Berberine Impairs the Survival of Triple Negative Breast Cancer Cells: Cellular and Molecular Analyses


Abstract
1. Introduction
2. Results
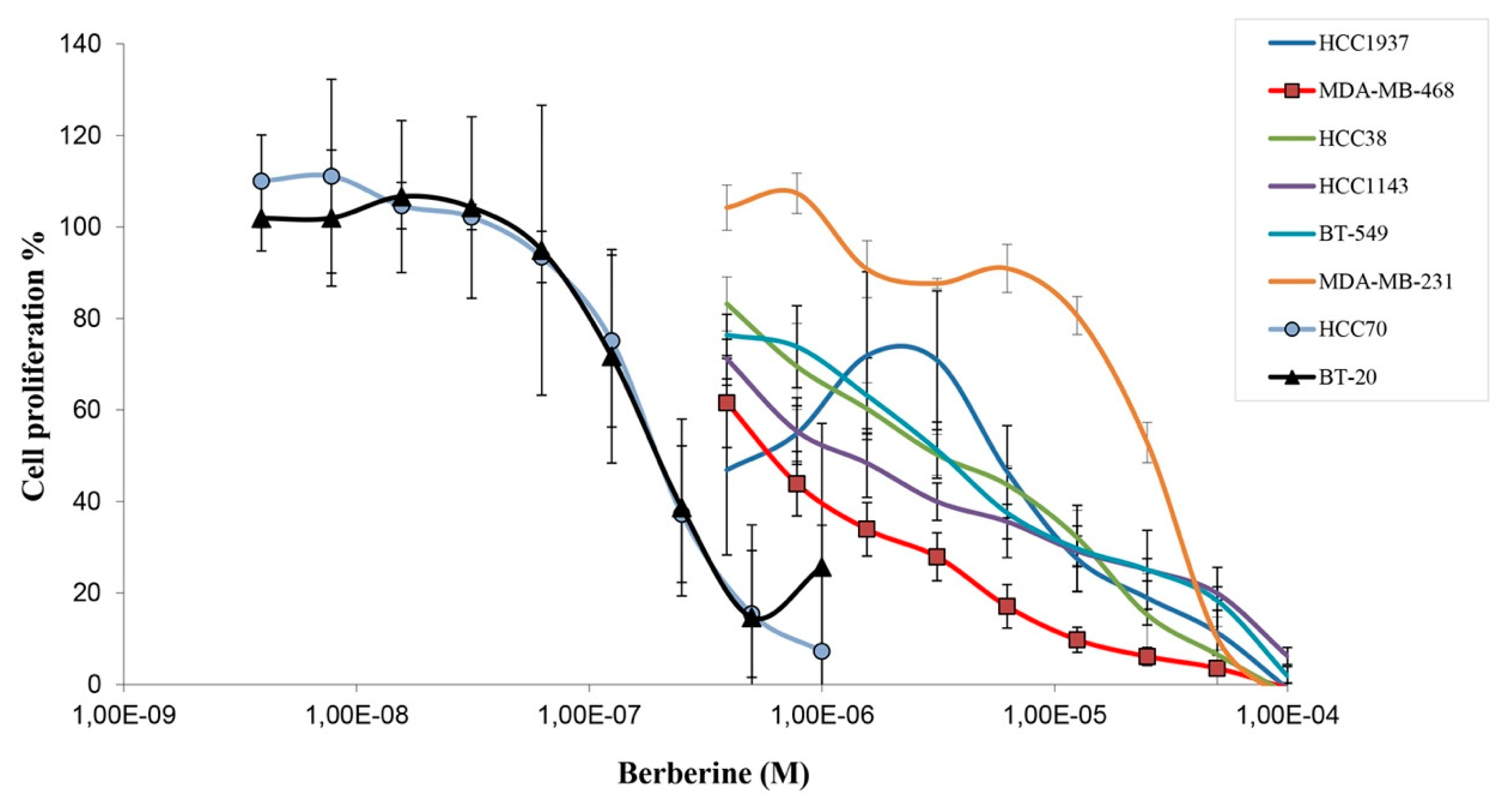
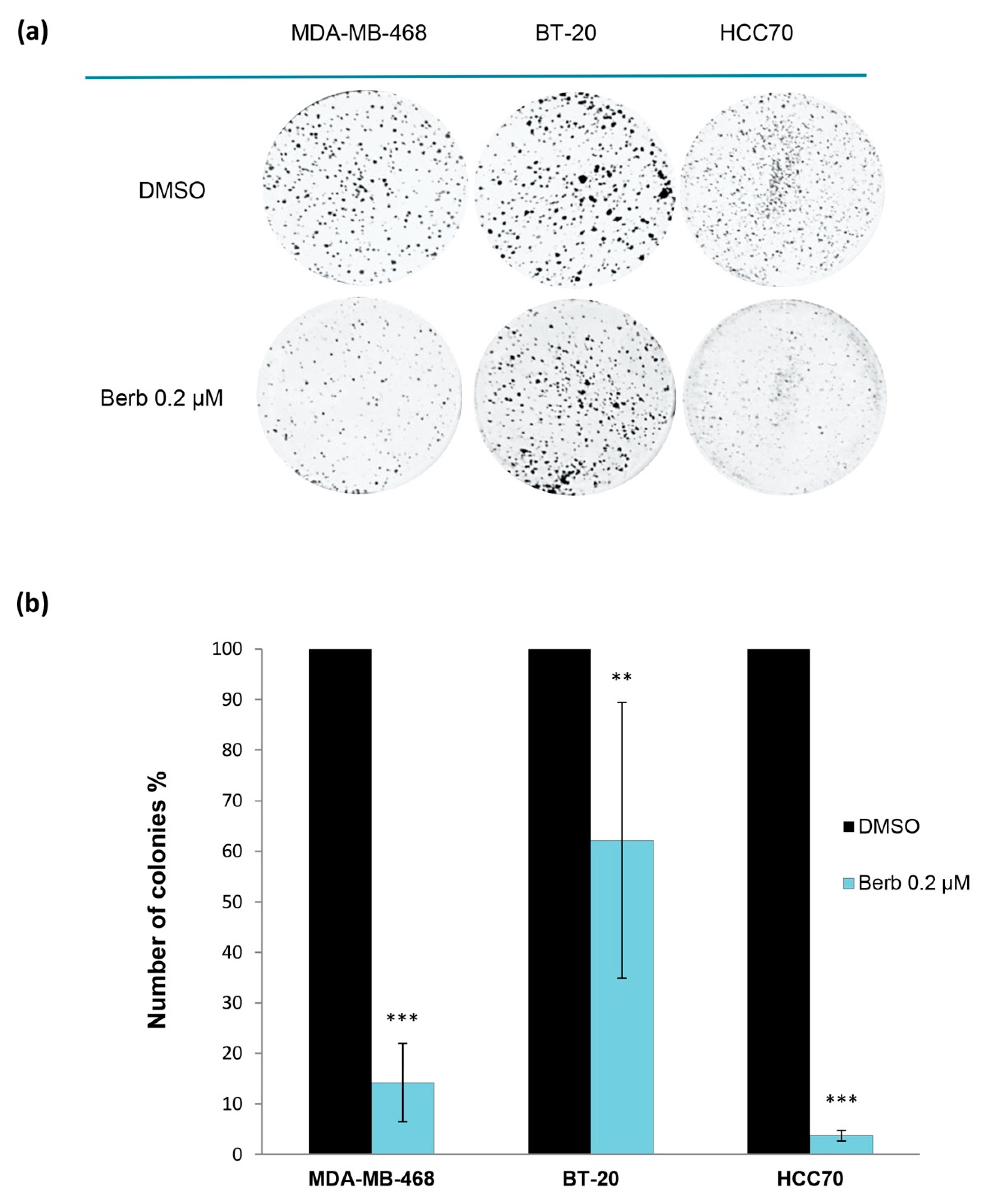
2.1. Berberine Inhibits Proliferation of Triple Negative Breast Cancer (TNBC) Cells
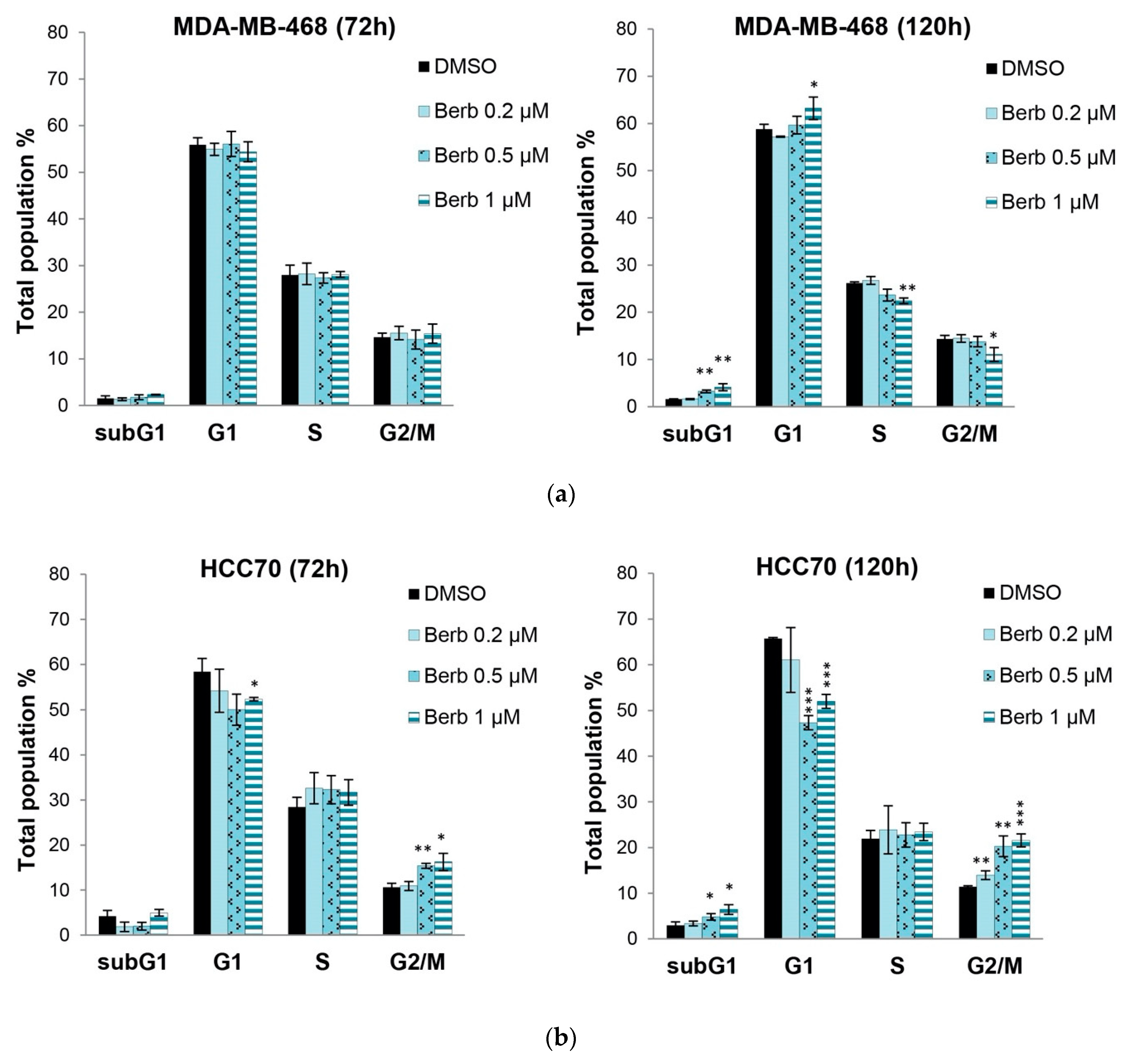
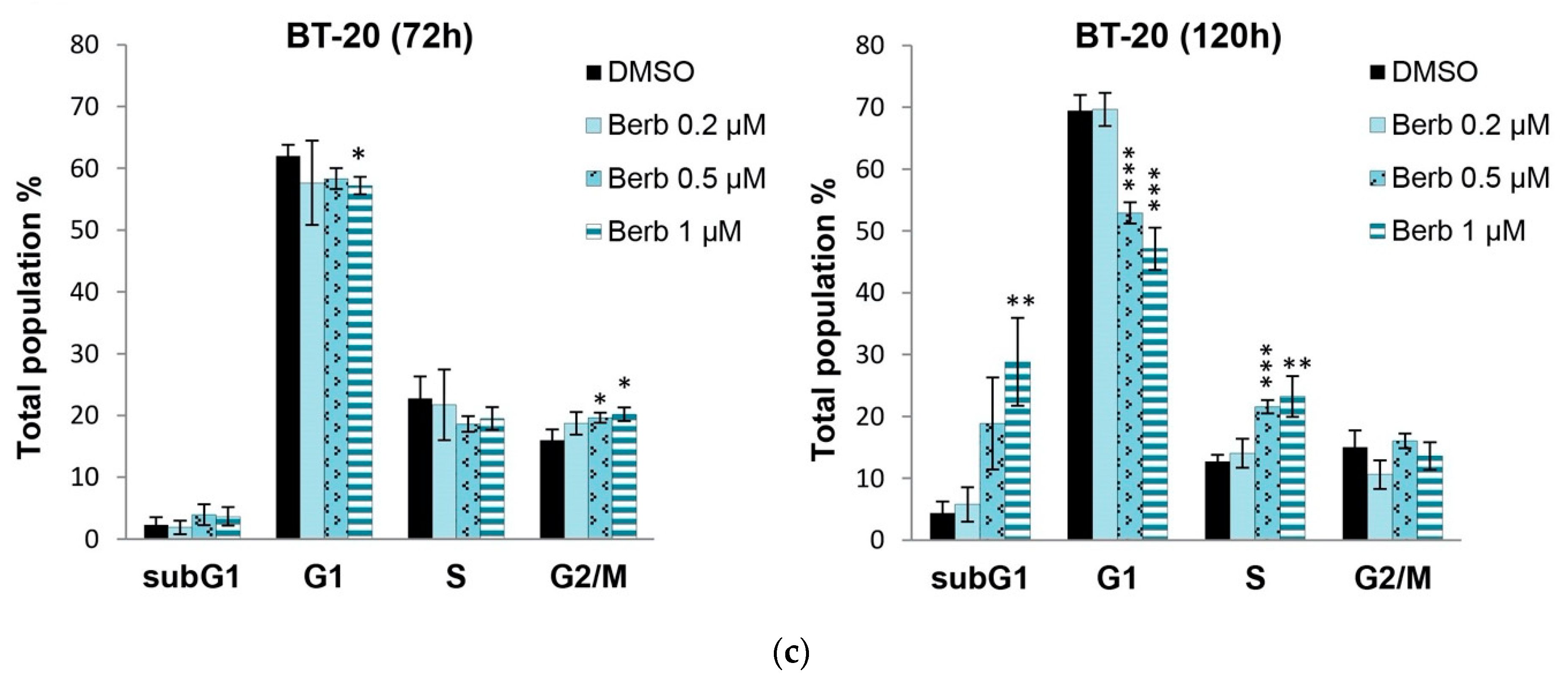
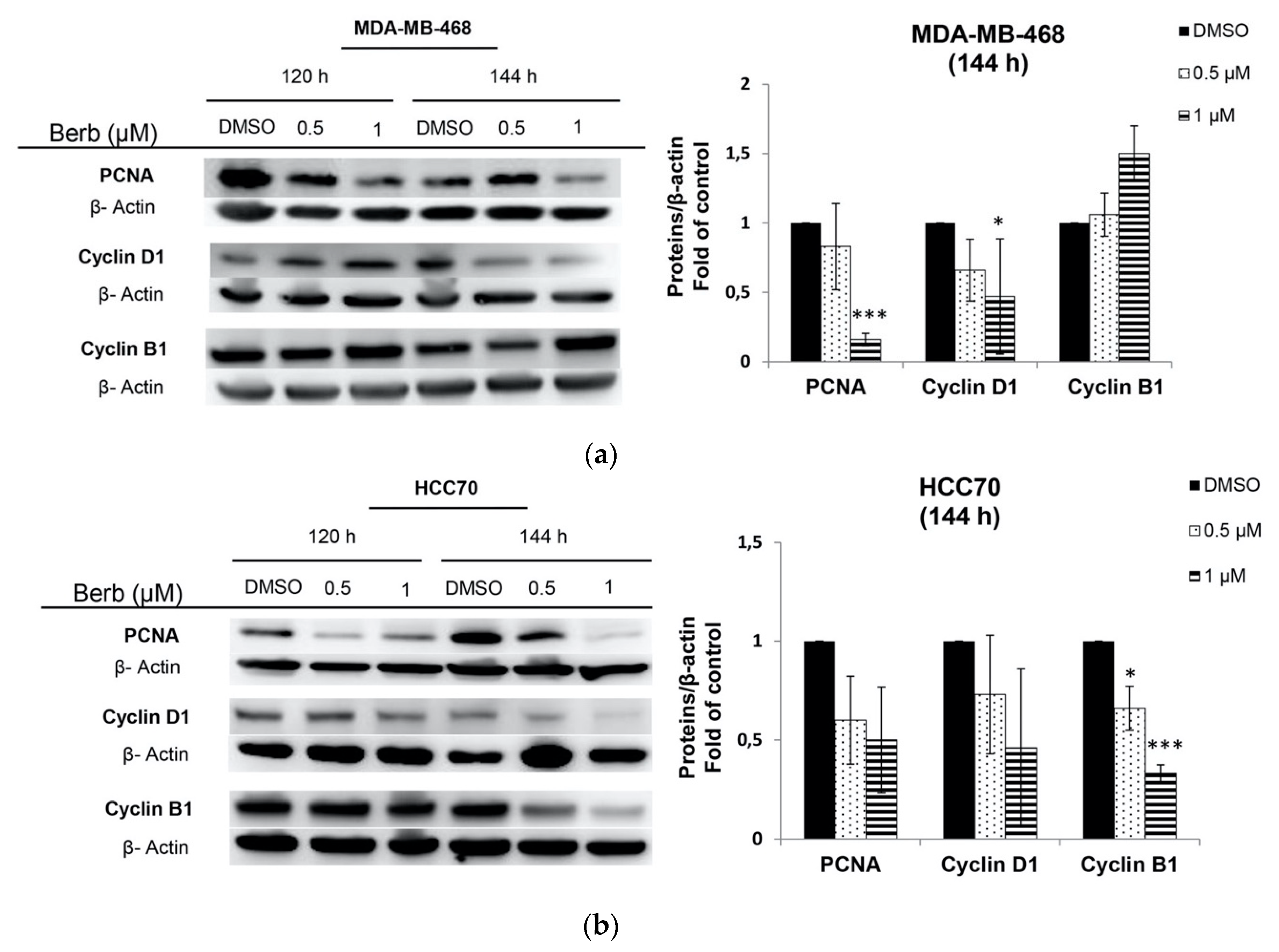
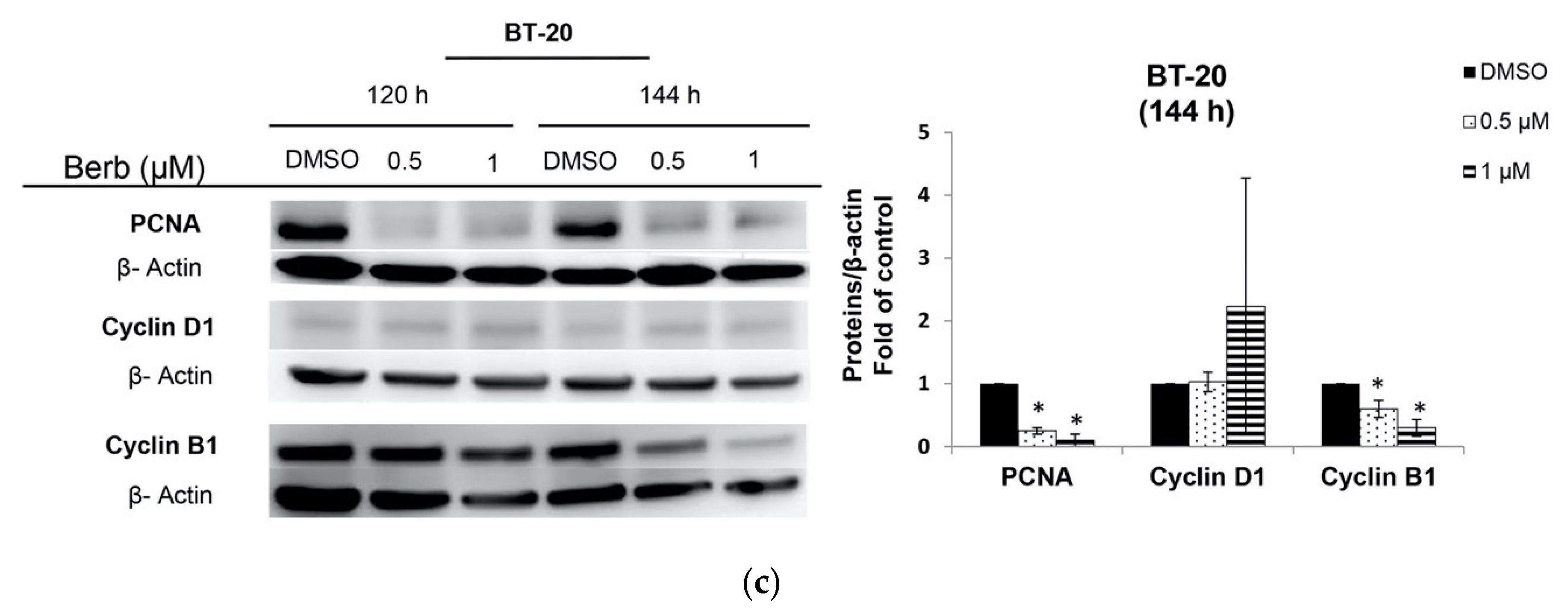
2.2. Berberine Differentially Affects TNBC Cell Cycle Progression
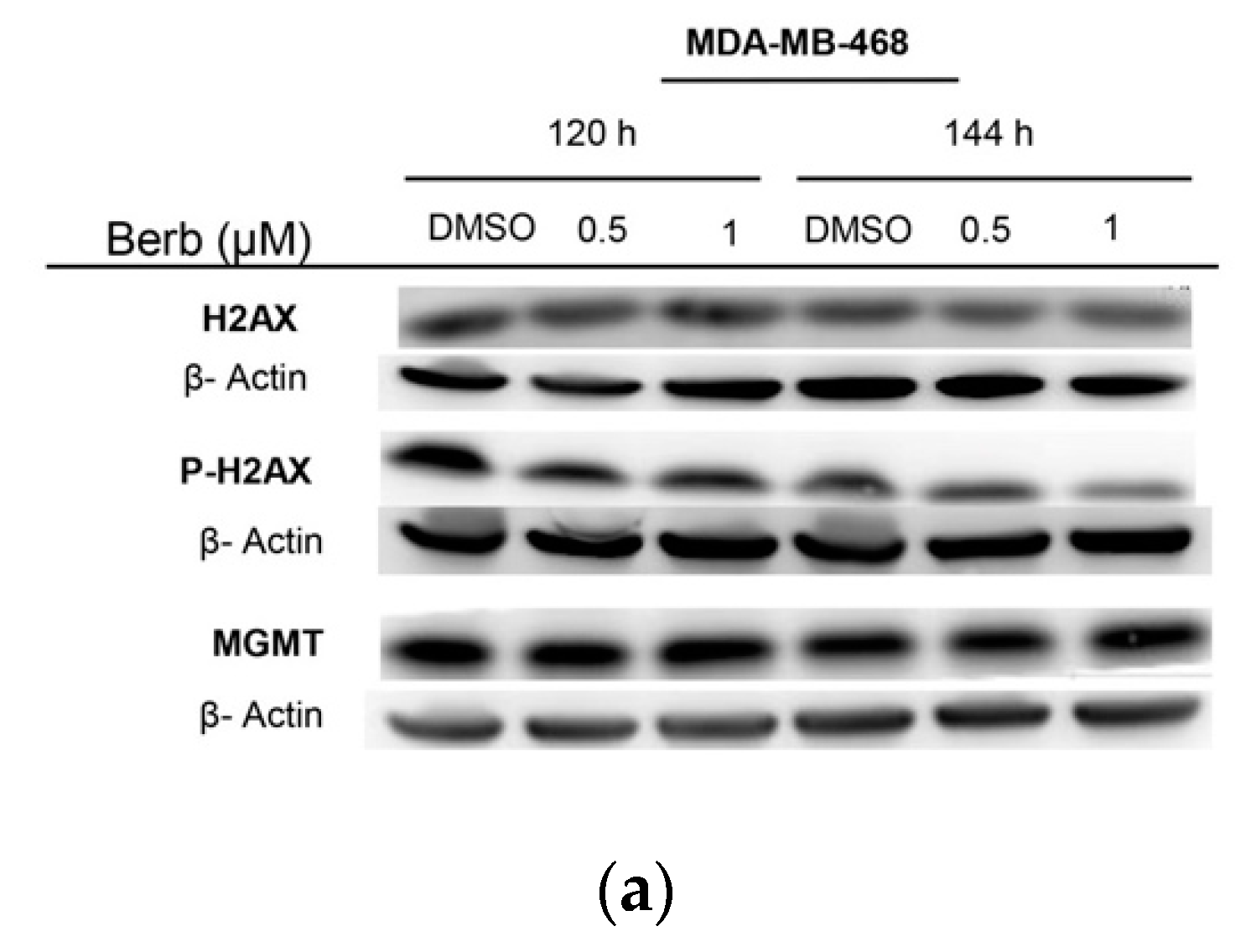
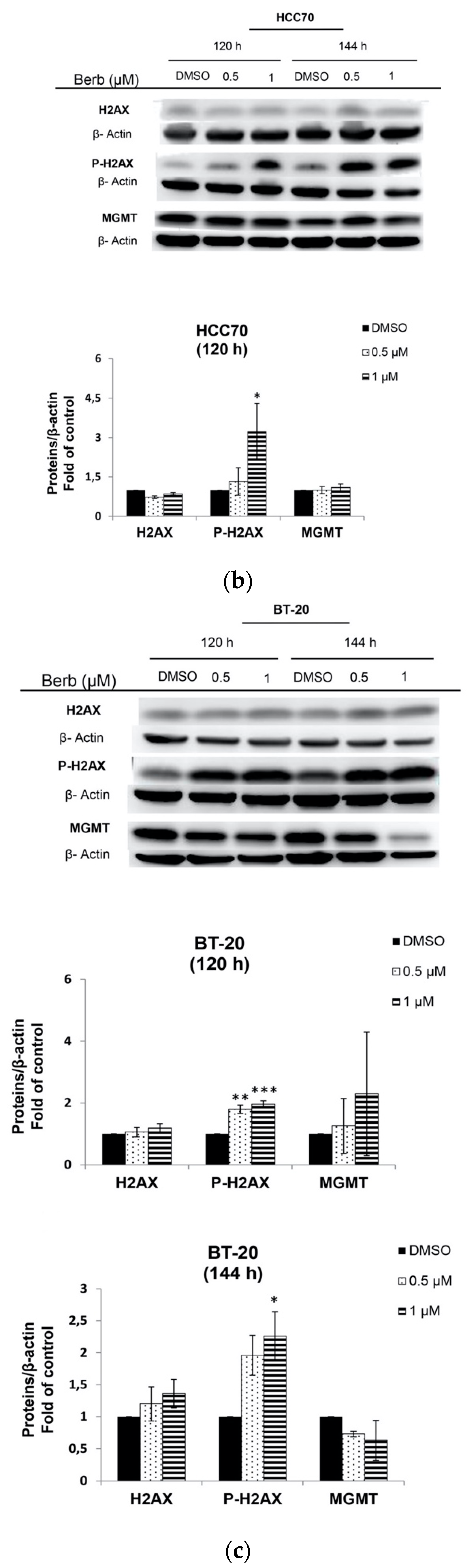
2.3. Berberine Induces DNA Damage in TNBC Cell Lines
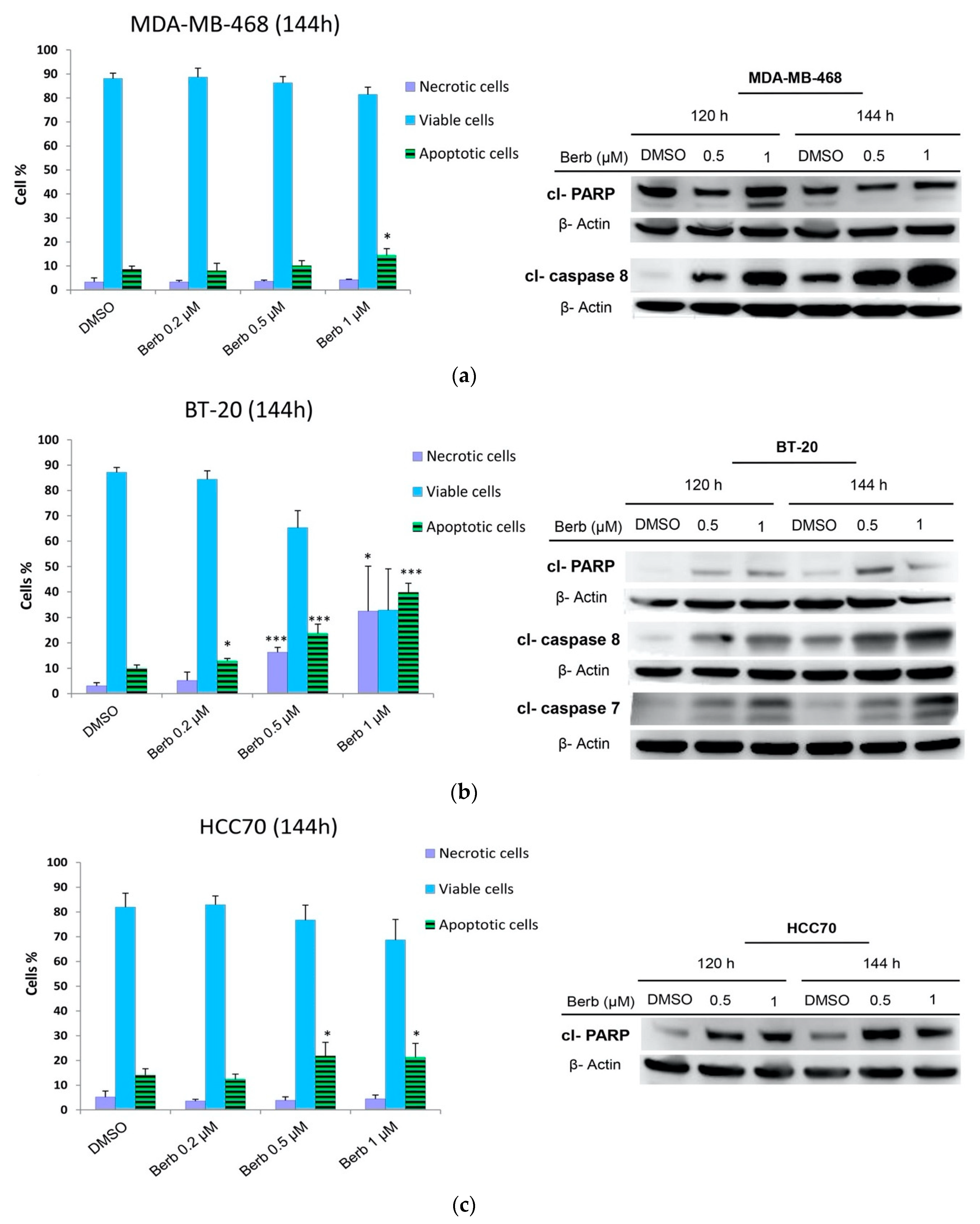
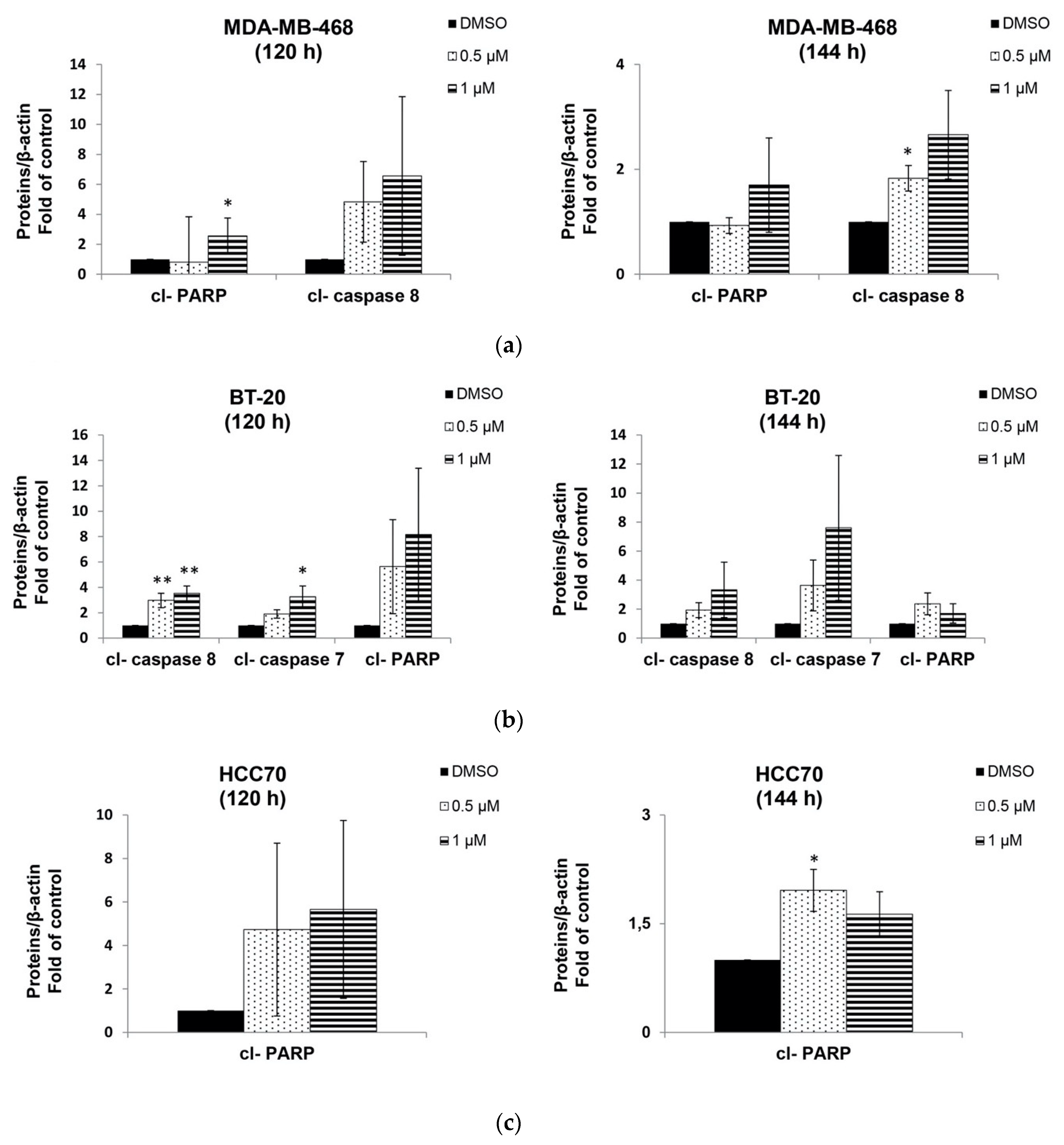
2.4. Berberine Induced Apoptosis on TNBC Cell Lines in a Dose and Time Dependent Manner
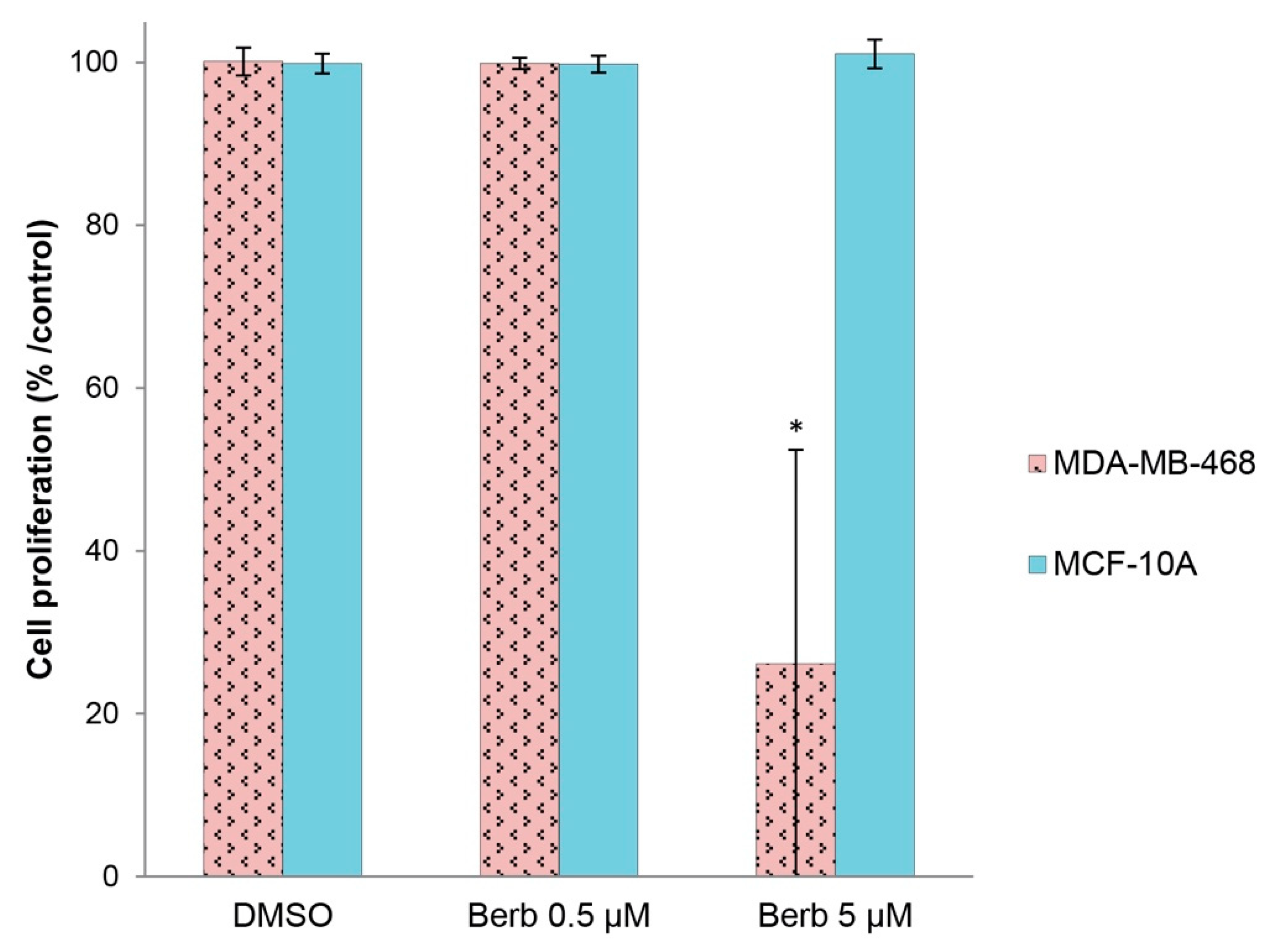
2.5. Berberine Does not Affect the Viability of “Normal” Human Breast MCF-10A Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Assessment of Cell Viability Using MTT Assay
4.4. Two-Dimensional Clonogenic Assay
4.5. Cell Cycle Analysis by Flow Cytometry
4.6. Apoptosis Annexin-V staining
4.7. Molecular Analysis by Western Blotting and Antibodies
4.8. Three-Dimensional Cell Culture
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Torre, L.; Rebecca Siegel, A.J. Global Cancer Facts & Figures, 3rd ed.; American Cancer Society: Atlanta, GA, USA, 2015; pp. 1–64. [Google Scholar]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23, 8–13. [Google Scholar] [CrossRef]
- Jia, L.Y.; Shanmugam, M.K.; Sethi, G.; Bishayee, A. Potential role of targeted therapies in the treatment of triple-negative breast cancer. Anticancer Drugs 2016, 27, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.S.; Agarwal, S.M. NPACT: Naturally occurring plant-based anti-cancer compound-activity-target database. Nucleic Acids Res. 2013, 41, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Feng, X.; Chai, L.; Cao, S.; Qiu, F. The metabolism of berberine and its contribution to the pharmacological effects. Drug Metab. Rev. 2017, 49, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Imanshahidi, M. Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother. Res. 2008, 22, 999–1012. [Google Scholar] [CrossRef]
- Kumar, A.; Ekavali, K.C.; Mukherjee, M.; Pottabathini, R.; Dhull, D.K. Current knowledge and pharmacological profile of berberine: An update. Eur. J. Pharmacol. 2015, 761, 288–297. [Google Scholar] [CrossRef]
- Liu, D.; Meng, X.; Wu, D.; Qiu, Z.; Luo, H. A natural isoquinoline alkaloid with antitumor activity: Studies of the biological activities of berberine. Front. Pharmacol. 2019, 10, 9. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of Poly(ADP-ribose) Polymerase (PARP) Cleavage in Apoptosis Caspase 3-resistant PARP mutant Increases Rates of Apoptosis in Transfected Cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2010, 4, 359–365. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef]
- Reddy, S.M.; Barcenas, C.H.; Sinha, A.K.; Hsu, L.; Moulder, S.L.; Tripathy, D.; Hortobagyi, G.N.; Valero, V. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease free at 5 years and relationship with low hormone receptor positivity. Br. J. Cancer 2018, 118, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.-M.; Ko, E.; Han, W.; Lee, J.; Shin, I.; Bae, J.Y.; Kim, S.; Noh, D.Y. Berberine Inhibits Growth of the Breast Cancer Cell Lines MCF-7 and MDA-MB-231. Planta Med. 2008, 74, 39–42. [Google Scholar] [CrossRef]
- Zhao, Y.; Jing, Z.; Lv, J.; Zhang, Z.; Lin, J.; Cao, X.; Zhao, Z.; Liu, P.; Mao, W. Berberine activates caspase-9/cytochrome c -mediated apoptosis to suppress triple-negative breast cancer cells in vitro and in vivo. Biomed. Pharmacother. 2017, 95, 18–24. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; You, D.; Jeong, Y.; Jeon, M.; Yu, J.; Kim, S.W.; Nam, S.J.; Lee, J.E. Berberine Suppresses Cell Motility Through Downregulation of TGF-β1 in Triple Negative Breast Cancer Cells. Cell Physiol. Biochem. 2018, 45, 795–807. [Google Scholar] [CrossRef]
- Kim, S.; You, D.; Jeong, Y.; Yu, J.; Kim, S.W.; Nam, S.J.; Lee, J.E. Berberine down-regulates IL-8 expression through inhibition of the EGFR/MEK/ERK pathway in triple-negative breast cancer cells. Phytomedicine 2018, 50, 43–49. [Google Scholar] [CrossRef]
- Yao, M.; Fan, X.; Yuan, B.; Takagi, N.; Liu, S.; Han, X.; Ren, J.; Liu, J. Berberine inhibits NLRP3 Inflammasome pathway in human triple-negative breast cancer MDA-MB-231 cell. BMC Complement. Altern. Med. 2019, 19, 216. [Google Scholar] [CrossRef]
- Lin, Y.S.; Chiu, Y.C.; Tsai, Y.H.; Tsai, Y.F.; Wang, J.Y.; Tseng, L.M.; Chiu, J.H. Different mechanisms involved in the berberine-induced antiproliferation effects in triple-negative breast cancer cell lines. J. Cell Biochem. 2019, 120, 13531–13544. [Google Scholar] [CrossRef]
- Kurki, P.; Vanderlaan, M.; Dolbeare, F.; Gray, J.; Tan, E.M. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp. Cell Res. 1986, 166, 209–219. [Google Scholar] [CrossRef]
- Nakayama, Y. Role of Cyclin B1 levels in DNA damage and DNA damage-induced senescence. Int. Rev. Cell. Mol. Biol. 2013, 305, 303–337. [Google Scholar] [PubMed]
- Zhao, W.; Liu, H.; Wang, J.; Wang, M.; Shao, R. Cyclizing-berberine A35 induces G2/M arrest and apoptosis by activating YAP phosphorylation (Ser127). J. Exp. Clin. Cancer Res. 2018, 37, 98. [Google Scholar] [CrossRef] [PubMed]
- Eo, S.H.; Kim, J.H.; Kim, S.J. Induction of G2 /M Arrest by Berberine via Activation of PI3K/Akt and p38 in Human Chondrosarcoma Cell Line. Oncol. Res. 2015, 22, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hao, Y.; Shi, T.; Deng, W.; Li, N. Berberine inhibits cyclin D1 expression via suppressed binding of AP-1 transcription factors to CCND1 AP-1 motif. Acta Pharmacol. Sin. 2008, 29, 628–633. [Google Scholar] [CrossRef]
- Chai, Y.S.; Hu, J.; Lei, F.; Wang, Y.G.; Yuan, Z.Y.; Lu, X.; Wang, X.P.; Du, F.; Zhang, D.; Xing, D.M.; et al. Effect of berberine on cell cycle arrest and cell survival during cerebral ischemia and reperfusion and correlations with p53/cyclin D1 and PI3K/Akt. Eur. J. Pharmacol. 2013, 708, 44–55. [Google Scholar] [CrossRef]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and Extrinsic Apoptotic Pathway Signaling as Determinants of Histone Deacetylase Inhibitor Antitumor Activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar]
- Letašiová, S.; Jantová, S.; Miko, M.; Ovádeková, R.; Horváthová, M. Effect of berberine on proliferation, biosynthesis of macromolecules, cell cycle and induction of intercalation with DNA, dsDNA damage and apoptosis in Ehrlich ascites carcinoma cells. J. Pharm. Pharmacol. 2006, 58, 263–270. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell. Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Patil, J.B.; Kim, J.; Jayaprakasha, G.K. Berberine induces apoptosis in breast cancer cells (MCF-7) through mitochondrial-dependent pathway. Eur. J. Pharmacol. 2010, 645, 70–78. [Google Scholar] [CrossRef]
- El Khalki, L.; Tilaoui, M.; Jaafari, A.; Ait Mouse, H.; Zyad, A. Studies on the Dual Cytotoxicity and Antioxidant Properties of Berberis vulgaris Extracts and Its Main Constituent Berberine. Adv. Pharmacol. Sci. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdès, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Maire, V.; Némati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G.; et al. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of berberine are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Berberine IC50 (µM) |
|---|---|
| HCC70 | 0.19 ± 0.06 |
| BT-20 | 0.23 ± 0.10 |
| MDA-MB-468 | 0.48 ± 0.25 |
| HCC1143 | 1.67 ± 0.74 |
| HCC38 | 3.24 ± 1.05 |
| BT-549 | 3.44 ± 1.29 |
| HCC1937 | 2.30 ± 2.09 |
| MDA-MB-231 | 16.7 ± 2.37 |
| Cell Lines | Cell Concentration × 103 (cell/mL) * | Cells Doubling Time (h) |
|---|---|---|
| HCC1937 | 10 | 45 |
| HCC70 | 20 | 50 |
| MDA-MB-468 | 10 | 45 |
| HCC38 | 20 | 60 |
| BT-20 | 10 | 50 |
| HCC1143 | 10 | 60 |
| BT-549 | 5 | 30 |
| MDA-MB-231 | 5 | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Khalki, L.; Maire, V.; Dubois, T.; Zyad, A. Berberine Impairs the Survival of Triple Negative Breast Cancer Cells: Cellular and Molecular Analyses. Molecules 2020, 25, 506. https://doi.org/10.3390/molecules25030506
El Khalki L, Maire V, Dubois T, Zyad A. Berberine Impairs the Survival of Triple Negative Breast Cancer Cells: Cellular and Molecular Analyses. Molecules. 2020; 25(3):506. https://doi.org/10.3390/molecules25030506
Chicago/Turabian StyleEl Khalki, Lamyae, Virginie Maire, Thierry Dubois, and Abdelmajid Zyad. 2020. "Berberine Impairs the Survival of Triple Negative Breast Cancer Cells: Cellular and Molecular Analyses" Molecules 25, no. 3: 506. https://doi.org/10.3390/molecules25030506
APA StyleEl Khalki, L., Maire, V., Dubois, T., & Zyad, A. (2020). Berberine Impairs the Survival of Triple Negative Breast Cancer Cells: Cellular and Molecular Analyses. Molecules, 25(3), 506. https://doi.org/10.3390/molecules25030506