, 77, 1217–1223. [Google Scholar] [CrossRef]
- Ross, P.D.; Hofrichter, J.; Eaton, W.A. Thermodynamics of gelation of sickle cell deoxyhemoglobin. J. Mol. Biol. 1977, 115, 111–134. [Google Scholar] [CrossRef]
- Noguchi, C.T.; Schechter, A.N. Effects of amino acids on gelation kinetics and solubility of sickle hemoglobin. Biochem. Biophys. Res. Commun. 1977, 74, 637–642. [Google Scholar] [CrossRef]
- Abraham, D.J.; Mokotoff, M.; Sheh, L.; Simmons, J.E. Design, synthesis, and testing of antisickling agents. 2. Proline derivatives designed for the donor site. J. Med. Chem. 1983, 26, 549–554. [Google Scholar] [CrossRef]
- Shamsuddin, M.; Mason, R.G.; Ritchey, J.M.; Honig, G.R.; Klotz, I.M. Sites of acetylation of sickle cell hemoglobin by aspirin. Proc. Natl. Acad. Sci. USA 1974, 71, 4693–4697. [Google Scholar] [CrossRef] [Green Version]
- Abraham, D.J.; Safo, M.K.; Boyiri, T.; Danso-Danquah, R.E.; Kister, J.; Poyart, C. How allosteric effectors can bind to the same protein residue and produce opposite shifts in the allosteric equilibrium. Biochemistry 1995, 34, 15006–15020. [Google Scholar] [CrossRef]
- Briehl, R.W.; Ewert, S. Effects of pH, 2,3-diphosphoglycerate and salts on gelation of sickle cell deoxyhemoglobin. J. Mol. Biol. 1973, 80, 445–458. [Google Scholar] [CrossRef]
- Poillon, W.N. Noncovalent inhibitors of sickle hemoglobin gelation: Effects of aryl-substituted alanines. Biochemistry 1982, 21, 1400–1406. [Google Scholar] [CrossRef]
- Noguchi, C.T.; Ackerman, S.; DiMaio, J.; Schiller, P.W.; Schechter, A.N. The effect of phenylalanine derivatives on the solubility of deoxyhemoglobin S. A model class of gelation inhibitors. Mol. Pharmacol. 1983, 23, 100–103. [Google Scholar]
- Ross, P.D.; Subramanian, S. Biochemical and Clinical Aspects of Hemoglobin Abnormalities; Caughey, W.S., Ed.; Academic Press: New York, NY, USA, 1978; pp. 629–646. [Google Scholar]
- Walder, J.A.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Armone, A. The interaction of hemoglobin with the cytoplasmic domain band 3 of the human erythrocyte membrane. J. Biol. Chem. 1984, 259, 10238–10246. [Google Scholar]
- Danish, E.H.; Lundren, D.W.; Harris, J.W. Inhibition of hemoglobin S polymerization by N-terminal band 3 peptides: New class of inhibitors: Solubility studies. Am. J. Hematol. 1994, 47, 106–112. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, X.; Shi, X.; Yang, F.; Cao, Y.; Qin, X.; Hou, Z.; Xie, M.; Liu, N.; Fang, Q.; et al. Alpha-helical motif as inhibitors of toxic amyloid-beta oligomer generation via highly specific recognition of amyloid surface. iScience 2019, 17, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saelices, L.; Nguyen, B.A.; Chung, K.; Wang, Y.; Ortega, A.; Lee, J.H.; Coelho, T.; Bijzet, J.; Benson, M.D.; Eisenberg, D.S. A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils. J. Biol. Chem. 2019, 294, 6130–6141. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pinter, M.; Tal, S.; Scherzer-Attali, R.; Abu-Hussien, M.; Alyagor, I.; Eisenbaum, T.; Gazit, E.; Segal, D. Naphthoquinone-Tryptophan Hybrid Inhibits Aggregation of the Tau-Derived Peptide PHF6 and Reduces Neurotoxicity. J. Alzheimer’s Dis. 2016, 51, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Marqus, S.; Pirogova, E.; Terrence, J.P. Evaluation of the use of therapeutic peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, L.; Quentin, J.B.; David, E.G. Protein Therapeutics: A summary and pharmacological classification. Nat. Rev. 2008, 7, 21–39. [Google Scholar]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic Peptides: Technological Advances Driving Peptides into Development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Thayer, A.M. Improving peptides. Chem. Eng. News Arch. 2011, 89, 13–20. [Google Scholar] [CrossRef]
- Werle, M.; Schnürch, B.A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Fasano, A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998, 16, 152–157. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.J.; Cho, C.H. Peptides as targeting probes against tumor vasculature for diagnosis and drug delivery. J. Transl. Med. 2012, 10, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, R.; Marbach, P.; Bauer, W.; Briner, U.; Fricker, G.; Bruns, C.; Pless, J. SDZ CO 611: A highly potent glycated analog of somatostatin with improved oral activity. Life Sci. 1993, 53, 517–525. [Google Scholar] [CrossRef]
- Kihlberg, J.; Ahman, J.; Walse, B.; Drakenberg, T.; Nilsson, A.; Söderberg-ahlm, C.; Bengtsson, B.; Olsson, H. Glycosylated peptide hormones: Pharmacological properties and conformational studies of analogues of [1-desamino,8-D-arginine]vasopressin. J. Med. Chem. 1995, 38, 161–169. [Google Scholar] [CrossRef]
- Varamini, P.; Mansfeld, F.M.; Blanchfield, J.T.; Wyse, B.D.; Smith, M.T.; Toth, I. Synthesis and biological evaluation of an orally active glycosylated endomorphin-1. J. Med. Chem. 2012, 55, 5859–5867. [Google Scholar] [CrossRef]
- Kovalszky, I.; Surmacz, E.; Scolaro, L.; Cassone, M.; Ferla, R.; Sztodola, A.; Olah, J.; Hatfield, M.P.; Lovas, S.; Otvos, L., Jr. Leptin-based glycopeptide induces weight loss and simultaneously restores fertility in animal models. Diabetes Obes. Metab. 2010, 12, 393–402. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Murthy, A.S.N.; Yang, H.; Im, J. Versatility of cell-penetrating peptides for intracellular delivery of siRNA. Drug Deliv. 2018, 25, 2005–2015. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, D.; Accardo, A.; Diaferia, C.; Milano, V.; Guillon, J.; Ronga, L.; Rossi1, F. Peptide-Based Drug-Delivery Systems in Biotechnological Applications: Recent Advances and Perspectives. Molecules 2019, 24, 351. [Google Scholar] [CrossRef] [Green Version]
- Schemmert, S.; Schartmann, E.; Zafiu, C.; Kass, B.; Hartwig, S.; Lehr, S.; Bannach, O.; Langen, K.-J.; Shah, N.J.; Kutzsche, J.; et al. Aβ oligomer elimination restores cognition in transgenic Alzheimer’s mice with full-blown pathology. Mol. Neurobiol. 2018, 56, 2211–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | MIMR |
|---|---|
| Suc-(L-Phe)-(L-Phe)-(L-Phe) | 9.5 ± 0.5 |
| Suc-(L-Phe)-(L-Phe)-(L-Phe)-(L-Arg) | 10.0 ± 1.0 |
| Suc-(L-Trp)-(L-Trp) | 10.0 ± 0.5 |
| Suc-(L-Trp)-(L-Phe) | 12.5 ± 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olubiyi, O.O.; Olagunju, M.O.; Strodel, B. Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease. Molecules 2019, 24, 4551. https://doi.org/10.3390/molecules24244551
Olubiyi OO, Olagunju MO, Strodel B. Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease. Molecules. 2019; 24(24):4551. https://doi.org/10.3390/molecules24244551
Chicago/Turabian StyleOlubiyi, Olujide O., Maryam O. Olagunju, and Birgit Strodel. 2019. "Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease" Molecules 24, no. 24: 4551. https://doi.org/10.3390/molecules24244551
APA StyleOlubiyi, O. O., Olagunju, M. O., & Strodel, B. (2019). Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease. Molecules, 24(24), 4551. https://doi.org/10.3390/molecules24244551