
Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages

Abstract
1. Introduction
2. Results
2.1. Changes in Body and Lung Weights
2.2. DEP Induces Neutrophilic Lung Inflammation in Mice
2.3. DEP Initiates ER Stress in DEP-Treated Mice
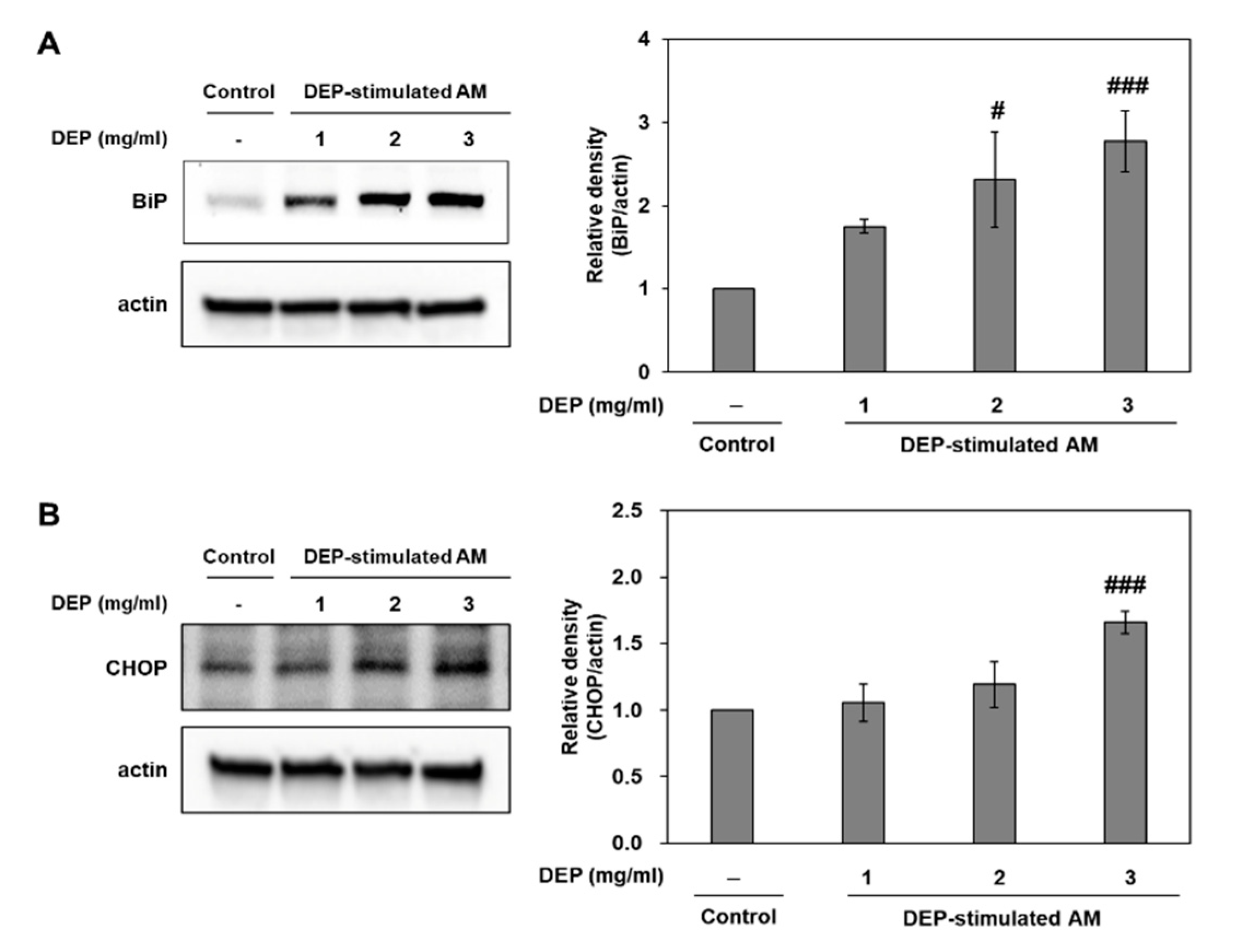
2.4. DEP Exposure Induces ER Stress and Activates the UPR Pathway in AM
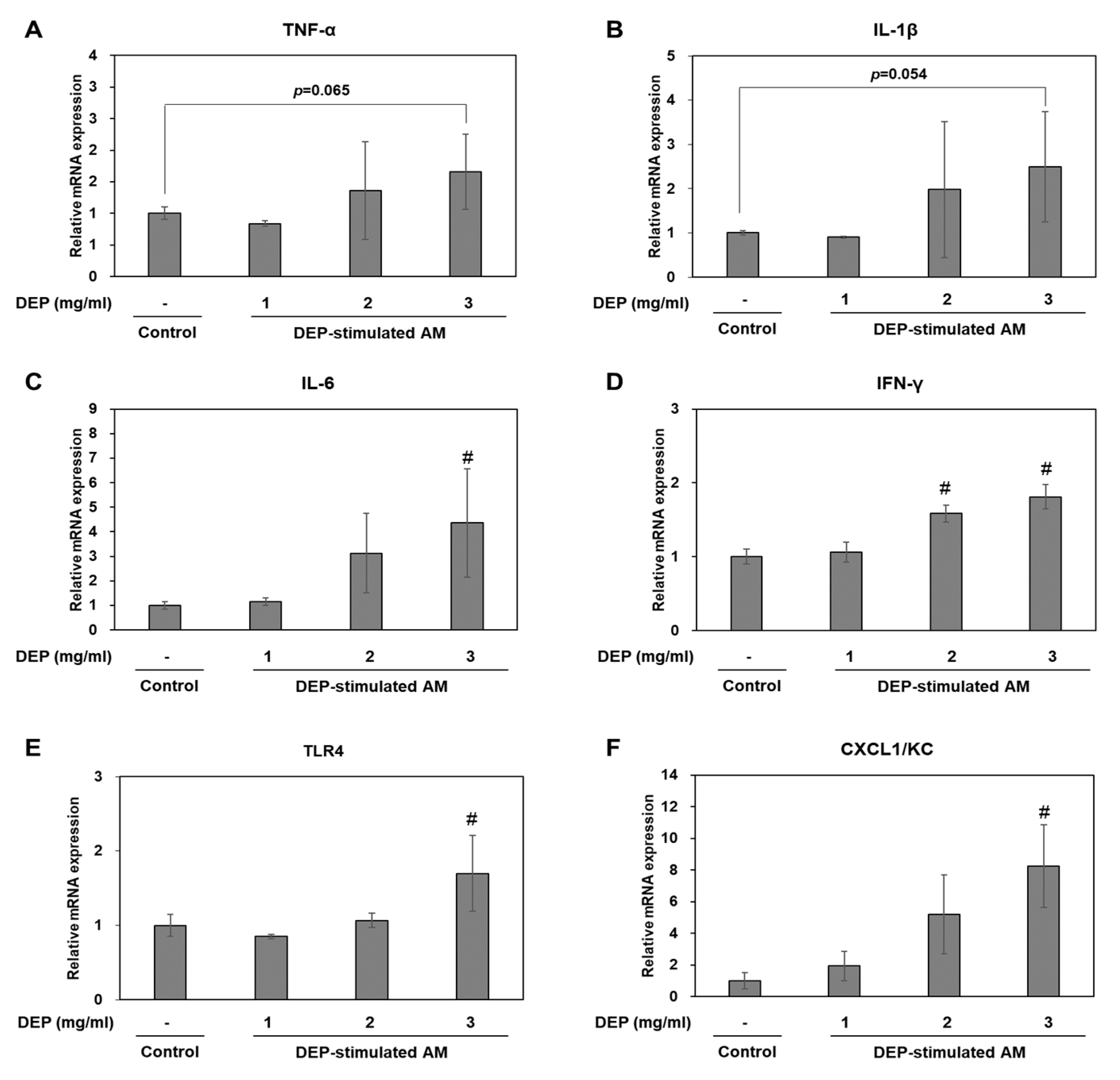
2.5. DEP-Stimulated AM Present with Inflammatory Responses
2.6. Effects of Cell Cytotoxicity and Oxidative Stress on DEP Stimulation in AM
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. DEP Instillation
4.3. Body and Lung Weight Measurements
4.4. BALF Preparation
4.5. Measurement of Cytokine and Chemokine Levels in BALF
4.6. Histological Analysis
4.7. Protein Extract Preparation and Western Blot Analysis
4.8. Murine AM Culture and DEP, H2O2, and NAC Treatment
4.9. Cell Viability and Reactive Oxygen Species (ROS) Measurements
4.10. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kurt, O.K.; Zhang, J.; Pinkerton, K.E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 2016, 22, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-M.; Kuschner, W.G.; Gokhale, J.; Shofer, S. Outdoor air pollution: Nitrogen dioxide, sulfur dioxide, and carbon monoxide health effects. Am. J. Med. Sci. 2007, 333, 249–256. [Google Scholar] [CrossRef]
- Falcon-Rodriguez, C.I.; Osornio-Vargas, A.R.; Sada-Ovalle, I.; Segura-Medina, P. Aeroparticles, composition, and lung diseases. Front. Immunol. 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.-Y.; Wang, Q.; Liu, T. Toxicity research of PM2.5 compositions in vitro. Int. J. Environ. Res. Pub. Health 2017, 14, 232. [Google Scholar] [CrossRef]
- Li, N.; Wang, M.; Oberley, T.D.; Sempf, J.M.; Nel, A.E. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J. Immunol. 2002, 169, 4531–4541. [Google Scholar] [CrossRef]
- Pant, P.; Shi, Z.; Pope, F.D.; Harrison, R.M. Characterization of traffic-related particulate matter emissions in a road tunnel in Birmingham, UK: Trace metals and organic molecular markers. Aerosol. Air Qual. Res. 2017, 17, 117–130. [Google Scholar] [CrossRef]
- Liu, N.M.; Grigg, J. Diesel, children and respiratory disease. BMJ Paediatr Open. 2018, 2, e000210. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Holme, J.A.; Rosas, I.; Schwarze, P.E.; Alfaro-Moreno, E. Recent advances in particulate matter and nanoparticle toxicology: A review of the in vivo and in vitro studies. BioMed Res. Int. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zanobetti, A.; Franklin, M.; Koutrakis, P.; Schwartz, J. Fine particulate air pollution and its components in association with cause-specific emergency admissions. Environ. Health 2009, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Dominici, F.; Peng, R.D.; Bell, M.L.; Pham, L.; McDermott, A.; Zeger, S.L.; Samet, J.M. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA 2006, 295, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.F.; Xu, Y.H.; Shi, M.H.; Lian, Y.X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, E69–E74. [Google Scholar] [CrossRef]
- Pablo-Romero, M.d.P.; Román, R.; Limón, J.M.G.; Praena-Crespo, M. Effects of fine particles on children’s hospital admissions for respiratory health in Seville, Spain. J. Air Waste Manag. Assoc. 2015, 65, 436–444. [Google Scholar] [CrossRef]
- Cao, Q.; Rui, G.; Liang, Y. Study on PM2.5 pollution and the mortality due to lung cancer in China based on geographic weighted regression model. BMC Publ. Health 2018, 18, 925. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, D.I.; Kang, M.R.; Lee, K.S.; Park, S.Y.; Jeong, J.S.; Lee, Y.C. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor κB activation. J. Allergy Clin. Immunol. 2013, 132, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.-H.; Burr, L.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in respiratory disease. Clin. Transl. Immunol. 2018, 7, e1019. [Google Scholar] [CrossRef]
- Hirai, K.E.; De Sousa, J.R.; Silva, L.M.; Junior, L.B.D.; Furlaneto, I.P.; Carneiro, F.R.O.; Aarão, T.L.D.S.; Sotto, M.N.; Quaresma, J.A.S. Endoplasmic Reticulum Stress Markers and Their Possible Implications in Leprosy’s Pathogenesis. Dis. Mark. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. Endoplasmic reticulum stress and allergic diseases. Curr. Allergy Asthma Rep. 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef]
- Burman, A.; Tanjore, H.; Blackwell, T.S. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018, 68–69, 355–365. [Google Scholar] [CrossRef]
- Marciniak, S.J. Endoplasmic reticulum stress in lung disease. Eur. Respir. Rev. 2017, 26, 170018. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Song, M.-K.; Kim, S.-H.; Park, C.Y.; Lee, K. TF-343 Alleviates Diesel Exhaust Particulate-Induced Lung Inflammation via Modulation of Nuclear Factor-κB Signaling. J. Immunol. Res. 2019, 2019, 8315845. [Google Scholar] [CrossRef] [PubMed]
- Imrich, A.; Ning, Y.; Lawrence, J.; Coull, B.; Gitin, E.; Knutson, M.; Kobzik, L. Alveolar macrophage cytokine response to air pollution particles: Oxidant mechanisms. Toxicol. Appl. Pharmacol. 2007, 218, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.C.; Townsley, M.I. Townsley. Evaluation of lung injury in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L231–L246. [Google Scholar] [CrossRef] [PubMed]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, Y.; Arashidani, K.; Yoshida, S.; Takano, H.; Sun, G.; Shibamoto, T. Urban PM2.5 exacerbates allergic inflammation in the murine lung via a TLR2/TLR4/MyD88-signaling pathway. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, H.J.; Kim, D.I.; Lee, K.B.; Park, H.J.; Jeong, J.S.; Cho, S.H.; Lee, Y.C. Blockade of interplay between IL-17A and endoplasmic reticulum stress attenuates LPS-induced lung injury. Theranostics 2015, 5, 1343–1362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, Y.C. Endoplasmic reticulum stress and the related signaling networks in severe asthma. Allergy Asthma Immunol. Res. 2015, 7, 106–117. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, S.; Nishikawa, M.; Mori, I.; Yanagisawa, R.; Takano, H.; Inoue, K.-I.; Sun, G.; Shibamoto, T. Urban particulate matter in Beijing, China, enhances allergen-induced murine lung eosinophilia. Inhal. Toxicol. 2010, 22, 709–718. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, D.; Ren, J.; Yang, J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Lindén, A.; Laan, M.; Anderson, G.P. Neutrophils, interleukin-17A and lung disease. Eur. Respir. J. 2005, 25, 159–172. [Google Scholar] [CrossRef]
- Cohen, A.J.; Pope, C.A. Lung cancer and air pollution. Environ. Heal. Perspect. 1995, 103, 219–224. [Google Scholar] [CrossRef]
- Cooper, D.M.; Loxham, M. Particulate matter and the airway epithelium: The special case of the underground? Eur. Respir. Rev. 2019, 28, 190066. [Google Scholar] [CrossRef]
- Mills, P.R.; Davies, R.J.; Devalia, J.L. Airway Epithelial Cells, Cytokines, and Pollutants. Am. J. Respir. Crit. Care Med. 1999, 160, S38–S43. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Rylance, J.; Fullerton, D.G.; Scriven, J.; Aljurayyan, A.N.; Mzinza, D.; Barrett, S.; Wright, A.K.A.; Wootton, D.G.; Glennie, S.J.; Baple, K.; et al. Household Air Pollution Causes Dose-Dependent Inflammation and Altered Phagocytosis in Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 52, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Kim, C.; Li, S.; Marchal, C.C.; Towe, J.; Atkinson, S.J.; Dinauer, M.C. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J. Immunol. 2004, 173, 5971–5979. [Google Scholar] [CrossRef]
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018, 108, 1866–1878. [Google Scholar] [CrossRef]
- Van Der Vliet, A.; Janssen-Heininger, Y.M.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Asp. Med. 2018, 63, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Okeson, C.D.; Riley, M.R.; Riley-Saxton, E. In Vitro Alveolar Cytotoxicity of Soluble Components of Airborne Particulate Matter: Effects of Serum on Toxicity of Transition Metals. Toxicol. In Vitro 2004, 18, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, I.-L.; Huang, Y.-J. Effects of serum on cytotoxicity of nano- and micro-sized ZnO particles. J. Nanoparticle Res. 2013, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Henderson, R.B.; Laschinger, M.; Hogg, N. Neutrophil chemokines KC and macrophage-inflammatory Protein-2 are newly synthesized by tissue macrophages using distinct TLR Signaling pathways. J. Immunol. 2008, 180, 4308–4315. [Google Scholar] [CrossRef]
- Kumar, K.P.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Beck-Schimmer, B.; Schlender, R.; Pasch, T.; Reyes, L.; Booy, C.; Schimmer, R.C. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respir. Res. 2005, 6, 61. [Google Scholar] [CrossRef]
- Zhao, C.; Pavicic, P.G., Jr.; Datta, S.; Sun, D.; Novotny, M.; Hamilton, T.A. Cellular stress amplifies TLR3/4-induced CXCL1/2 gene transcription in mononuclear phagocytes via RIPK1. J. Immunol. 2014, 193, 879–888. [Google Scholar] [CrossRef]
- Alexander, D.J.; Collins, C.J.; Coombs, D.W.; Gilkison, I.S.; Hardy, C.J.; Healey, G.; Karantabias, G.; Johnson, N.; Karlsson, A.; Kilgour, J.D.; et al. Association of Inhalation Toxicologists (AIT) Working Party Recommendation for Standard Delivered Dose Calculation and Expression in Non-Clinical Aerosol Inhalation Toxicology Studies with Pharmaceuticals. Inhal. Toxicol. 2008, 20, 1179–1189. [Google Scholar] [CrossRef]
- Renne, R.; Brix, A.; Harkema, J.; Herbert, R.; Kittel, B.; Lewis, D.; March, T.; Nagano, K.; Pino, M.; Rittinghausen, S.; et al. Proliferative and Nonproliferative Lesions of the Rat and Mouse Respiratory Tract. Toxicol. Pathol. 2009, 37, 5S–73S. [Google Scholar] [CrossRef]
- Mackie, J. Background Lesions in Laboratory Animals: A Color Atlas; McInness, E.F., Ed.; Saunders Elsevier: Edinburgh, UK, 2012; pp. 45–72. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Naive Control | Vehicle Control | DEP 25 | DEP 50 | DEP 100 | |
|---|---|---|---|---|---|---|
| Accumulation of black particle-laden | Minimal | 0 | 0 | 1 | 0 | 0 |
| alveolar macrophages and black | Mild | 0 | 0 | 4 | 2 | 0 |
| particles in alveolar lumen | Moderate | 0 | 0 | 0 | 3 | 2 |
| Marked | 0 | 0 | 0 | 0 | 3 | |
| Mean ± SD | 0 | 0 | 1.8 ± 0.45 ## | 2.6 ± 0.55 ## | 3.6 ± 0.55 ## | |
| Inflammatory cell infiltration, | Minimal | 0 | 0 | 4 | 3 | 0 |
| peribronchiolar/perivascular/interstitial | Mild | 0 | 0 | 0 | 2 | 5 |
| Mean ± SD | 0 | 0 | 0.8 ± 0.45 # | 1.4 ± 0.55 ## | 2 ± 0.0 ## |
| Primer Name | Forward Primer | Reverse Primer |
|---|---|---|
| BiP | TTCAGCCAATTATCAGCAAACTCT | TTTTCTGATGTATCCTCTTCACCAGT |
| CHOP | CCACCACACCTGAAAGCAGAA | AGGTGAAAGGCAGGGACTCA |
| sXBP-1 | CTGAGTCCGAATCAGGTGCAG | GTCCATGGGAAGATGTTCTGG |
| ATF4 | GGGTTCTGTCTTCCACTCCA | AAGCAGCAGAGTCAGGCTTTC |
| TNF-α | ATGAGCACAGAAAGCATGA | AGTAGACAGAAGAGCGTGGT |
| IL-1β | CAACCAACAAGTGATATTCTCCATG | ATCCACACTCTCCAGCTGCA |
| IL-6 | GCTACCAAACTGGATATAATCAGGA | CCAGGTAGCTATGGTACTCCAGAA |
| IFN-γ | TTCTTCAGCAACAGCAAGGC | TCAGCAGCGACTCCTTTTCC |
| TLR4 | AAACGGCAACTTGGACCTGA | AGCTTAGCAGCCATGTGTTCCA |
| CXCL1/KC | CGCTCGCTTCTCTGTGCA | ATTTTCTGAACCAAGGGAGCT |
| actin | GGCACCACACCTTCTACAATG | GGGGTGTTGAAGGTCTCAAAC |
Sample Availability: Data used and/or analyzed during the current study are available from the corresponding author on reasonable request. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.I.; Song, M.-K.; Kim, H.-I.; Han, K.M.; Lee, K. Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages. Molecules 2020, 25, 6046. https://doi.org/10.3390/molecules25246046
Kim DI, Song M-K, Kim H-I, Han KM, Lee K. Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages. Molecules. 2020; 25(24):6046. https://doi.org/10.3390/molecules25246046
Chicago/Turabian StyleKim, Dong Im, Mi-Kyung Song, Hye-In Kim, Kang Min Han, and Kyuhong Lee. 2020. "Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages" Molecules 25, no. 24: 6046. https://doi.org/10.3390/molecules25246046
APA StyleKim, D. I., Song, M.-K., Kim, H.-I., Han, K. M., & Lee, K. (2020). Diesel Exhaust Particulates Induce Neutrophilic Lung Inflammation by Modulating Endoplasmic Reticulum Stress-Mediated CXCL1/KC Expression in Alveolar Macrophages. Molecules, 25(24), 6046. https://doi.org/10.3390/molecules25246046