A Many-Faced Alkaloid: Polymorphism of (–)-Monophyllidin

 ,
,
Abstract
1. Introduction
2. Results and Discussion
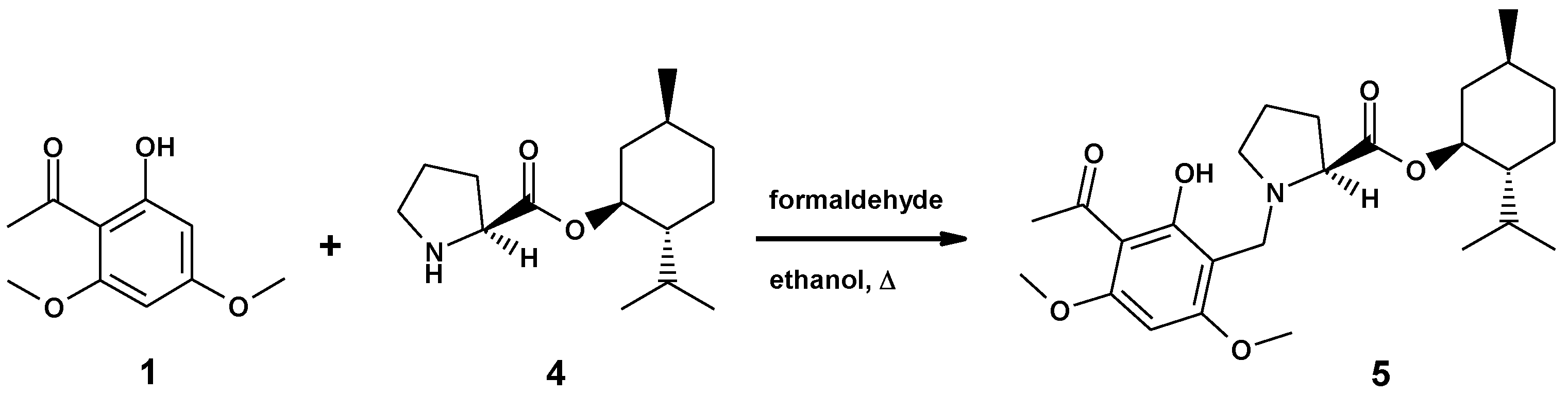
2.1. Synthesis of (–)- and (+)-Monophyllidin
2.2. Synthesis of the Ester of (–)-Monophyllidin and (+)-Menthol
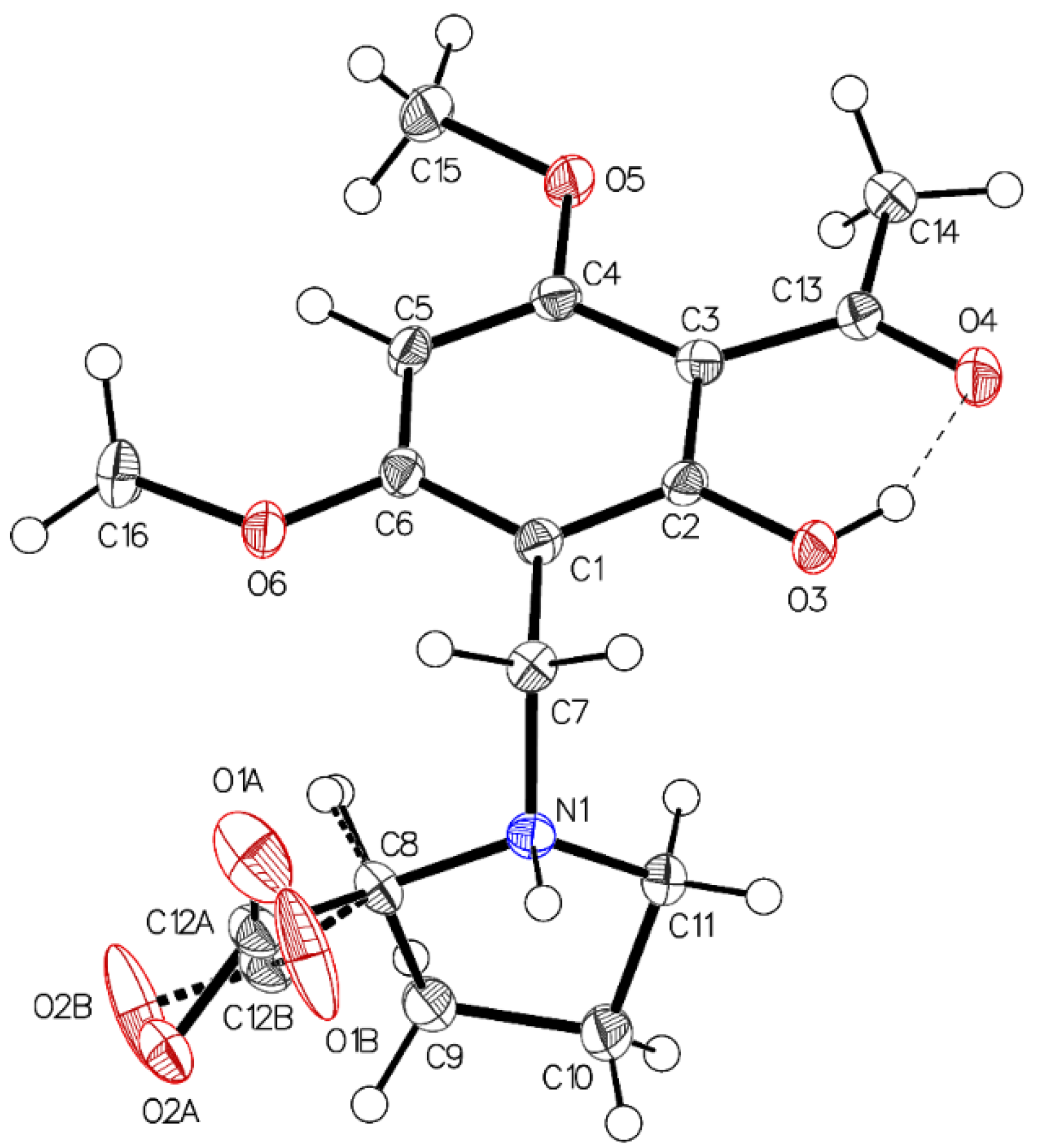
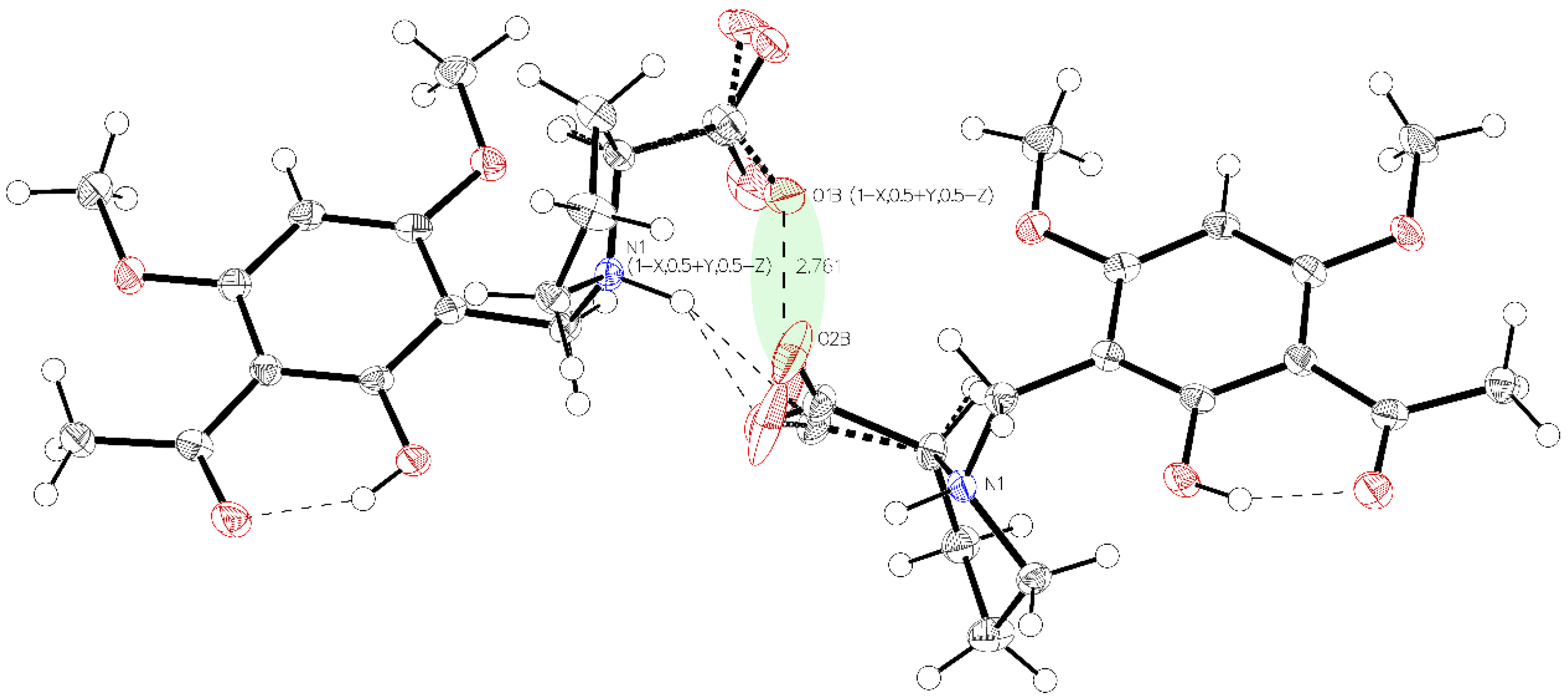
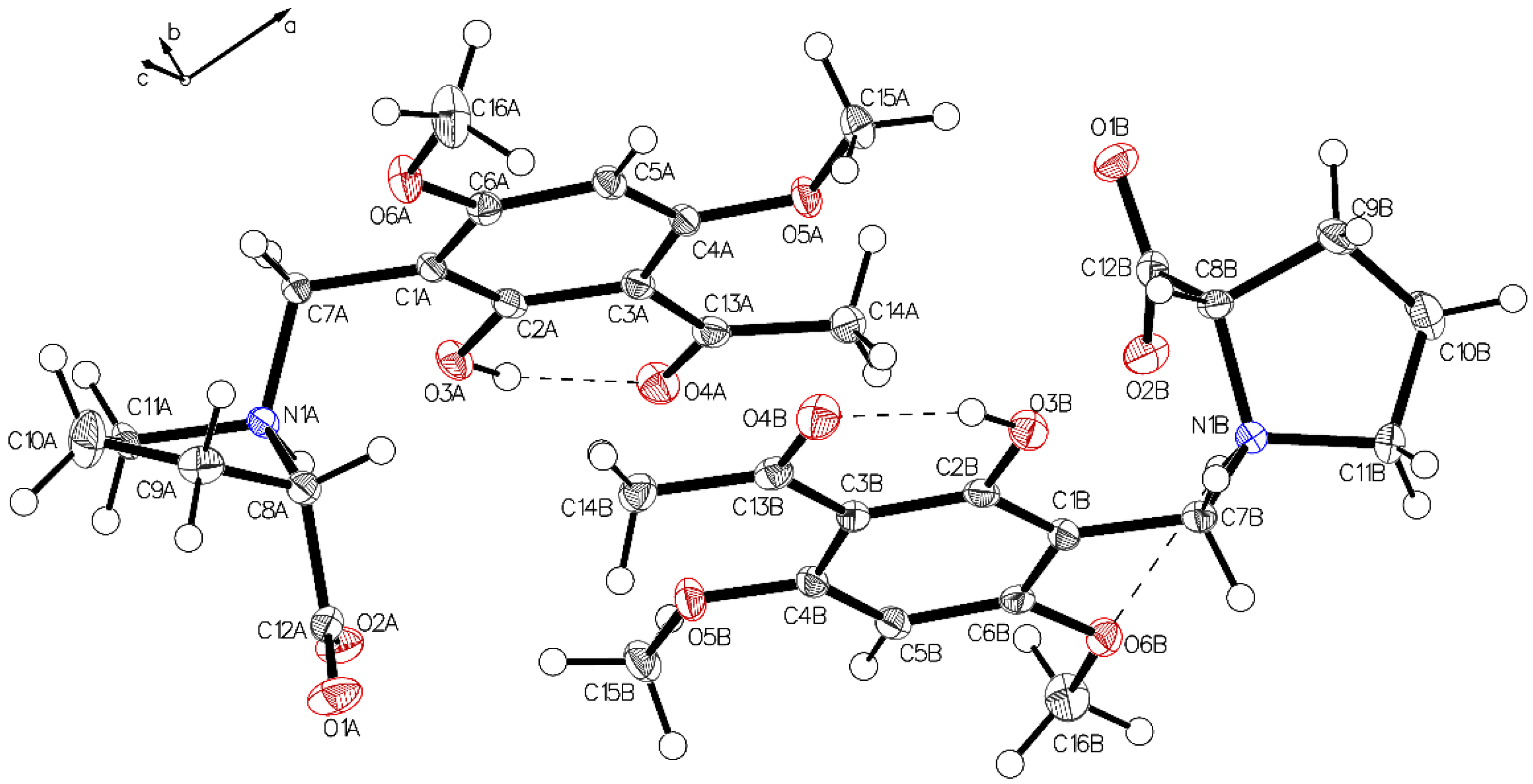
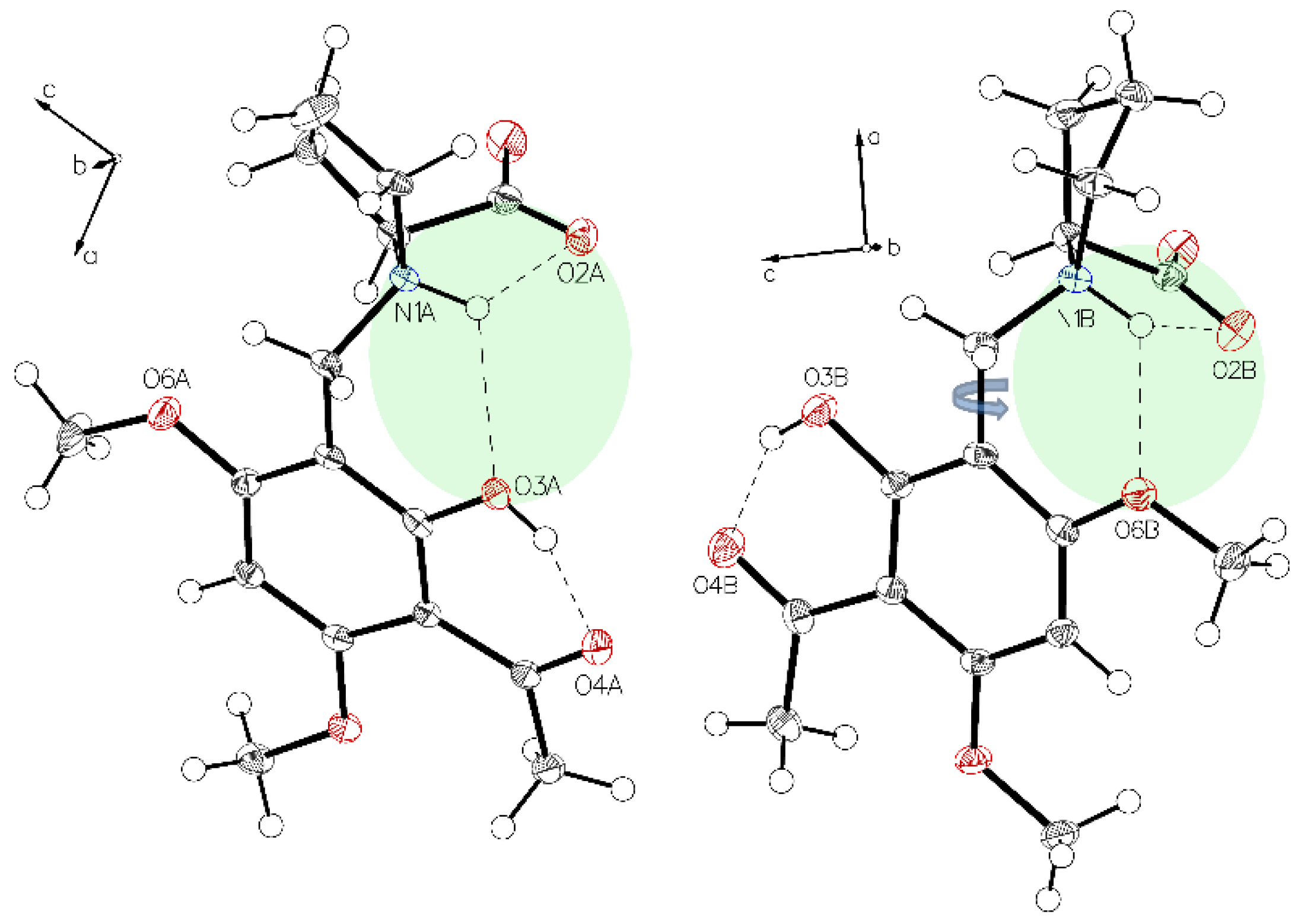
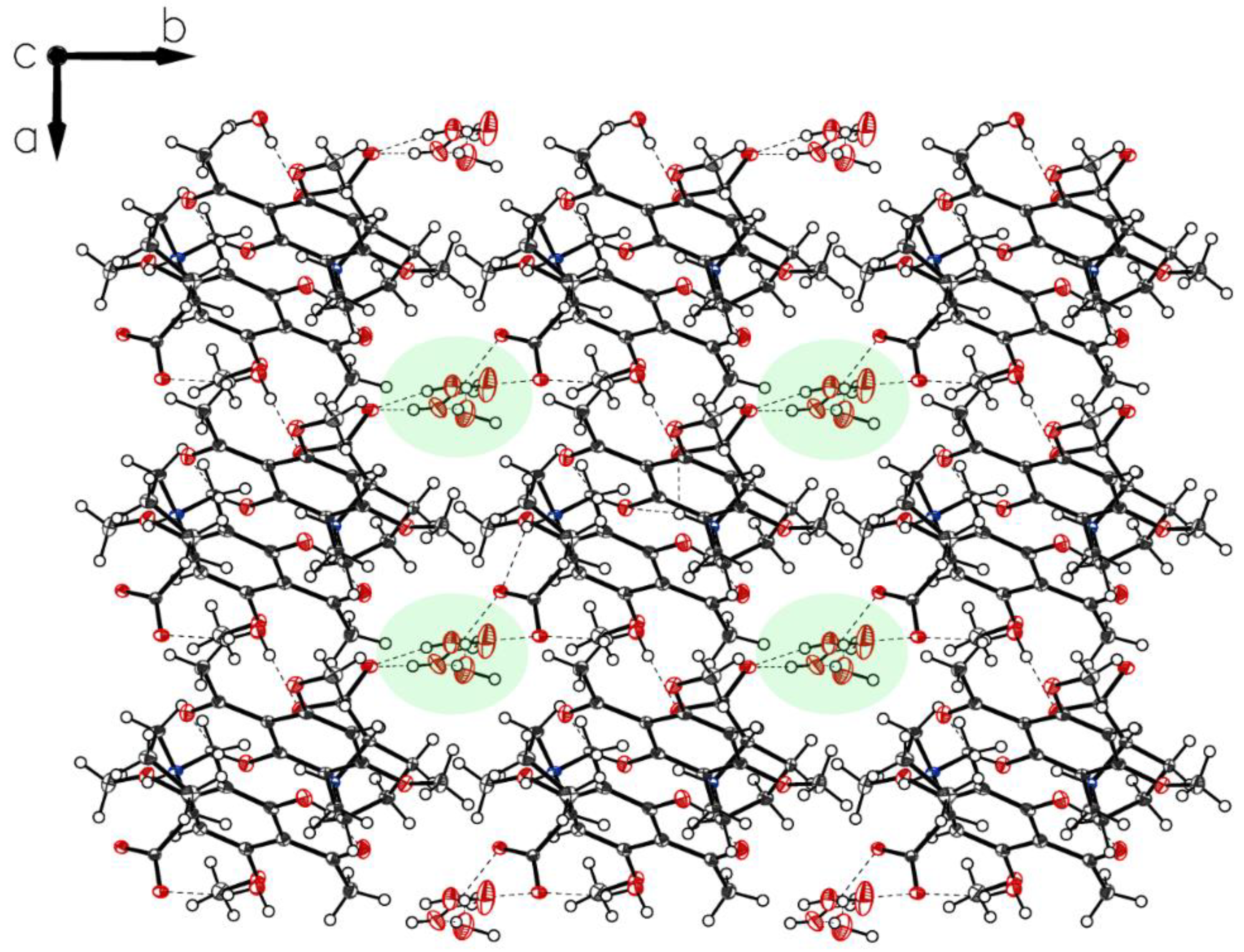
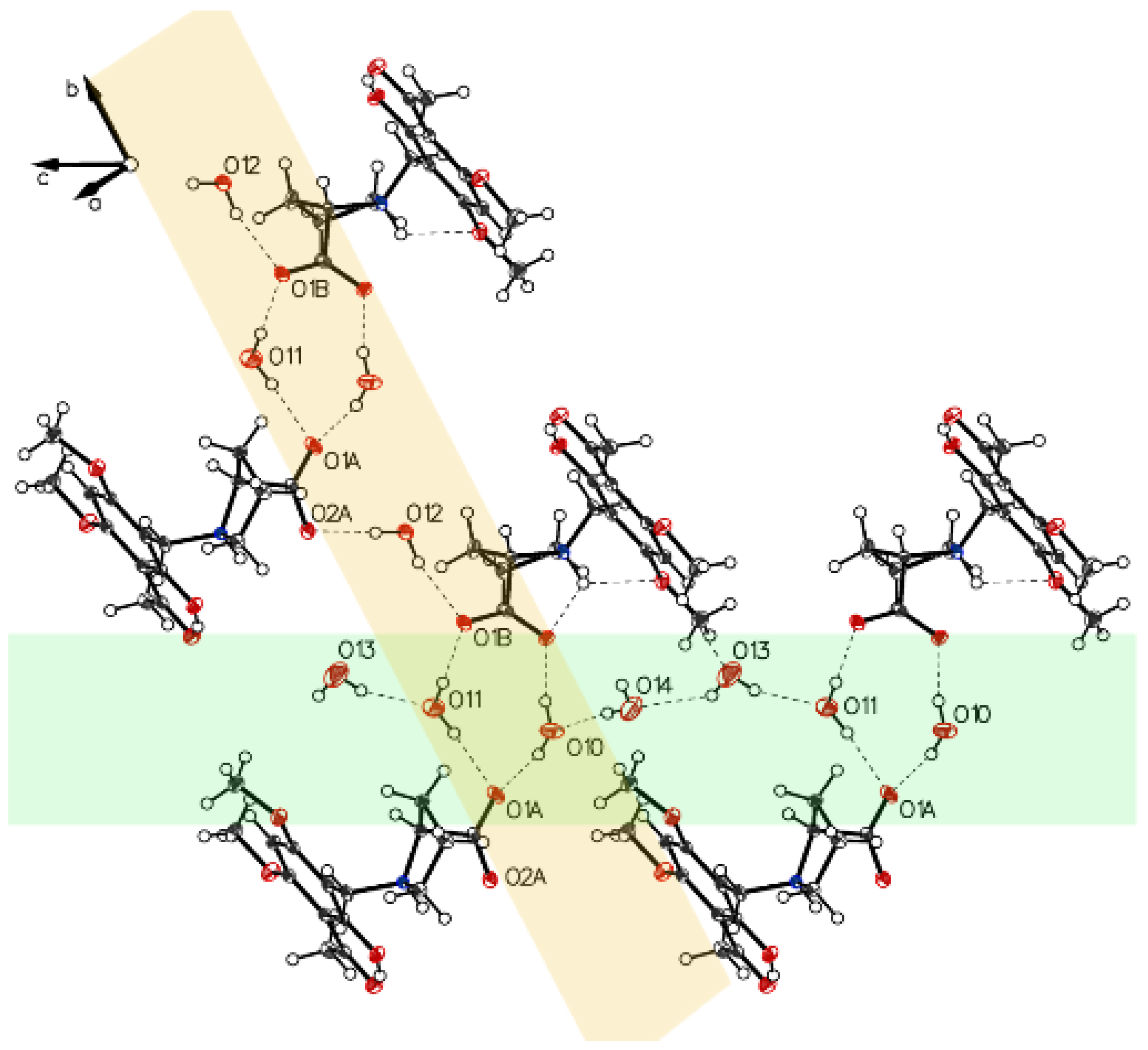
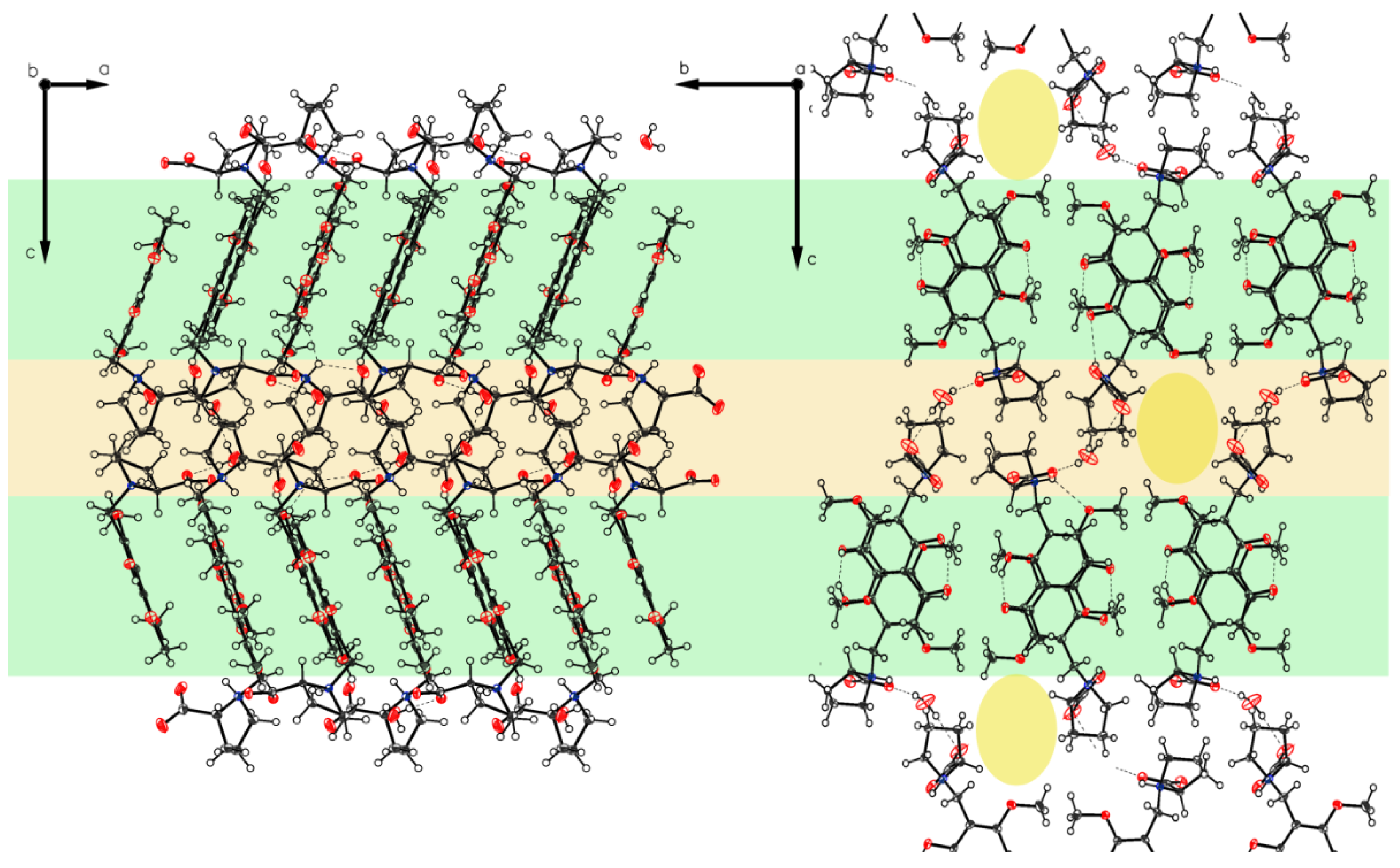
3. Crystallographic Characterization of (–)-Monophyllidin Polymorphs (P-I to P-VI)
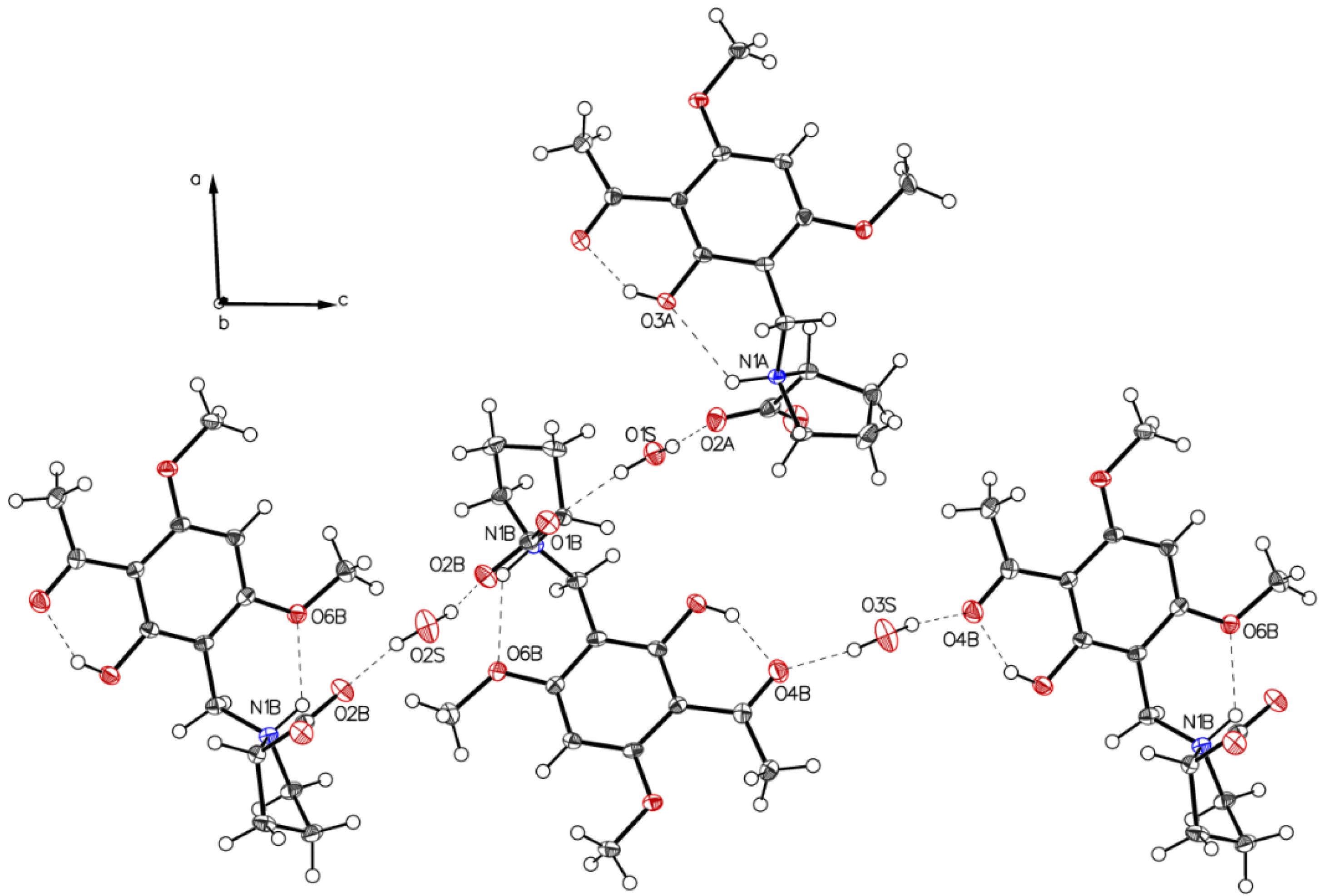
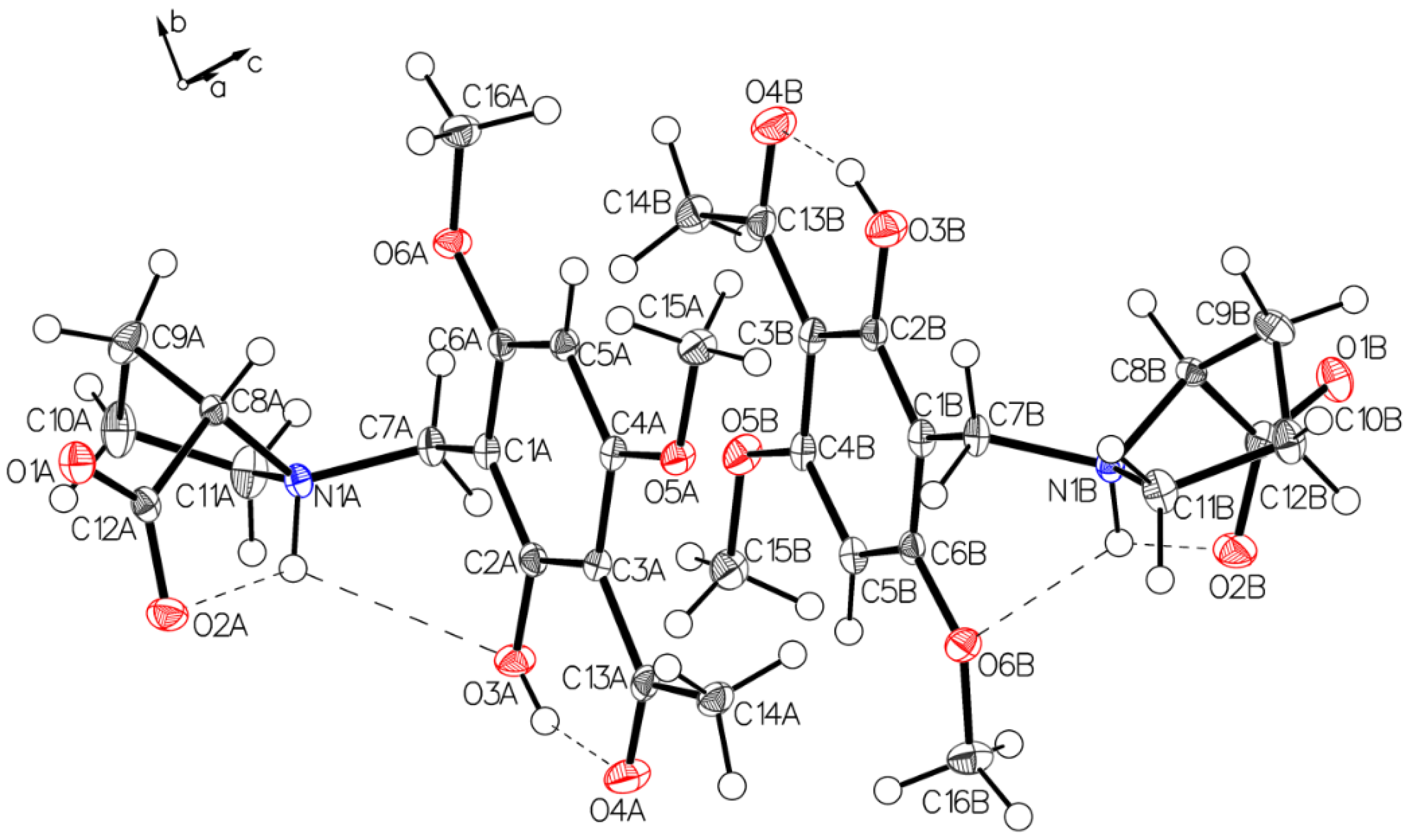
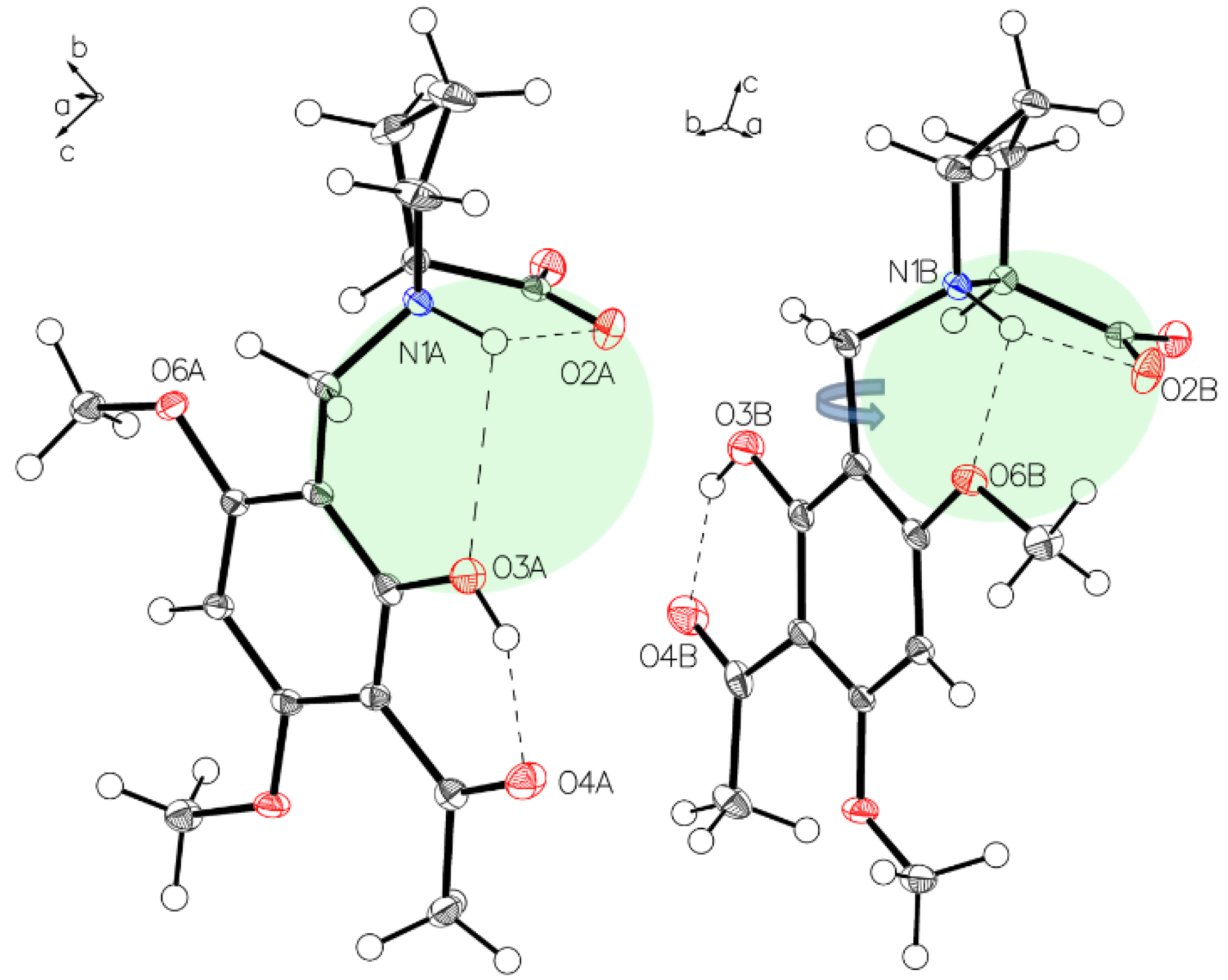
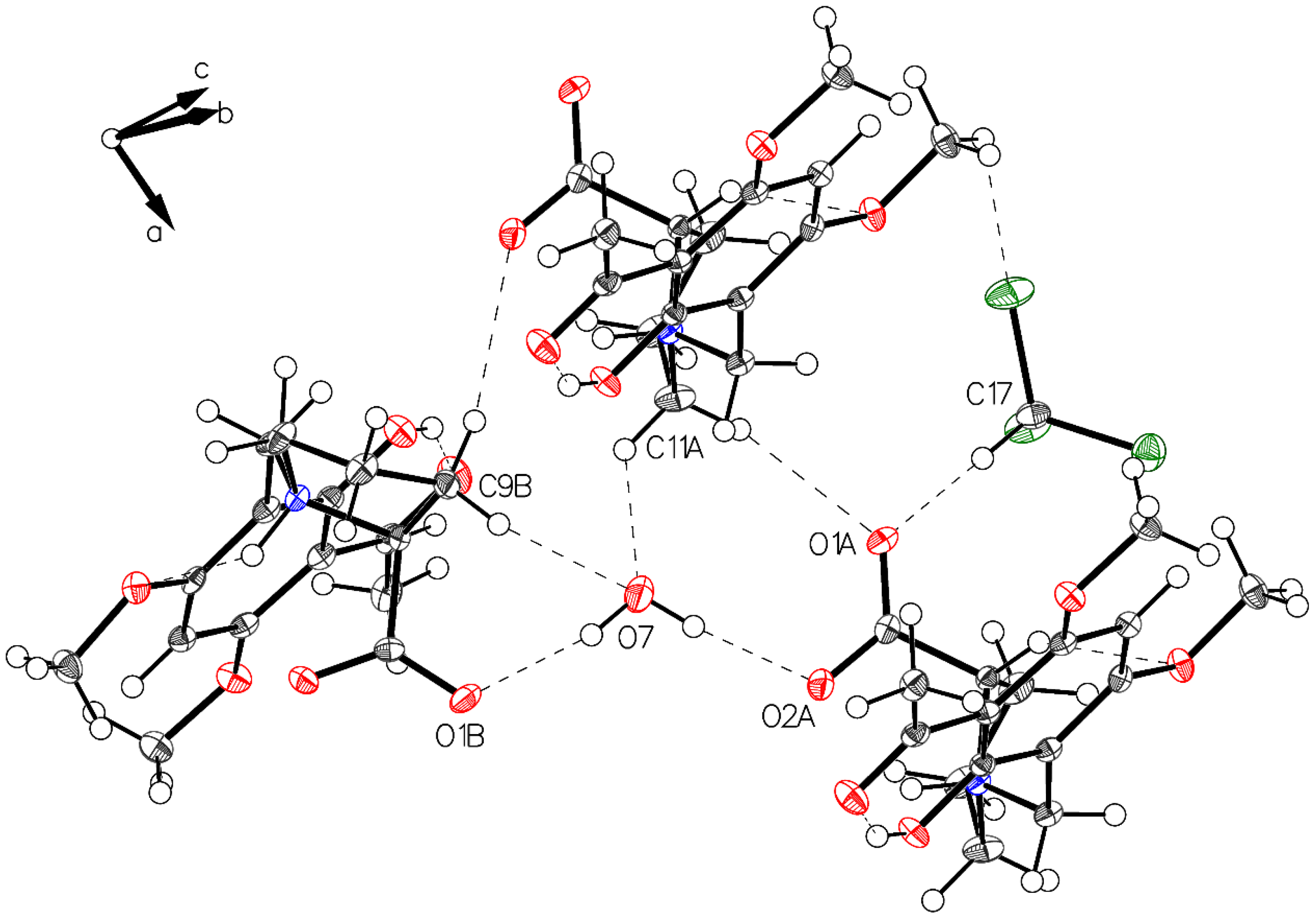
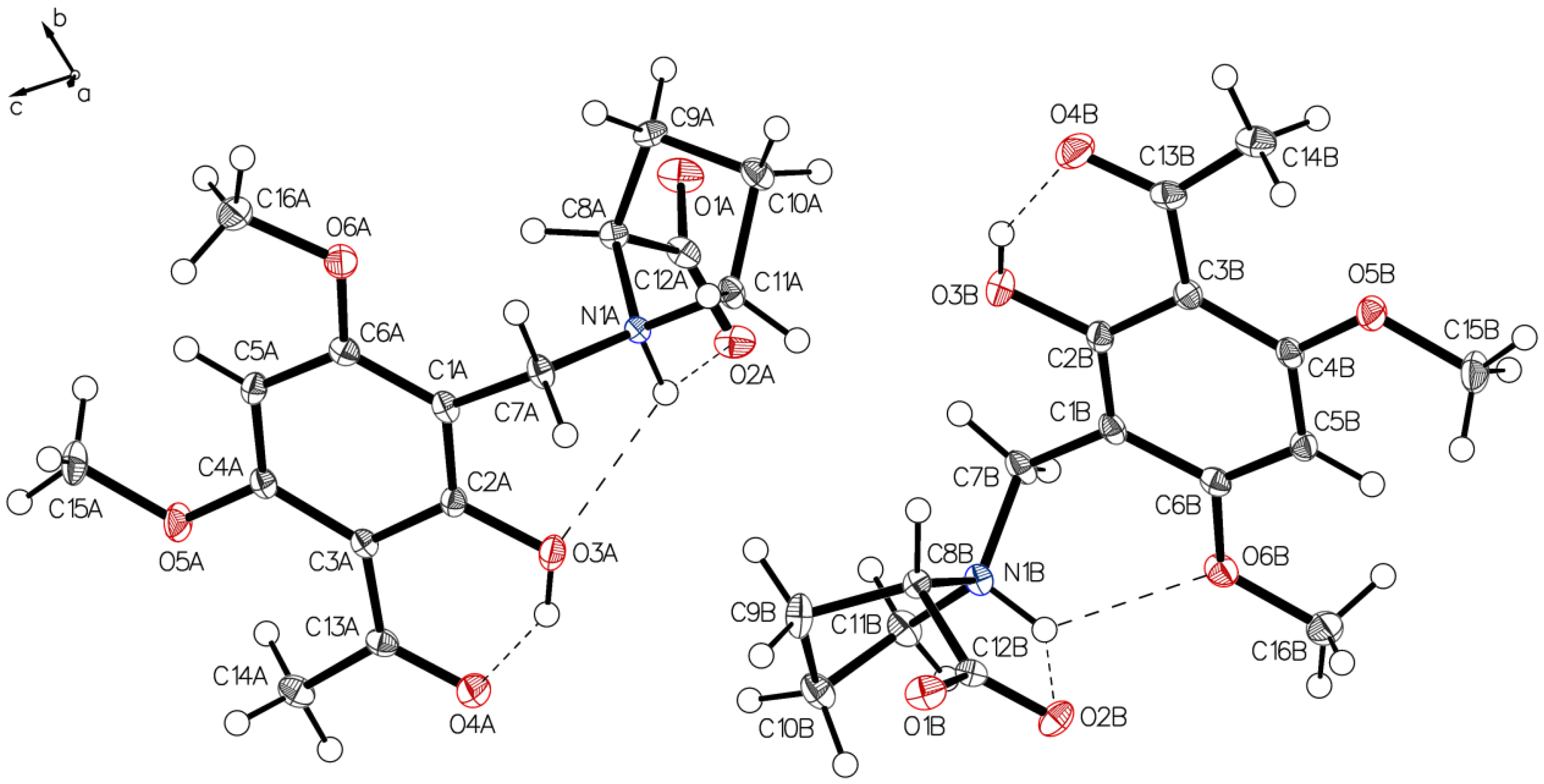
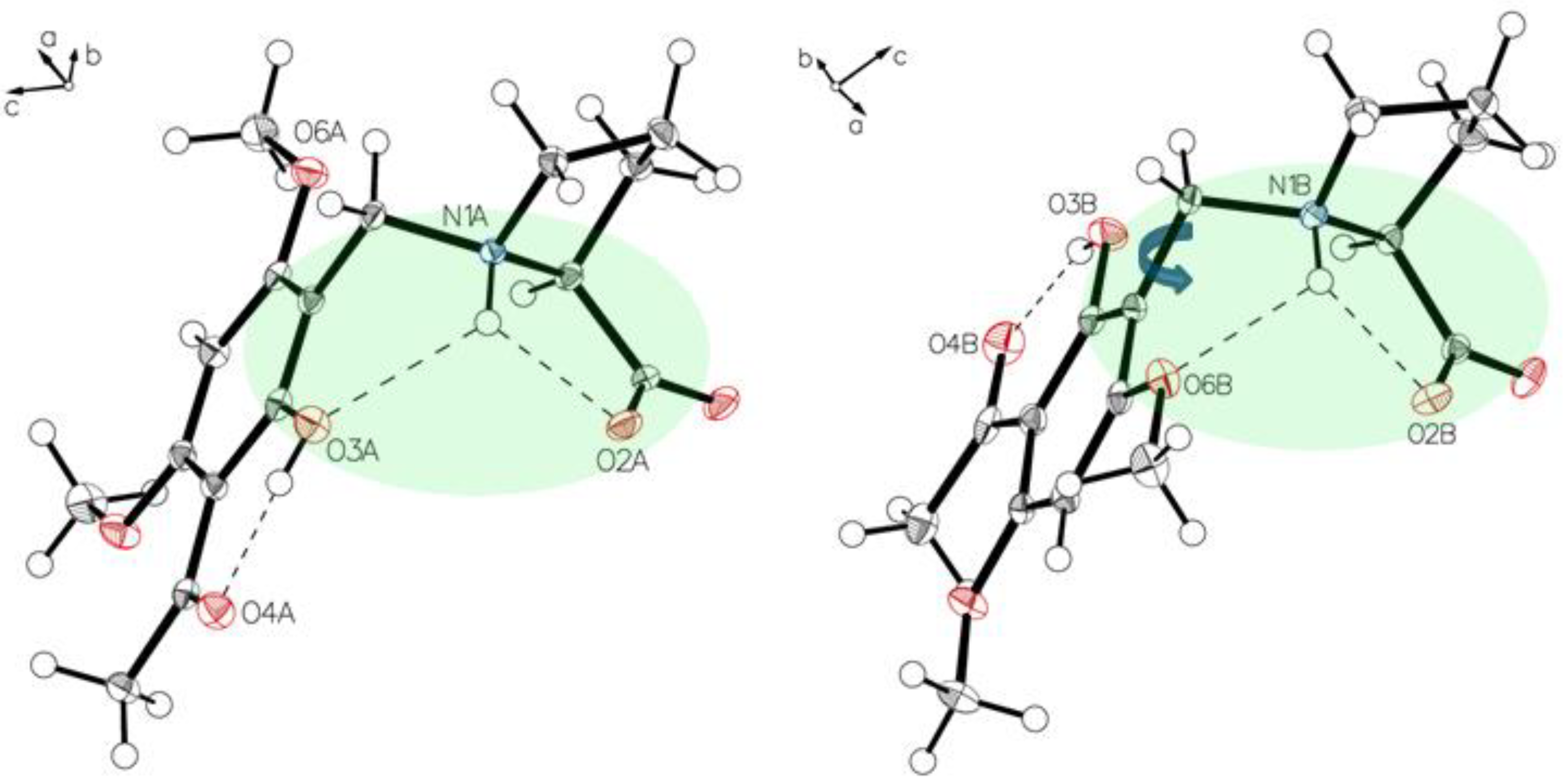
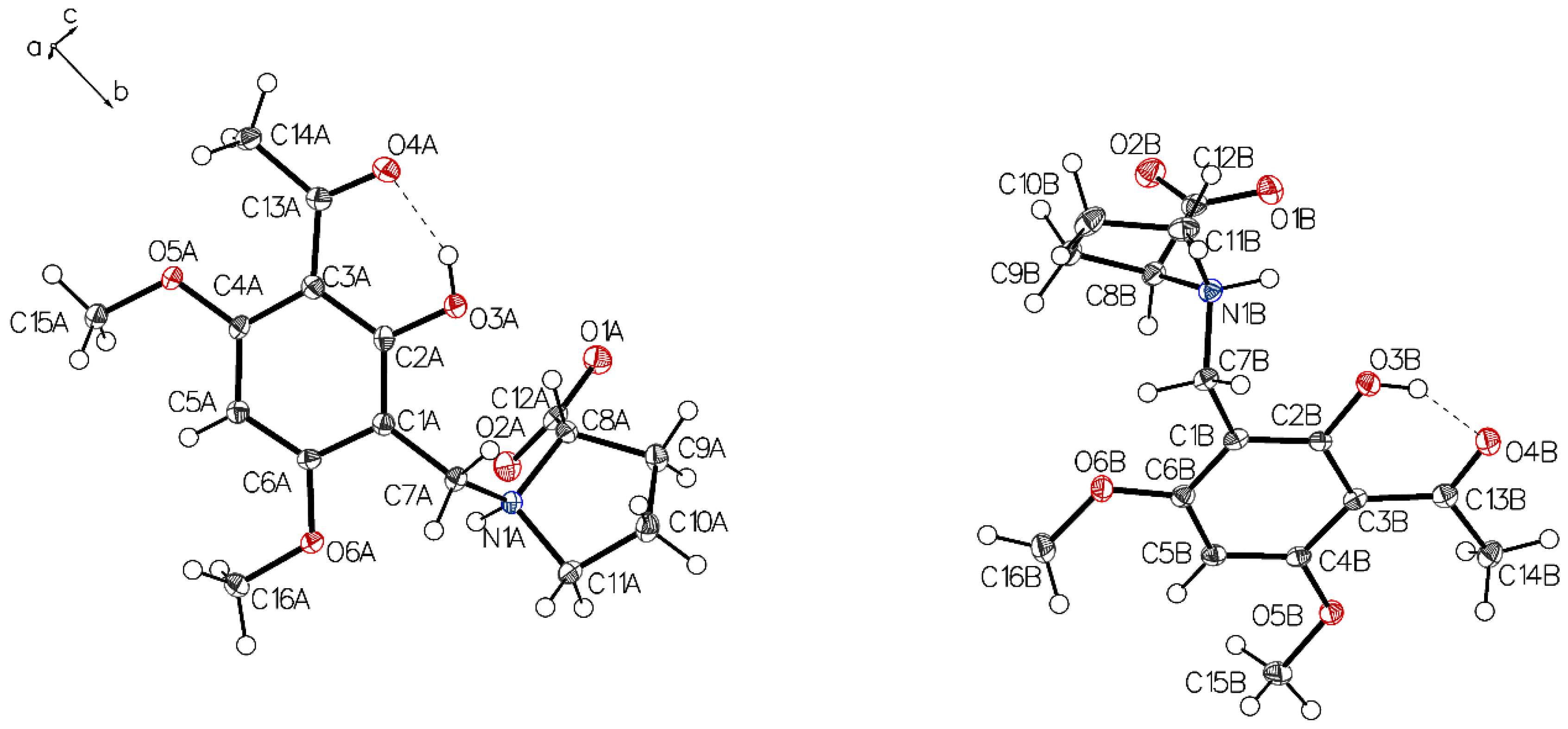
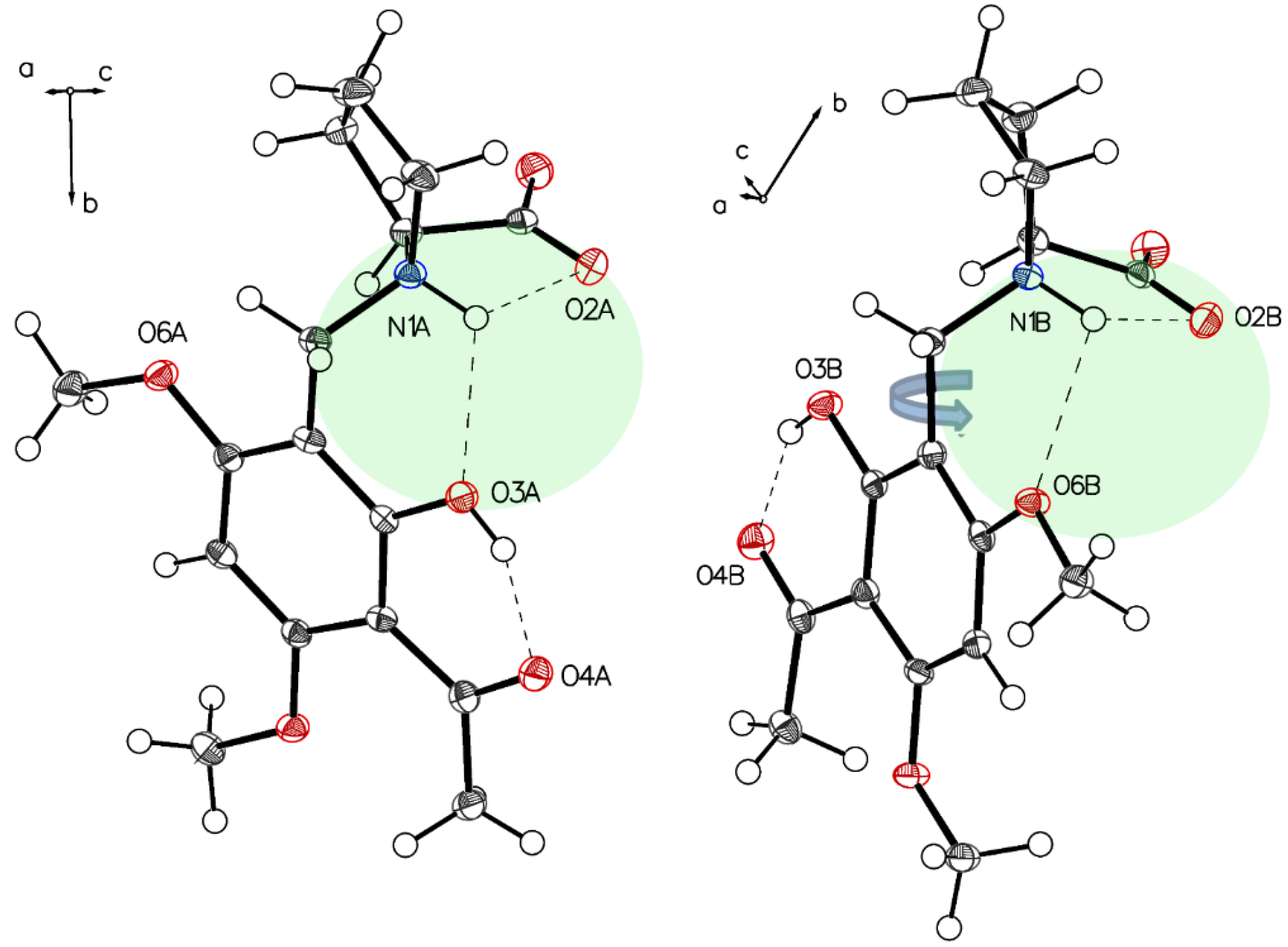
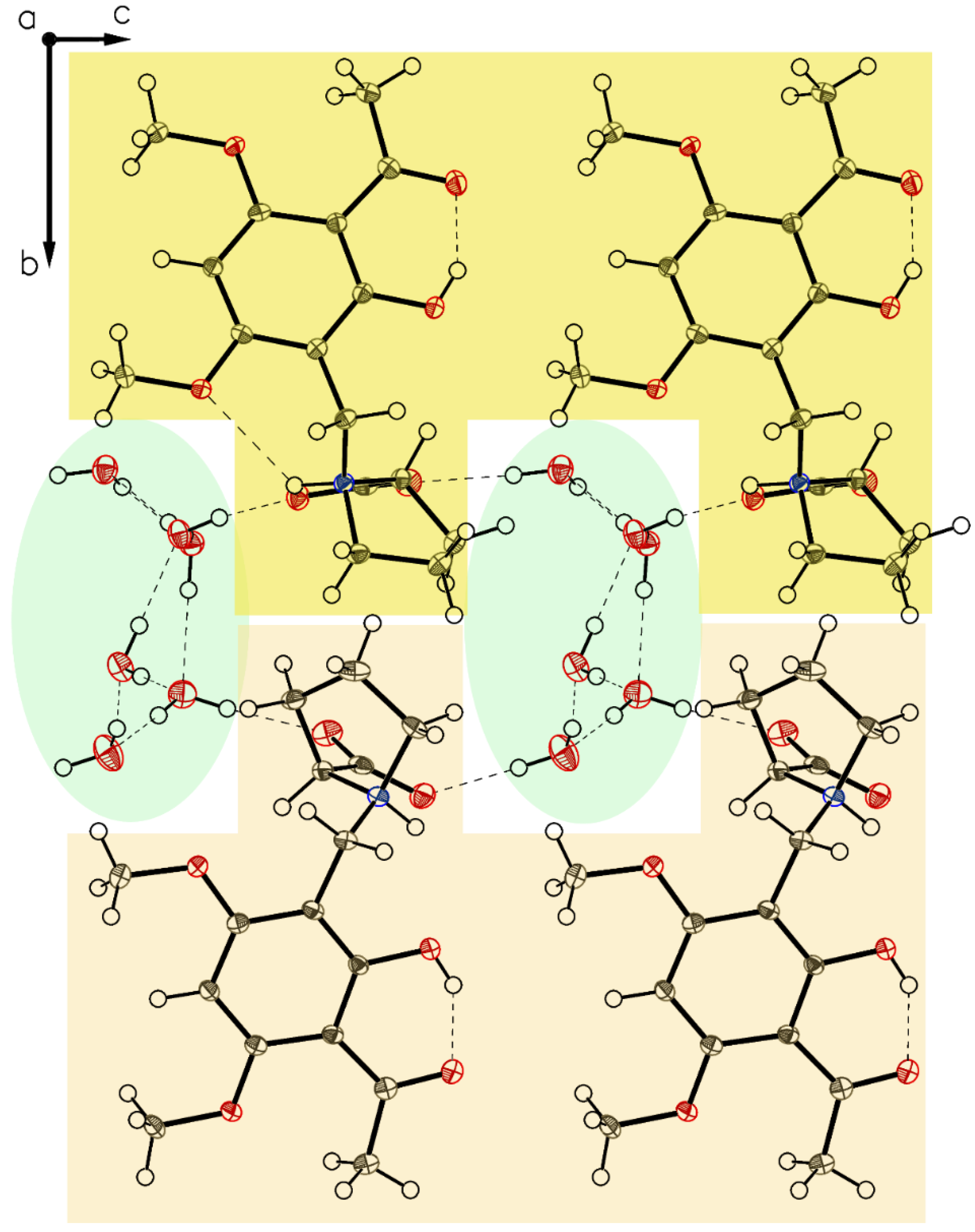
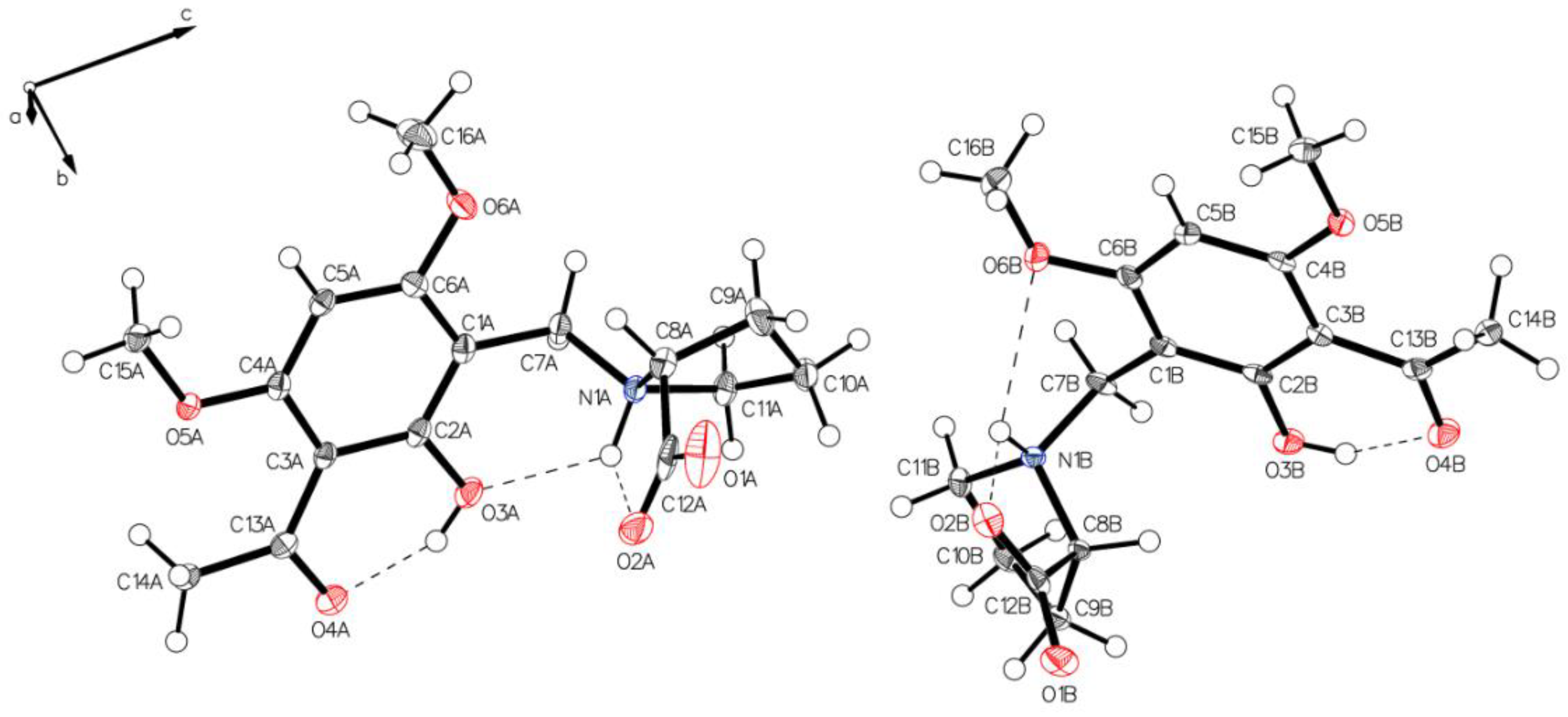
3.1. (–)-Monophyllidin Polymorph-I (P-I) (Crystallized from CH3CN; Space Group P212121; Crystal System Orthorhombic)
3.2. (–)-Monophyllidin Polymorph-II (P-II) (Crystallized from CH3CN; Space Group C2; Crystal System Monoclinic)
3.3. (–)-Monophyllidin Polymorph-III (P-III) (Crystallized from CHCl3; Space Group P1; Crystal System Triclinic)
3.4. (–)-Monophyllidin Polymorph-IV (P-IV) (Obtained from Recrystallization in Ethanol Followed by Acetonitrile; Space Group P1; Crystal System Triclinic)
3.5. (–)-Monophyllidin Polymorph-V (P-V) (Obtained from Water; Space Group P21; Crystal System Monoclinic)
3.6. (–)-Monophyllidin Polymorph-VI (P-VI) (Plate Isolated from the Primary Crystal Mixture; Space Group P212121; Crystal System Orthorhombic)
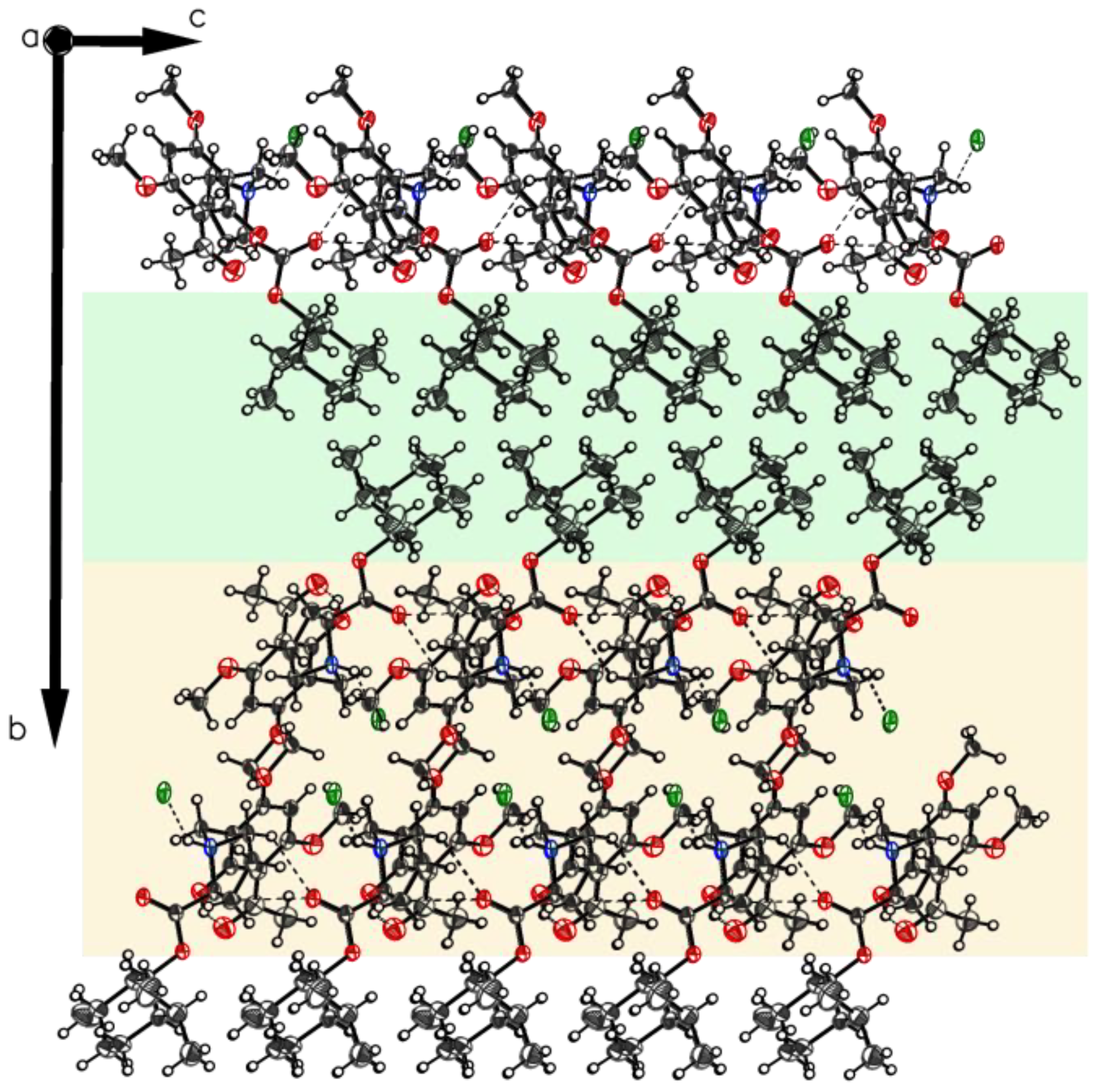
3.7. (S)-(1S,2R,5S)-2-Isopropyl-5-methylcyclohexyl1-(3-acetyl-2-hydroxy-4,6-dimethoxy-benzyl) pyrrolidine-2-carboxylate Hydrochloride (5xHCl).
3.8. Summary and Conclusions
4. Materials and Methods
Chemistry
General Information
Synthetic procedures (for NMR interpretation and spectra see Supplementary Materials)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patino, O.J.; Cuca, L.E. Monophyllidin, a new alkaloid L-proline derivative from Zanthoxylum monophyllum. Phytochem. Lett. 2011, 4, 22–25. [Google Scholar] [CrossRef]
- Roskov, Y.; Kunze, T.; Orrell, T.; Abucay, L.; Paglinawan, L.; Culham, A.; Bailly, N.; Kirk, P.; Bourgoin, T.; Baillargeon, G.; et al. Species 2000 & ITIS Catalogue of Life, 2019 Annual Checklist; Species 2000: Naturalis, Leiden, The Netherlands, 2014; Available online: www.catalogueoflife.org/annual-checklist/2019 (accessed on 4 December 2019).
- Wang, C.; Wan, J.F.; Mei, Z.N.; Yang, X.Z. Acridone alkaloids with cytotoxic and antimalarial activities from Zanthoxylum simullans Hance. Pharm. Mag. 2014, 10, 73–76. [Google Scholar] [CrossRef]
- Talontsi, F.M.; Matasyoh, J.C.; Ngoumfo, R.M.; Chepkorir, R. Mosquito larvicidal activity of alkaloids from Zanthoxylum lemairei against the malaria vector Anopheles gambiae. Pestic. Biochem. Phys. 2011, 99, 82–85. [Google Scholar] [CrossRef]
- Mazzini, C.; Sambri, L.; Regeling, H.; Zwanenburg, B.; Chittenden, G.J.F. Enantiospecific syntheses of (R)- and (S)-proline and some derivatives from D-glucono-1,5-lactone. J. Chem. Soc. Perk. T. 1997, 1, 3351–3356. [Google Scholar] [CrossRef][Green Version]
- Kaoru, H.; Tadao, H. Syntheses of α-Amino Acid Menthyl Esters. Bull. Chem. Soc. Jpn. 1964, 37, 191–194. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS; University of Gottingen: Gottingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.C.; Sheen, J.F.; Hwang, L.S.; Wei, G.J. Identification of 5,7,3’,4’-tetramethoxyflavone metabolites in rat urine by the isotope-labeling method and ultrahigh-performance liquid chromatography-electrospray ionization-mass spectrometry. J. Agric. Food Chem. 2012, 60, 8123–8128. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–5 are available from HG. |






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | ID | Space Group | mp [°C] | Crystal Habitus |
|---|---|---|---|---|
| acetonitrile | P-I | P212121 | 161–162 | block-like |
| acetonitrile | P-II | C2 | 158–160 | needles |
| acetonitrile | P-VI | P212121 | nd (singleton) | plates |
| chloroform | P-III | P1 | 158–159 | needles |
| ethanol-acetonitrile | P-IV | P1 | 158–159 | needles |
| water | P-V | P21 | 155–156 | blocks |
| ID | Space Group | mp [°C] | Crystal Habitus | Guest Molecules | (–)-3a: Guest(s) |
|---|---|---|---|---|---|
| P-I | P212121 | 161–162 | blocks | CH3CN | 1:1 |
| P-II | C2 | 158–160 | needles | CH3CN:water | 2:1:2 |
| P-III | P1 | 158–159 | needles | CHCl3:water | 2:2:1 |
| P-IV | P1 | 158–159 | needles | H2O | 2:5 |
| P-V | P21 | 155–156 | blocks | H2O | 2:1 |
| P-VI | P212121 | nd | plates | H2O | 2:1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dank, C.; Wurzer, R.; Felsinger, S.; Bugl, R.; Kählig, H.; Hela, W.; Roller, A.; Gstach, H. A Many-Faced Alkaloid: Polymorphism of (–)-Monophyllidin. Molecules 2020, 25, 449. https://doi.org/10.3390/molecules25030449
Dank C, Wurzer R, Felsinger S, Bugl R, Kählig H, Hela W, Roller A, Gstach H. A Many-Faced Alkaloid: Polymorphism of (–)-Monophyllidin. Molecules. 2020; 25(3):449. https://doi.org/10.3390/molecules25030449
Chicago/Turabian StyleDank, Christian, Richard Wurzer, Susanne Felsinger, Ricarda Bugl, Hanspeter Kählig, Wolfgang Hela, Alexander Roller, and Hubert Gstach. 2020. "A Many-Faced Alkaloid: Polymorphism of (–)-Monophyllidin" Molecules 25, no. 3: 449. https://doi.org/10.3390/molecules25030449
APA StyleDank, C., Wurzer, R., Felsinger, S., Bugl, R., Kählig, H., Hela, W., Roller, A., & Gstach, H. (2020). A Many-Faced Alkaloid: Polymorphism of (–)-Monophyllidin. Molecules, 25(3), 449. https://doi.org/10.3390/molecules25030449