
A Self-Healing Polymer with Fast Elastic Recovery upon Stretching


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
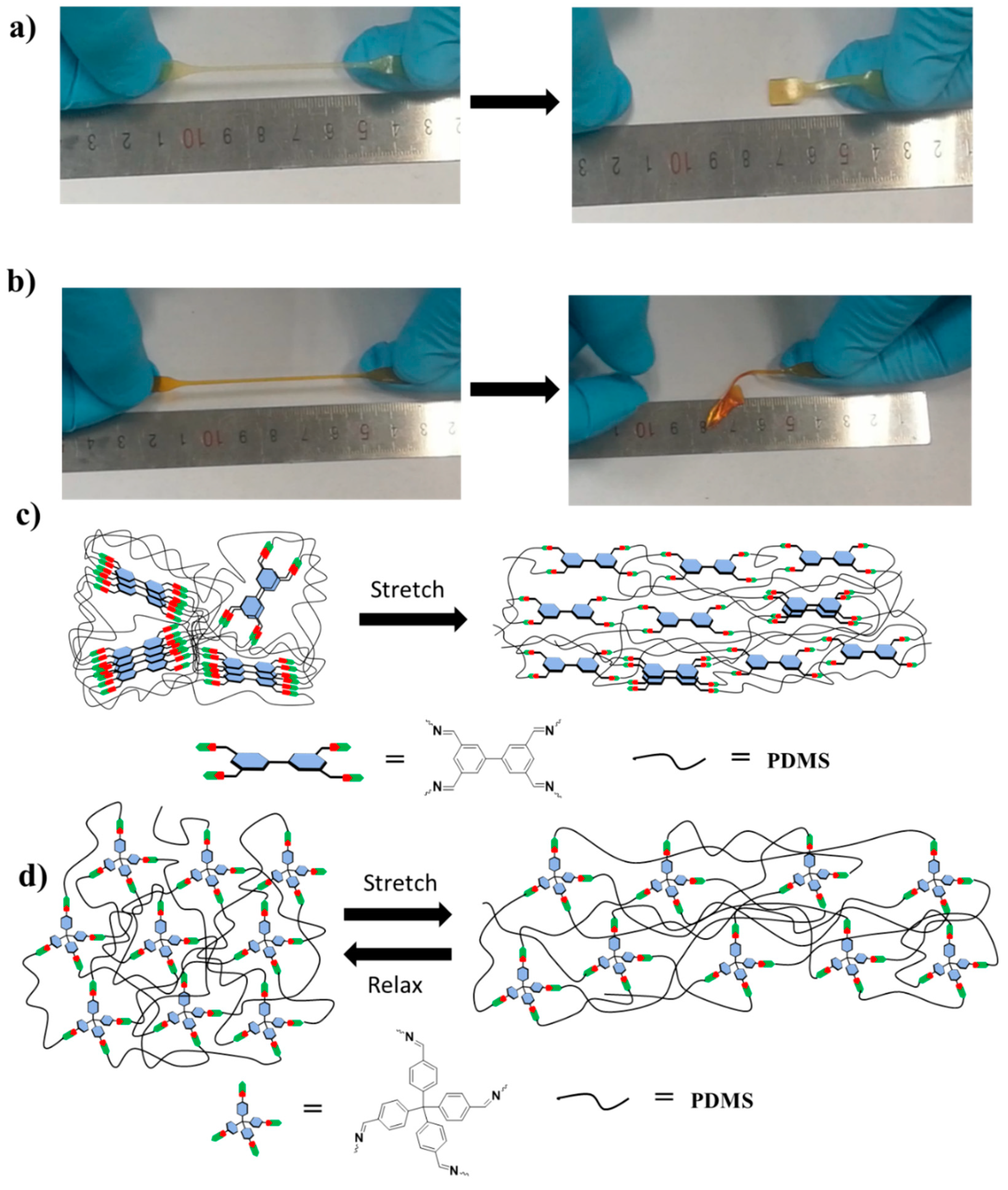
1. Introduction
2. Results and Discussion
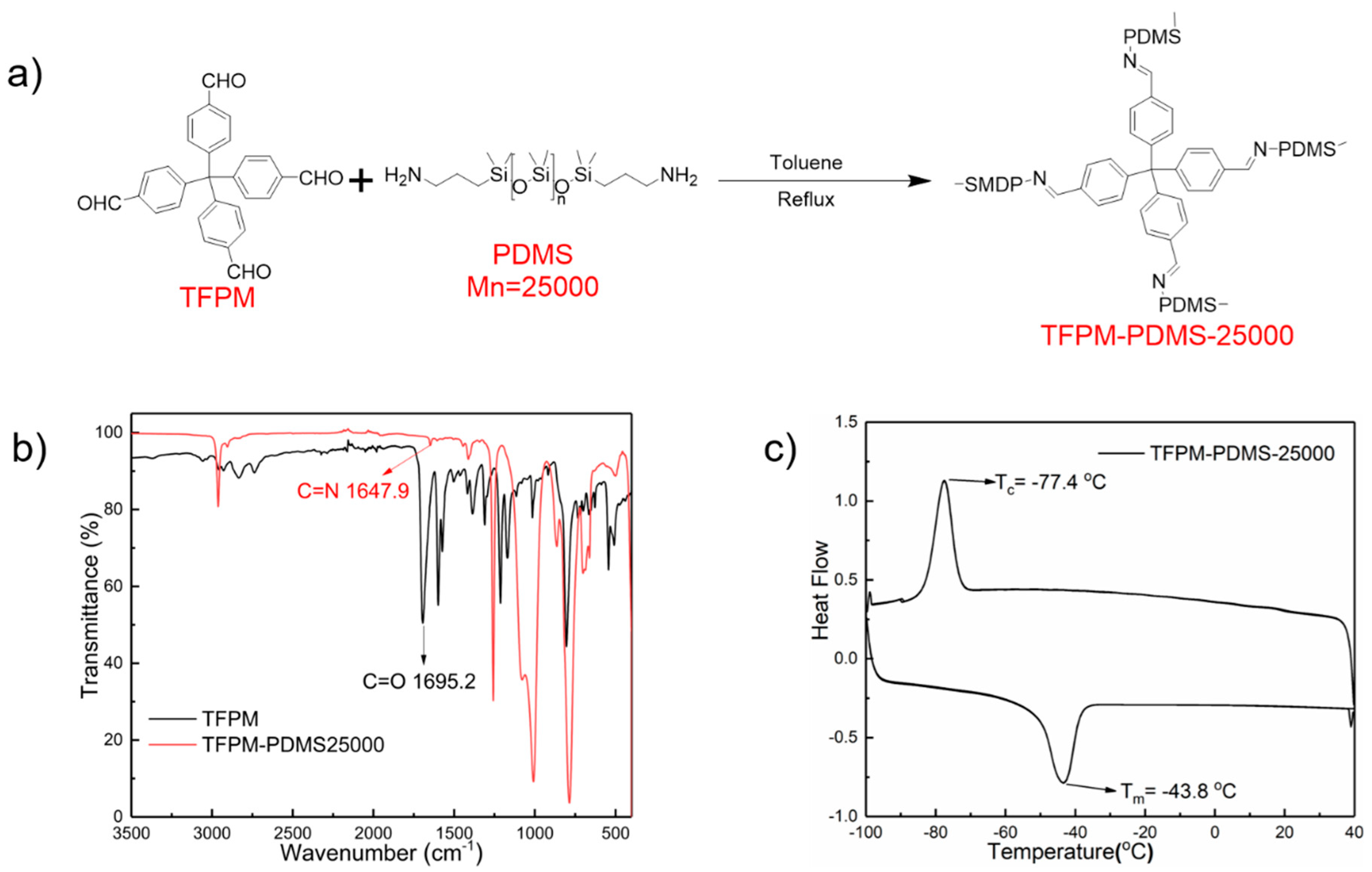
2.1. Synthesis and General Characterization
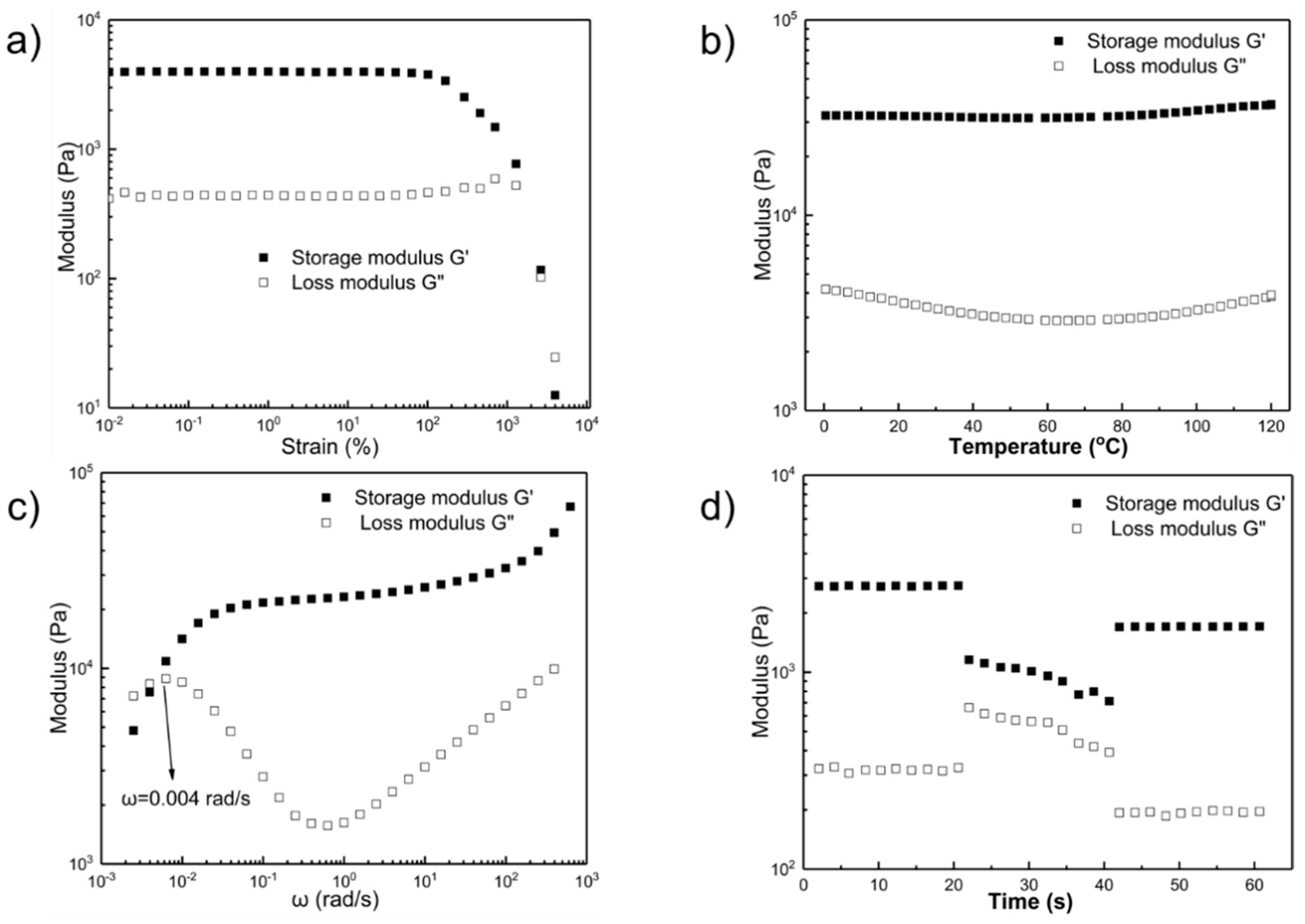
2.2. Rheological Studies
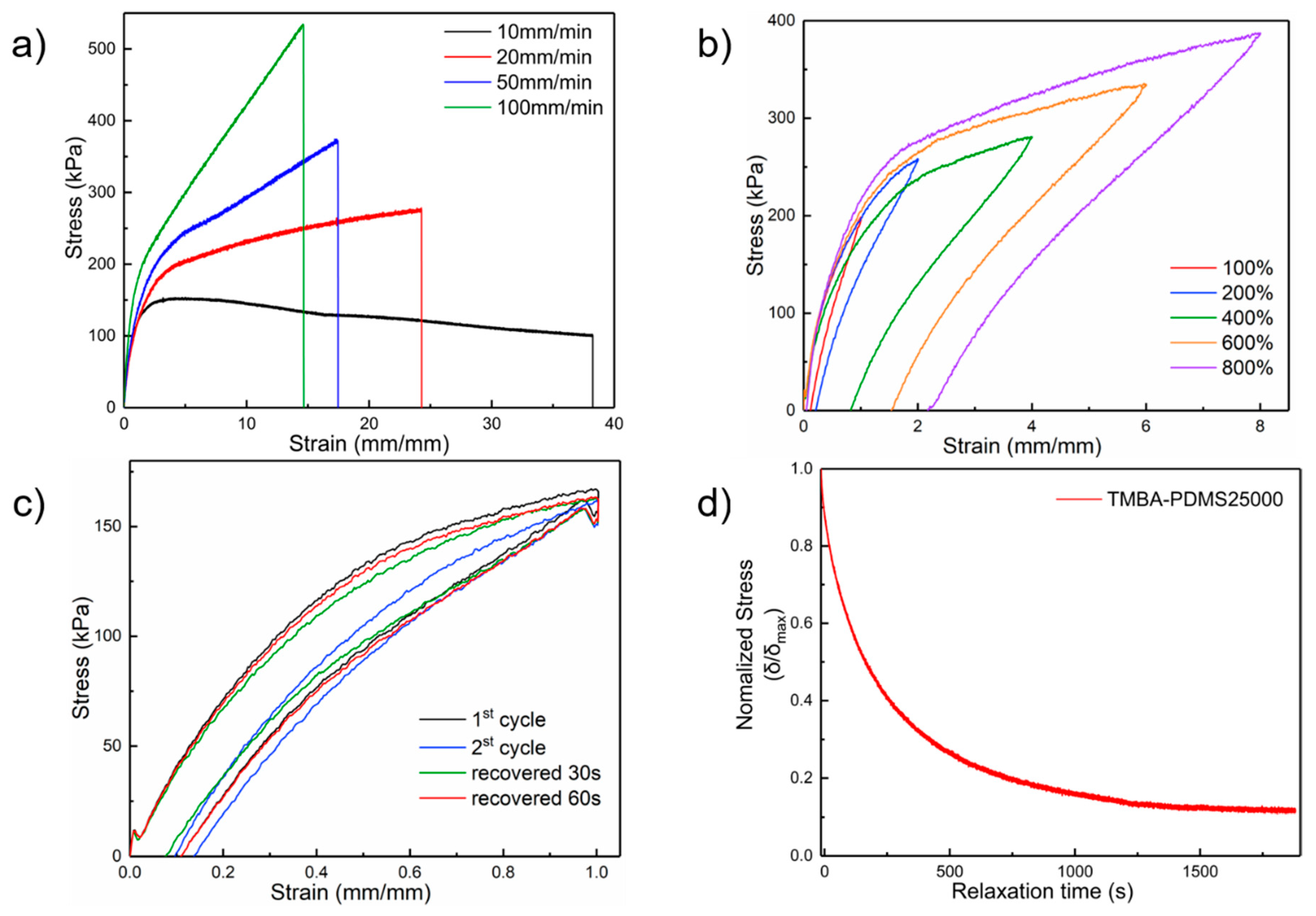
2.3. Mechanical Property
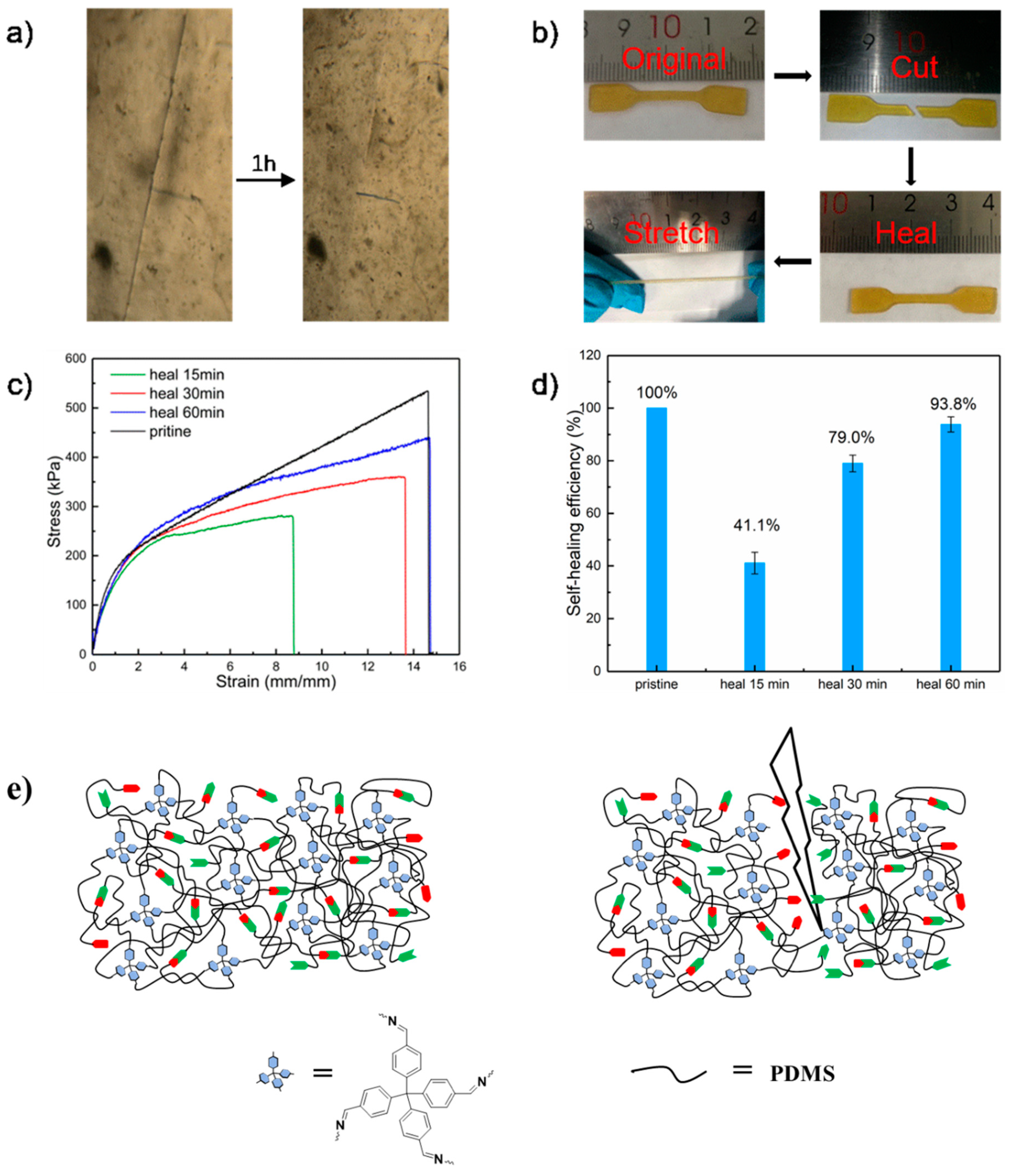
2.4. Self-Healing Properties
2.5. Mechanism for the Fast Elastic Recovery
3. Experimental Section
3.1. Materials
3.2. General Measurements
3.3. Preparation of TFPM-PDMS-25000 Polymer Films
3.4. Rheological Test
3.5. Mechanical and Self-Healing Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Binder, W.H. Self-Healing Polymers: From Principles to Applications; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2013. [Google Scholar]
- AbdolahZadeh, M.; van der Zwaag, S.; Garcia, S.J. Self-healing corrosion-protective sol-gel coatings based on extrinsic and intrinsic healing approaches. In Self-Healing Materials; Hager, M.D., van der Zwaag, S., Schubert, U.S., Eds.; Springer International Publishing: Cham, Germany, 2016; pp. 185–218. [Google Scholar]
- Hia, I.-L.; Vahedi, V.; Pasbakhsh, P. Self-healing polymer composites: Prospects, challenges, and applications. Polym. Rev. 2016, 56, 225–261. [Google Scholar] [CrossRef]
- Syrett, J.A.; Becer, C.R.; Haddleton, D. Self-healing and self-mendable polymers. Polym. Chem. 2010, 1, 978–987. [Google Scholar] [CrossRef]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Samadzadeh, M.; Boura, S.H.; Peikari, M.; Kasiriha, S.; Ashrafi, A. A review on self-healing coatings based on micro/nanocapsules. Prog. Org. Coat. 2010, 68, 159–164. [Google Scholar] [CrossRef]
- Seongpil, A.; Min, W.L.; Alexander, L.Y.; Sam, S.Y. A review on corrosion-protective extrinsic self-healing: Comparison of microcapsule-based systems and those based on core-shell vascular networks. Chem. Eng. J. 2018, 344, 206–220. [Google Scholar]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 749–797. [Google Scholar] [CrossRef]
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.S.; White, S.R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. [Google Scholar] [CrossRef]
- Yu, F.; Cao, X.; Du, J.; Wang, G.; Chen, X. Multifunctional hydrogel with good structure integrity, self-healing, and tissue-adhesive property formed by combining diels-alder click reaction and acylhydrazone bond. ACS Appl. Mater. Interfaces 2015, 7, 24023–24031. [Google Scholar] [CrossRef]
- Turkenburg, D.H.; Fischer, H.R. Diels-alder based, thermo-reversible cross-linked epoxies for use in self-healing composites. Polymer 2015, 79, 187–194. [Google Scholar] [CrossRef]
- Tanasi, P.; Santana, M.H.; Carretero-González, J.; Verdejo, R.; López-Manchado, M.A. Thermo-reversible crosslinked natural rubber: A diels-alder route for reuse and self-healing properties in elastomers. Polymer 2019, 175, 15–24. [Google Scholar] [CrossRef]
- Abdallh, M.; He, P.; Hearn, M.T.W.; Simon, G.P.; Saito, K. Light-switchable self-healing dynamic linear polymers: Reversible cycloaddition reactions of thymine-containing units. ChemPlusChem 2019, 84, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, B.-H.; Oh, M.; Park, D.H.; Lee, S. Repeatable crack self-healing by photochemical [2 + 2] cycloaddition of TCE-co-DCE monomers enclosed in homopolymer microcapsules. Polymers 2019, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Tang, C.; Li, F.; Jiang, H.; Chen, Y. Covalent crosslinked polymer gels with reversible sol-gel transition and self-healing properties. Macromolecules 2010, 43, 1191–1194. [Google Scholar] [CrossRef]
- Wang, P.; Deng, G.; Zhou, L.; Li, Z.; Chen, Y. Ultrastretchable, self-healable hydrogels based on dynamic covalent bonding and triblock copolymer micellization. ACS Macro Lett. 2017, 6, 881–886. [Google Scholar] [CrossRef]
- Dong, P.; Cui, K.; Xu, F.; Jiang, T.; Ma, Z. Synthesis of new ionic crosslinked polymer hydrogel combining polystyrene and poly (4-vinyl pyridine) and its self-healing through a reshuffling reaction of the trithiocarbonate moiety under irradiation of ultraviolet light. Polym. Int. 2018, 67, 868–873. [Google Scholar] [CrossRef]
- Yu, L.-X.; Zhuo, D.; Ran, R. Repeatable self-healing of gels based on a dynamic covalent trithiocarbonate cross-linker under microwave irradiation. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 749–754. [Google Scholar] [CrossRef]
- Chang, K.; Han, J.; Gu, S.-Y. A transparent, highly stretchable, self-healing polyurethane based on disulfide bonds. Eur. Polym. J. 2019, 112, 822–831. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Li, G.; Ling, L.; Zhang, G.; Sun, R.; Wong, C.-P. Heat-triggered poly(siloxane-urethane)s based on disulfide bonds for self-healing application. J. Appl. Polym. Sci. 2018, 135, 46532–46539. [Google Scholar] [CrossRef]
- Na, S.; Kim, Y.; Lee, C.; Liechti, K.M.; Suk, J. Adhesion and self-healing between monolayer molybdenum disulfide and silicon oxide. Sci. Rep. 2017, 7, 14740. [Google Scholar] [CrossRef]
- Imato, K.; Natterodt, J.C.; Sapkota, J.; Goseki, R.; Weder, C.; Takahara, A.; Otsuka, H. Dynamic covalent diarylbibenzofuranone-modified nanocellulose: Mechanochromic behaviour and application in self-healing polymer composites. Polym. Chem. 2017, 8, 2115–2122. [Google Scholar] [CrossRef]
- Imato, K.; Nishihara, M.; Kanehara, T.; Amamoto, Y.; Takahara, A.; Otsuka, H. Self-healing of chemical gels cross-linked by diarylbibenzofuranone-based trigger-free dynamic covalent bonds at room temperature. Angew. Chem. Int. Ed. 2012, 51, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, P.; Zhang, H.; Yan, C.; Zheng, Z.; Wu, B.; Yu, Y. A transparent, highly stretchable, autonomous self-healing poly(dimethylsilox-ane) elastomer. Macromol. Rapid Commun. 2017, 38, 1700110. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Negulescu, I.; Zhang, D. Dynamic covalent polymer networks based on degenerative imine bond exchange: Tuning the malleability and self-healing properties by solvent. Macromolecules 2016, 49, 6277–6284. [Google Scholar] [CrossRef]
- Kim, S.-M.; Jeon, H.; Shin, S.-H.; Park, S.-A.; Jegal, J.; Hwang, S.-Y.; Oh, D.X.; Park, J. Superior toughness and fast self-healing at room temperature engineered by transparent elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef]
- Neal, J.A.; Mozhdehi, D.; Guan, Z. Enhancing mechanical performance of a covalent self-healing material by sacrificial noncovalent bonds. J. Am. Chem. Soc. 2015, 137, 4846–4850. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Z.; Zhang, Q.; Lopez, J.; Wang, H.; Wu, H.-C.; Niu, S.; Yan, H.; Wang, S.; Lei, T.; et al. Quadruple H-bonding cross-linked supramolecular polymeric materials as substrates for stretchable, antitearing, and self-healable thin film electrodes. J. Am. Chem. Soc. 2018, 140, 5280–5289. [Google Scholar] [CrossRef]
- Herbst, F.; Döhler, D.; Michael, P.; Binder, W.H. Self-healing polymers via supramolecular forces. Macromol. Rapid Commun. 2013, 34, 203–220. [Google Scholar] [CrossRef]
- Jiang, H.; Duan, L.; Ren, X.; Gao, G. Hydrophobic association hydrogels with excellent mechanical and self-healing properties. Eur. Polym. J. 2019, 112, 660–669. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A healable supramolecular polymer blend based on aromatic π-π stacking and hydrogen-bonding Interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Hayes, W.; Mackay, M.E.; Rowan, S.J.; Colquhoun, H.M. A supramolecular polymer based on tweezer-type π-π stacking interactions: Molecular design for healability and enhanced toughness. Chem. Mater. 2011, 23, 6–8. [Google Scholar] [CrossRef]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.F.; Hayes, W.; Mackay, M.E.; Rowan, S.J. A self-repairing, supramolecular polymer system: Healability as a consequence of donor-acceptor π-π stacking interactions. Chem. Commun. 2009, 319, 6717–6719. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Kong, W.; Zhang, Z.; Wang, L.; Hu, Y.; Zhu, G.; Chen, R.; Ma, L.; Yan, W.; Wang, Y.; et al. Ionic liquid-immobilized polymer gel electrolyte with self-healing capability, high ionic conductivity and heat resistance for dendrite-free lithium metal batteries. Nano Energy 2018, 54, 17–25. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, Y.; Wu, Q.; Wang, S.; Huang, G.; Wu, J. Strong and tough self-healing elastomers enabled by dual reversible networks formed by ionic interactions and dynamic covalent bonds. Polymer 2018, 157, 172–179. [Google Scholar] [CrossRef]
- Weng, W.; Beck, J.B.; Jamieson, A.M.; Rowan, S.J. Understanding the mechanism of gelation and stimuli-responsive nature of a class of metallo-supramolecular gels. J. Am. Chem. Soc. 2006, 128, 11663–11672. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.-F.; Jia, X.-Y.; Lai, J.-C.; Sun, Y.; Li, C.-H.; Wu, J.-H.; Cao, Y.; You, X.-Z.; Bao, Z. A highly stretchable and autonomous self-healing polymer based on combination of Pt...Pt and π-π interactions. Macromol. Rapid Commun. 2016, 37, 1667–1675. [Google Scholar] [CrossRef]
- Li, C.-H.; Wang, C.; Keplinger, C.; Zuo, J.-L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C.; et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.-Y.; Mei, J.-F.; Lai, J.-C.; Li, C.-H.; You, X.-Z. A self-healing PDMS polymer with solvatochromic properties. Chem. Commun. 2015, 51, 8928–8930. [Google Scholar] [CrossRef]
- Jia, X.-Y.; Mei, J.-F.; Lai, J.-C.; Li, C.-H.; You, X.-Z. A highly stretchable polymer that can be thermally healed at mild temperature. Macromol. Rapid Commun. 2016, 37, 952–956. [Google Scholar] [CrossRef]
- Wang, D.-P.; Lai, J.-C.; Lai, H.-Y.; Mo, S.-R.; Zeng, K.-Y.; Li, C.-H.; Zuo, J.-L. Distinct mechanical and self-healing properties in two polydimethylsiloxane coordination polymers with fine-tuned bond strength. Inorg. Chem. 2018, 57, 3232–3242. [Google Scholar] [CrossRef]
- Lai, J.-C.; Li, L.; Wang, D.-P.; Zhang, M.-H.; Mo, S.-R.; Wang, X.; Zeng, K.-Y.; Li, C.-H.; Jiang, Q.; You, X.-Z.; et al. A rigid and healable polymer cross-linked by weak but abundant Zn(II)-carboxylate interactions. Nat. Commun. 2018, 9, 2725. [Google Scholar] [CrossRef]
- Mo, S.-R.; Lai, J.-C.; Zeng, K.-Y.; Wang, D.-P.; Li, C.-H.; Zuo, J.-L. New insights into the mechanical and self-healing properties of polymers cross-linked by Fe(III)-2, 6-pyridinedicarboxamide coordination complexes. Polym. Chem. 2019, 10, 362–371. [Google Scholar] [CrossRef]
- Lai, J.-C.; Jia, X.-Y.; Wang, D.-P.; Deng, Y.-B.; Zheng, P.; Li, C.-H.; Zuo, J.-L.; Bao, Z. Thermodynamically stable whilst kinetically labile coordination bonds lead to strong and tough self-healing polymers. Nat. Commun. 2019, 10, 1164. [Google Scholar] [CrossRef] [PubMed]
- Sadd, M.H. Elasticity Theory: Applications, and Numerics; Elsevier Butterworth Heinemann: New York, NY, USA, 2005; p. 69. [Google Scholar]
- Chen, Y.; Kushner, A.M.; Williams, G.A.; Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Mozhdehi, D.; Ayala, S.; Cromwell, O.R.; Guan, Z. Self-healing multiphase polymers via dynamic metal-ligand interactions. J. Am. Chem. Soc. 2014, 136, 16128–16131. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, C.; Tang, J.; Suo, Z. Strong and degradable adhesion of hydrogels. ACS Appl. Bio Mater. 2019, 25, 1781–1786. [Google Scholar] [CrossRef]
- Yang, H.; Li, C.; Yang, M.; Pan, Y.; Yin, Q.; Tang, J.; Qi, H.; Suo, Z. Printing hydrogels and elastomers in arbitrary sequence with strong adhesion. Adv. Funct. Mater. 2019, 29, 1901721. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, X.; Wang, J.; Tang, J.; Hu, J.; Lu, T.; Suo, Z. Fatigue of double-network hydrogels. Eng. Fract. Mech. 2018, 187, 74–93. [Google Scholar] [CrossRef]
- Wang, D.-P.; Zhao, Z.-H.; Li, C.-H.; Zuo, J.-L. An ultrafast self-healing polydimethylsiloxane elastomer with persistent sealing performance. Mater. Chem. Front. 2019, 3, 1411–1421. [Google Scholar] [CrossRef]
- Clarke, S.M.; Hotta, A.; Tajbakhsh, A.R.; Terentjev, E.M. Effect of cross-linker geometry on equilibrium thermal and mechanical properties of nematic elastomers. Phys. Rev. E 2001, 64, 061702. [Google Scholar] [CrossRef]
- Xua, J.; Liu, Z.; Erhan, S.Z.; Carriere, C.J. Cross-linkers control the viscoelastic properties of soybean oil-based biomaterials. J. Am. Oil Chem. Soc. 2004, 81, 813–816. [Google Scholar] [CrossRef]
- Tsuchitani, A.; Ashida, H.; Urayama, K. Pronounced effects of cross-linker geometries on the orientation coupling between dangling mesogens and network backbones in side-chain type liquid crystal elastomers. Polymer 2015, 61, 29–35. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Guan, X.; Tang, J.; Yusran, Y.; Li, Z.; Xue, M.; Fang, Q.; Yan, Y.; Valtchev, V.; et al. Three-dimensional ionic covalent organic frameworks for rapid, reversible, and selective ion exchange. J. Am. Chem. Soc. 2017, 139, 17771–17774. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Ma, Y.; Li, H.; Yusran, Y.; Xue, M.; Fang, Q.; Yan, Y.; Valtchev, V.; Qiu, S. Fast, ambient temperature and pressure ionothermal synthesis of three-dimensional covalent organic frameworks. J. Am. Chem. Soc. 2018, 140, 4494–4498. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ma, Y.; Li, H.; Guan, X.; Yusran, Y.; Xue, M.; Fang, Q.; Yan, Y.; Qiu, S.; Valtchev, V. Postsynthetic functionalization of three-dimensional covalent organic frameworks for selective extraction of lanthanide ions. Angew. Chem. Int. Ed. 2018, 57, 6042–6048. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound TFPM-PDMS-25000 are available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, P.-C.; Li, W.; Huang, W.; Li, C.-H. A Self-Healing Polymer with Fast Elastic Recovery upon Stretching. Molecules 2020, 25, 597. https://doi.org/10.3390/molecules25030597
Zhao P-C, Li W, Huang W, Li C-H. A Self-Healing Polymer with Fast Elastic Recovery upon Stretching. Molecules. 2020; 25(3):597. https://doi.org/10.3390/molecules25030597
Chicago/Turabian StyleZhao, Pei-Chen, Wen Li, Wei Huang, and Cheng-Hui Li. 2020. "A Self-Healing Polymer with Fast Elastic Recovery upon Stretching" Molecules 25, no. 3: 597. https://doi.org/10.3390/molecules25030597
APA StyleZhao, P.-C., Li, W., Huang, W., & Li, C.-H. (2020). A Self-Healing Polymer with Fast Elastic Recovery upon Stretching. Molecules, 25(3), 597. https://doi.org/10.3390/molecules25030597