1. Introduction
Theranostics provide a cutting-edge approach to cancer therapy by utilizing a single agent that can diagnose, monitor, and treat a patient’s disease. Auger and alpha emitting radionuclides have high-linear energy transfer (high-LET) properties capable of inducing DNA damage more efficiently than low-LET gamma or beta radiation [
1]. Although alpha-emitters are known to cause high levels of DNA damage, very few radionuclides have imageable photons in their decay chain [
2]. For these reasons, theranostic Auger emitters offer a unique portfolio of radionuclides that have imageable properties through positron or single-photon emission tomography and therapeutic properties from Auger electrons [
3].
Several small molecules have been investigated to deliver therapeutic Auger radionuclides, including iodine-123, iodine-125, and bromine-77. These agents target various proteins or processes, including direct DNA incorporation ([
125I]IUDR, [
77Br]BrUDR), prostate-specific antigens ([
125I]DCIBzL), and estrogen receptors ([
123I]iodo-1,1-bis(4-hydroxyphenyl)-2-phenylethylene) [
4,
5,
6,
7]. These studies have shown the initial proof of concept for utilizing Auger emitters as theranostic radionuclides; however, with the exception of [
125I]IUDR and [
77Br]BrUDR, the molecular targets are not located within the nucleus or in close proximity to DNA. Furthermore, deoxyuridine analogues have shown in-vivo deiodonation/debromination, which suggests instability of the construct. Recently, poly (ADP-ribose) polymerase (PARP) targeted beta and Auger emitting radiopharmaceuticals have been reported for the treatment of glioblastoma and showed anti-tumor effects both in-vitro and in-vivo [
8,
9]. Furthermore, other bromine-77 PARP inhibitors have been reported, although pre-clinical evaluation is ongoing [
10,
11].
The path-length of Auger electrons is 10–500 nm [
3], requiring delivery of Augur emitters directly or in close proximity to DNA to induce damage and cancer cell death. Poly (ADP-ribose) polymerase 1 (PARP-1) is an ADP-ribosylation enzyme that is essential for DNA damage response and is overexpressed in most tumor types [
12]. PARP-1 subcellular localization on the chromatin provides an ideal target for tumor cell specific delivery of Auger emitters to DNA [
13].
In the present study, we investigated PARP-1 as a unique molecular target that is highly expressed in cancer nuclei for the delivery of theranostic Auger emitting radionuclides. Recent work has demonstrated excellent visualization of nuclear localization of the PARP inhibitor rucaparib through fluorescent imaging, which is a close analogue to KX1 reported in this work, supporting a strong rationale for this approach [
14]. Herein, we report in-vitro mechanistic studies exploring the specificity of DNA damage induced by a previously reported radioiodinated PARP inhibitor, KX1, labeled with Auger emitting radionuclides iodine-125 and iodine-123. Studies were performed using viable patient tumor slice cultures as a means for screening therapeutic efficacy ex-vivo. Furthermore, we performed in-vivo proof of concept studies to highlight the theranostic potential of PARP inhibitors radiolabeled with theranostic Auger emitters.
3. Discussion
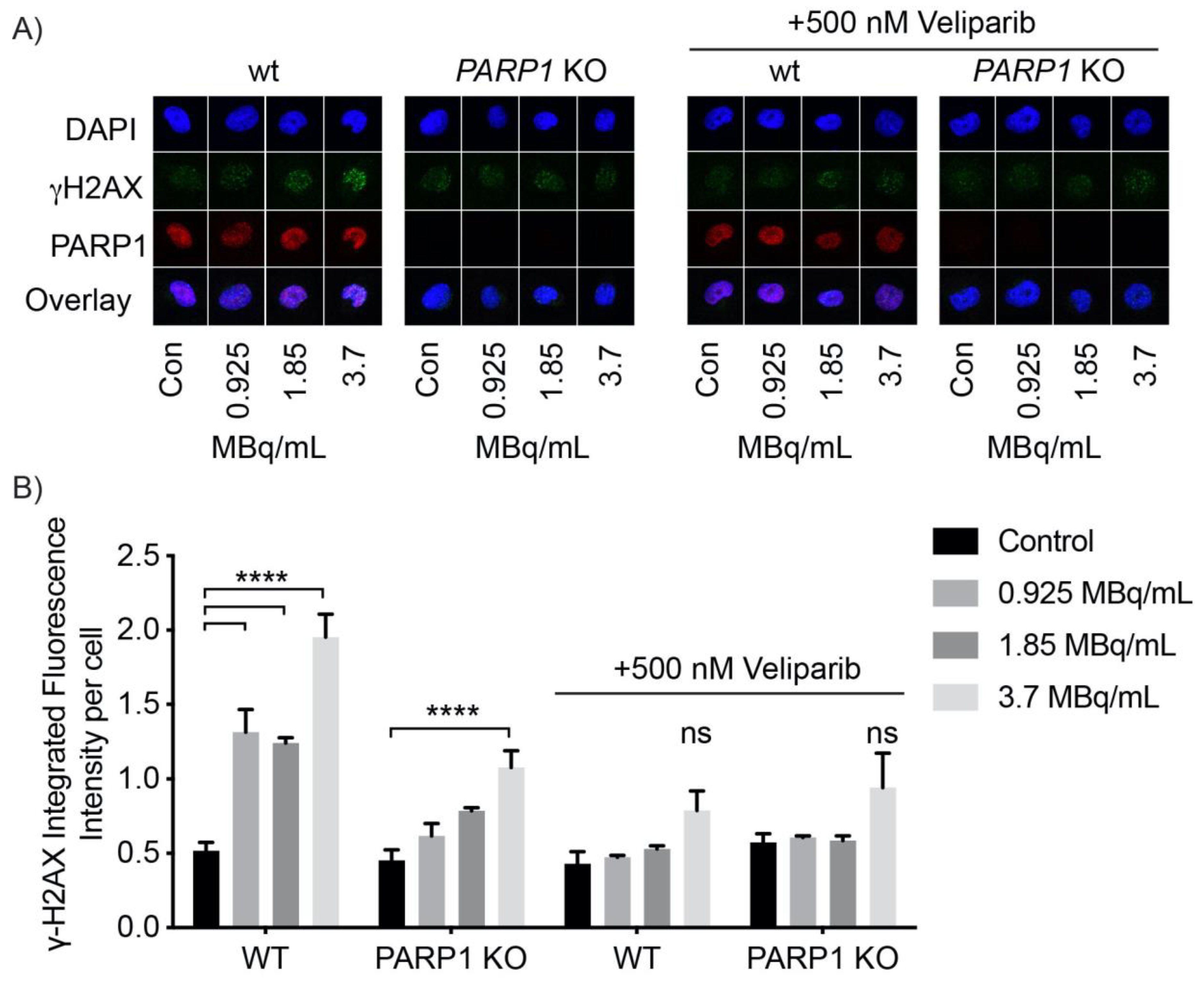
The present study showed the feasibility of using Auger emitters as theranostic agents. Specifically, we were able to show that the radiolabeled PARP inhibitor KX1 is capable of delivering iodine-125 and iodine-123 Auger emitting radionuclides to the nucleus of cells to induce DNA damage. Reduced cytotoxicity, and γH2AX foci observed after treating OVCAR8 PARP1 knockout cell lines demonstrated that both Auger emitters caused DNA damage as a result of specifically targeting PARP-1. These data provide evidence that radiolabeled PARP inhibitors such as KX1 can deliver Auger emitting radionuclides within close proximity to DNA to maximize the high-LET effects of Auger electrons for inducing DNA damage and cell death, building on our previous work [
19].
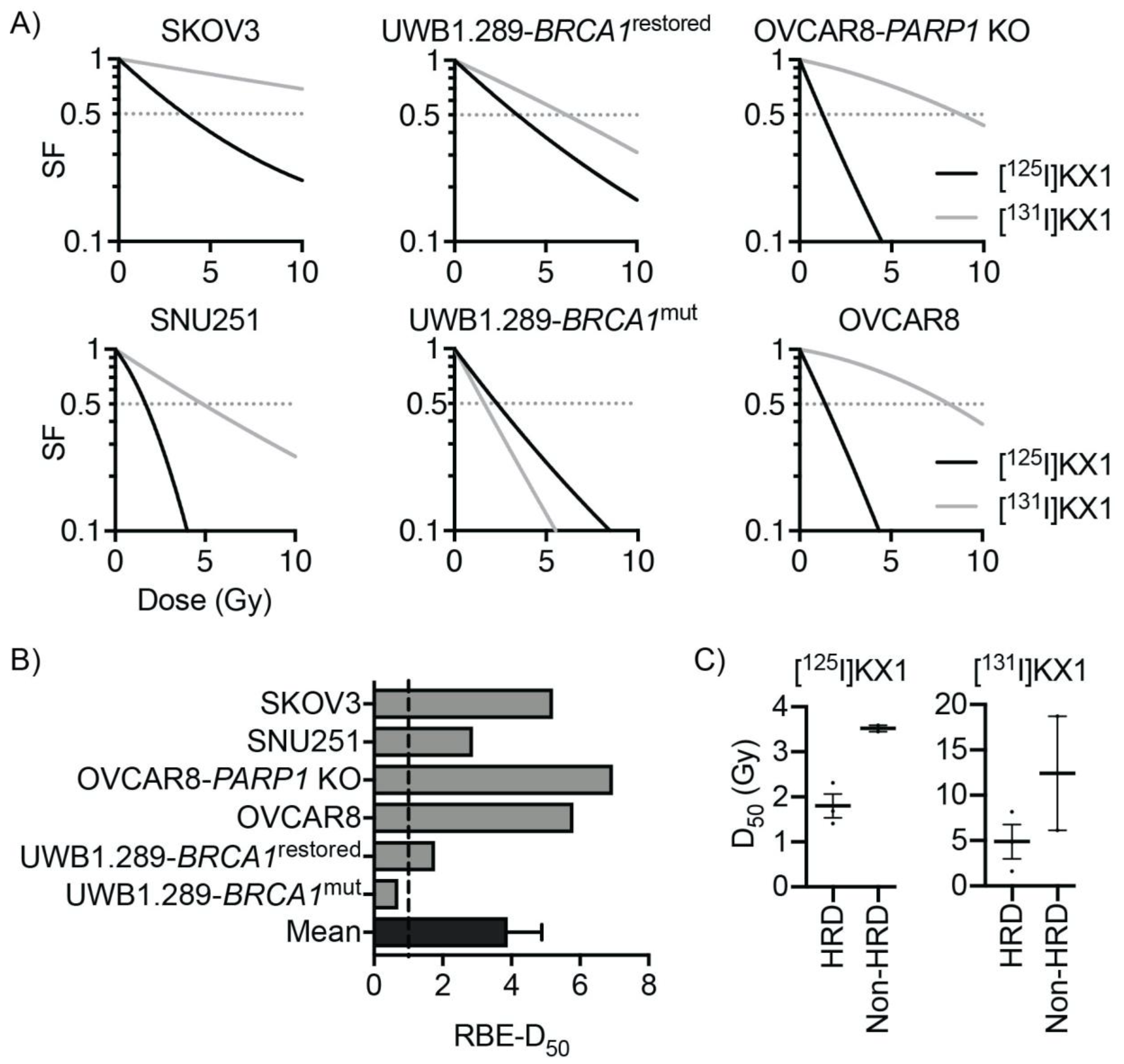
Another interesting result found in our study is that DNA damage induced by KX1 is enzymatically independent of PARP-1 inhibition. The primary difference between radiolabeled PARP inhibitors and conventional PARP inhibitors as anticancer drugs is that radiolabeled PARP inhibitors induce DNA damage using ionizing radiation and that they do not enzymatically inhibit PARP-1. This is in contrast to conventional PARP inhibitors that primarily work through synthetic lethality, where loss of primary homologous recombination (HR) genes such as BRCA1 or BRCA2 combined with PARP inhibition results in cell death. Although PARP inhibition and BRCA mutations may confer synthetic lethality, the loss of HR also reduced cellular fitness against DNA damage caused by Auger radiation. This effect enabled [125I]KX1 to induce DNA damage and cell death dependent on BRCA1 mutation, as shown in dose–response studies where restoration of BRCA1 in UWB1.289 ovarian cancer cells decreased radiosensitivity. Indeed, BRCA1 mutant ovarian cancer cell lines were the most sensitive to [125I]KX1; however, dosimetric analysis revealed interesting differences in RBE, which suggests [125I]KX1 is more effective than low-LET beta-emitting analogue [131I]KX1 in ovarian cancer cell lines with functional BRCA1. We propose that increased DNA repair capacity of ovarian cancer cell lines provides a better cellular fitness capable of mitigating DNA damage induced by low-LET radiation, whereas a known property of high-LET radiation is that cell kill effects are independent of DNA repair capacity. Together these data further support Auger electrons as a form of high-LET radiation that are less susceptible to common resistance mechanisms to low-LET radiation, even clinical PARP inhibitors. We found the BRCA1 mutation resulted in a resistance enhancement of only 1.59 for [125I]KX1, in contrast to [131I]KX1, which showed 3.79, and is in stark contrast with the 100 to 1000 times difference for clinically used PARP inhibitors. Furthermore, BRCA1 reversion mutations are the most common cause of clinical resistance where BRCA1 regains partial or complete functions and tumors no longer respond. Our work shows [125I]KX1 has the potential to remain effective even after a BRCA1 reversion mutation and possibly have efficacy in non-BRCA mutant tumors.
In this study, we showed that it is feasible to evaluate the radiosensitivity of viable patient tumor samples ex-vivo. This not only increases the translational potential of evaluating Auger-emitting radiopharmaceuticals for therapy but also enables the direct evaluation of anti-tumor effects before early phase clinical trials take place. Future work will be directed towards ex-vivo analysis of response to better understand tumor radiobiology and rigorously test the appropriateness for Auger-emitting radiopharmaceutical therapy. In summary, we have shown that radiolabeled PARP inhibitors can effectively and specifically serve as targeting vectors for the delivery of therapeutic Auger emitting radionuclides as cancer theranostics. Future studies will focus on the theranostic application of [123I]KX1, evaluating the predictive capability of SPECT imaging-based dosimetry on anti-tumor response and long-term survival in pre-clinical models.
4. Materials and Methods
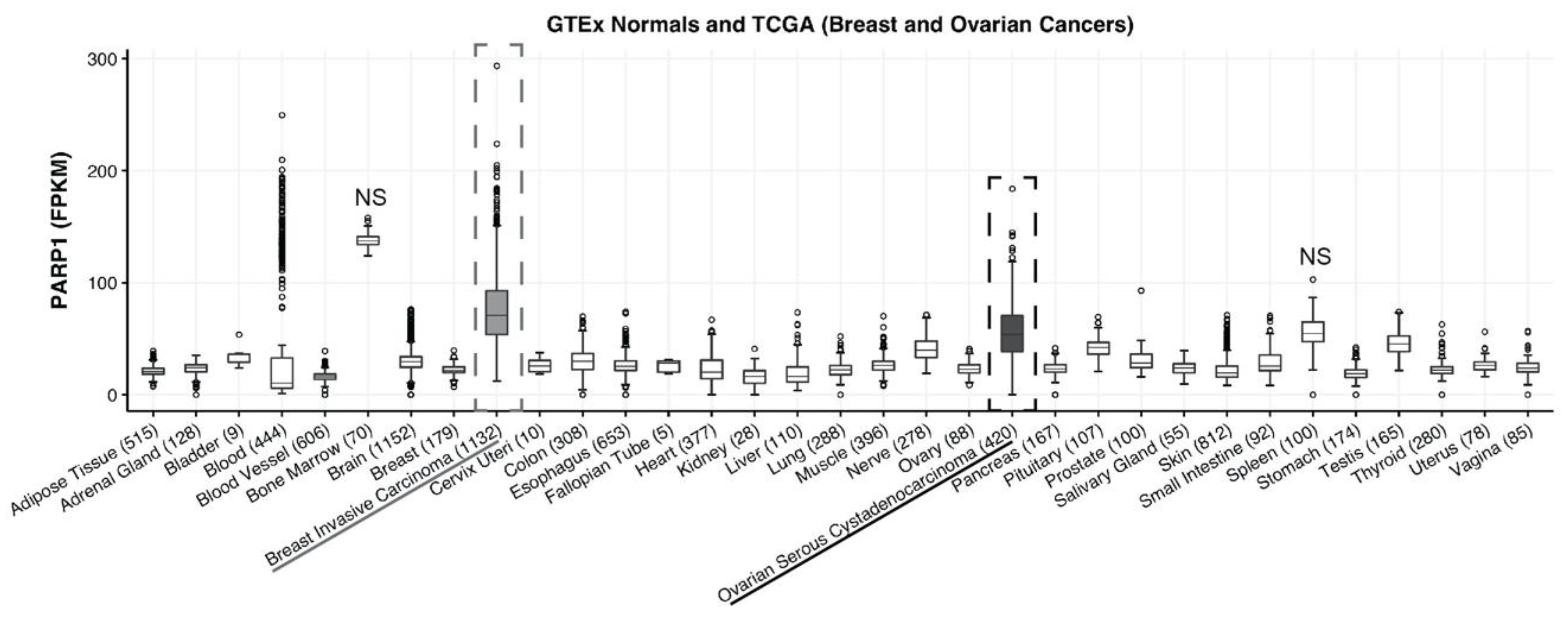
4.1. PARP-1 Expression in Breast and Ovarian Cancer
To determine the relative expression of PARP-1 in breast or ovarian cancer vs. adult normal tissue, we used RNA sequencing data from The Cancer Genome Atlas (
https://cancergenome.nih.gov/) and the Genotype Tissue Expression (GTEx) project.
4.2. Cell Culture
Cell lines were cultured using standard techniques under 5% CO
2 and 10% O
2 at 37 °C. OVCAR8 and OVCAR8 PARP1 knockout (KO) cell lines were cultured in RPMI 1640 with 10% FBS and 1% penicillin/streptomycin. Cell lines with stable knockout of PARP1 were previously reported [
16]. Cells were selected for stable expression of each sgRNA using 2µg/mL puromycin for one week. PARP1 deletion was confirmed by Western blot protein analysis. OVCAR8 PARP1 KO were continuously selected with 2 µg/mL of puromycin. UWB1.289 and UWB1.289
BRCA1 restored cell lines were cultured in a 1:1 mixture of MEGM with bullet kit (ATCC) and RPMI 1640 with 10% FBS and 1% penicillin/streptomycin.
4.3. Radiochemistry
Iodine-123 and iodine-125 were purchased from Nuclear Diagnostic Products Radiopharmacy (Cherry Hill, NJ) and Perkin Elmer (Waltham, MA). [
123/125I]KX1 radiolabeling was performed as previously described by electrophilic destannylation of a tin precursor under mild oxidative conditions, although newer improved approaches have been reported for copper-mediated halodeboronation of boronic pinacol ester precursors [
15]. Briefly, 100 µg of stannous-KX1 precursor material was dissolved in 50 µL of MeOH followed by the addition of 100 µL of 3:1 acetic acid:hydrogen peroxide. The reaction was heated at 100 °C for 30 min and purified by reverse phase chromatography under isocratic conditions (40% MeCN:60 % 1 M ammonium formate pH 4.5) on a semi-preparative HPLC using a Phenomenex Luna
® 5 µm C18 100 Å column 250 × 4.6 mm (Waters, Milford, MA, USA). The product peak was collected, diluted with water to <10% MeCN, and concentrated on a Sep-Pak C18 Plus Light Cartridge, 130 mg, 55–105 µm (Waters, Milford, MA, USA). The final product was diluted in 200 proof ethanol and diluted to a final concentration of <1% ethanol for respective biological studies.
4.4. Western Blot Analysis
OVCAR8 cells were treated with 3.7 MBq/mL of [123I]KX1 for either 2 or 24 h to determine whether [123I]KX1 treatment resulted in biochemical inhibition of PARP-1. OVCAR8 cells were treated with the FDA approved PARP inhibitor, olaparib, as a positive control. Cell lysates were prepared by first lysing cells in RIPA buffer (Thermo Fisher, Philadelphia, PA, USA) with protease and phosphatase inhibitors (Sigma Aldrich, St. Louis MO) on ice for 30 min followed by sonication. Lysates were then centrifuged at 14,000× g for 20 min at 4 °C to remove cellular debris. Next, solutions were diluted with 4× Laemmli buffer and heated at 100 °C for 5 min. Gel electrophoresis was performed on a BioRad system and transferred to a PDVF membrane using a BioRad Turbo transfer at 1.3 A 25V for 7 min. Membranes were then blocked in LiCor Odyssey blocking buffer for 1 h, followed by 1 h incubation at 37 °C in Odyssey blocking buffer with primary antibodies for PARP-1 (1:1000, Cell Signaling and Technologies 46D11), and PAR/poly (ADP-ribose) (1:1000, Enzo 10H), γH2AX (1:5000, Millipore JBW301). Membranes were then washed 3 times for 5 min per wash in 0.2% PBST. Secondary antibodies corresponding to primary species (Thermo Fisher, Philadelphia, PA, USA) were added at a 1:10,000 dilution in Odyssey blocking buffer and incubated at 37 °C for 1 h. Membranes were then washed 3 times for 5 min per wash in PBST followed by 1 wash in PBS. Membranes were then read on a LiCor imager.
4.5. Single Cell Immunofluorescence
OVCAR8 cells were analyzed by immunofluorescent cell microscopy to measure PARP-1 inhibition and the isogenic pair, OVCAR8 and OVCAR8 PARP1 KO, cell lines to evaluate [123/125I]KX1 induced DNA damage. Cells were first seeded on round cover slips in 24-well plates at 50,000 cells/well and 8-well chamber slides at 10,000 cells/well for 48 h before treatment with [123/125I]KX1 at concentrations from 0.74–4.44 MBq/mL for 1, 2, or 24 h. After treatment, cells were washed once with PBS, then fixed with 2% PFA for 10 min, washed with PBS 3 times, and permeabilized in 0.4% triton X on ice for 10 min. Cells were incubated with primary antibody incubation for 1 h, followed by 3 washes with 0.2% PBST, then incubated with corresponding secondary antibodies (goat anti-mouse IgG Alexa Fluor Plus 555 cat # A32727, goat anti-rabbit IgG Alexa Fluor Plus 488 cat # A327731) for 1 h. Cells were mounted with ProLong Glass mounting media containing NucBlue (Invitrogen). Primary antibodies used for analysis included DNA damage marker γH2AX (dilution: 1:5000, 05-636, anti-phospho-Histone H2A.X ser139, Millipore). The biochemical function of PARP-1 was analyzed through measuring the biochemical product poly(ADP-ribose) (PAR) (dilution: 1:1000, poly(ADP-ribose) monoclonal antibody 10H, Enzo, Farmingdale, NY, USA) after treatment with [123/125I]KX1. PARP-1 (dilution: 1:1000, PARP rabbit mAb 46D11, Cell Signaling Technology, Danvars, MA, USA) was assessed to confirm the absence of PARP-1 in OVCAR8 PARP1 KO cells. Nuclear staining of DAPI was used for identifying the nuclei of cells for quantifying DNA damage response markers. Images were taken on a Zeiss Observer Microscope and fluorescent intensity was quantified using Zeiss Zen software (Zeiss, Netherlands). For confocal microscopy experiments, images were acquired on a Leica SP8 Confocal Microscope and fluorescence intensity was quantified using Cell Profiler software.
4.6. Cell Viability
OVCAR8, OVCAR8 PARP1 KO Guide 1(G1), OVCAR8 PARP1 KO G2, OVCAR8 PARP1 KO G3, UWB1.289, and UWB1.289-BRCA1 restored cell lines were seeded at 1000 cells/well in a 96-well plate for 24 h and subsequently treated with [125I]KX1 at doses ranging from 37–3700 MBq/mL for 1 h. Following the treatment period, treatment medium was aspirated, and cell culture media was replenished. Cells were then allowed to regrow for 5–7 days and cell viability was quantified by measuring ATP using the bioluminescent assay CellTiter Glo (Promega, Waltham, MA, USA). Plates were read on a Perkin Elmer EnSpire multimode plate reader (Waltham, MA, USA). Dose–response curves were fitted using a non-linear sigmoidal dose–response curve in Prism GraphPad v 7.0. Effective concentrations for 50% reductions in cell viability were calculated.
4.7. Radiation Dosimetry
To calculate the radiation dosimetry of [
131I]KX1 and [
125I]KX1, methods were performed as previously published [
19]. Briefly, the radiation dose to the cell nucleus was derived from the radiopharmacology data (
Supplementary Table S3) and calculated using Monte Carlo simulation with Medical Internal Radiation Dosimetry Cell (MIRDcell) V2.1, as previously described [
20]. The radii of the cell and its nucleus were measured using phase contrast and fluorescent microscopy with DAPI. The cytotoxic dose–response curves for the radiopharmaceuticals were transformed to radiation dose–response curves based on the linear-quadratic model; 50% survival was used as the reference endpoint. The relative biological effectiveness (RBE) of each type of radiation was calculated, and data are reported as [
125I]KX1 RBE/[
131I]KX1 RBE.
4.8. SPECT/CT Imaging, Ex-Vivo Autoradiography and Histology
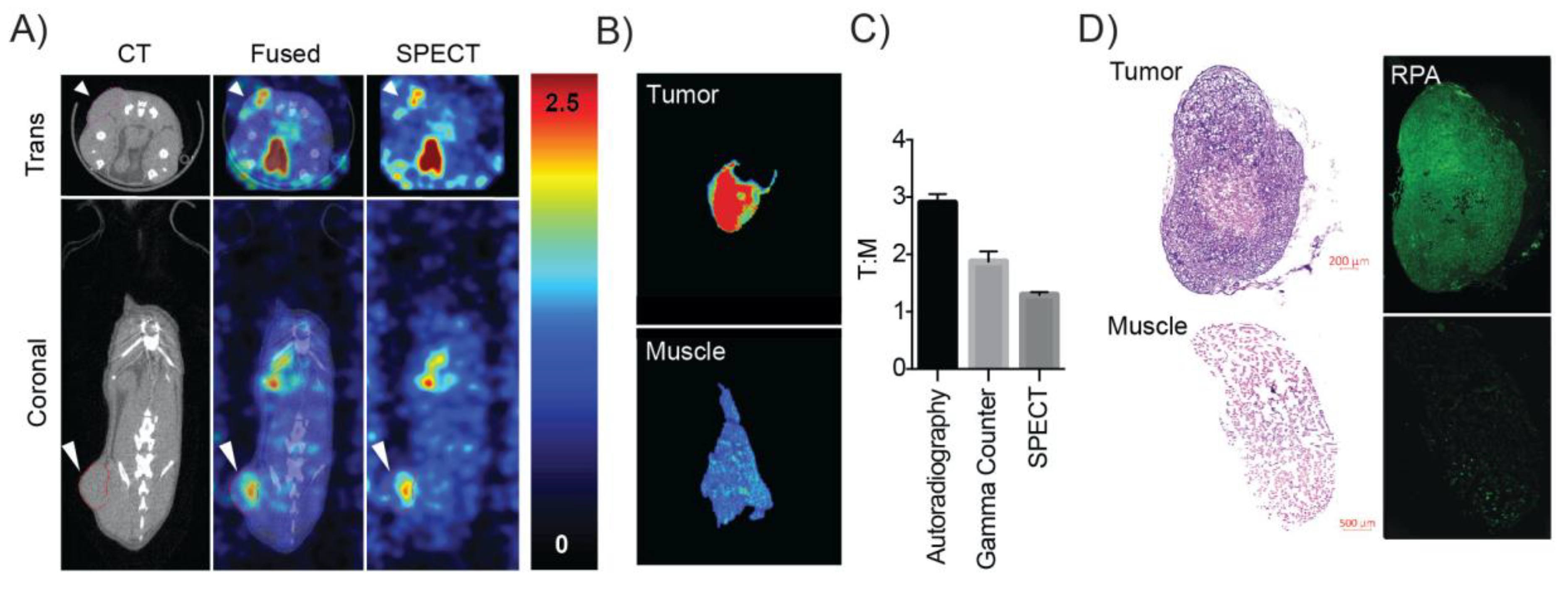
All animal studies were conducted under protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Tumor xenografts were generated by injecting 1 × 107 OVCAR8 cells subcutaneously into the right flank of 10 week-old female SCID mice that weighed between 20–25 grams. Tumors were allowed to reach 100 mm3 over 4 weeks before SPECT/CT imaging studies were performed. On the day of the study, [123I]KX1 was injected at 29.6 MBq/mouse (n = 3). SPECT/CT imaging was performed on the U-SPECT and U-CT (Netherlands) from 40–120 min post injection. Images were co-registered using PMOD version 3.7 and tumor to muscle ratios were calculated. Immediately following the completion of imaging, tumors and muscle controls were resected, snap frozen, and sectioned at 30 microns on a Leica cryostat (Wetzlar, Germany). Sections were then exposed to phosphor films for 24 h and then the film was read on a Perkin Elmer Typhoon (Waltham, MA, USA). Tumor to muscle ratios were then calculated using Perkin Elmer software. Adjacent portions of the tumors were immediately fixed post resection for histological analysis.
4.9. Viable Tumor Slice Cultures
Tumors were obtained from patients at the time of ovarian cancer biopsy (IRB #702679). Fresh viable tumor samples were obtained from the PENN Ovarian Cancer Research Center Biotrust Collection (
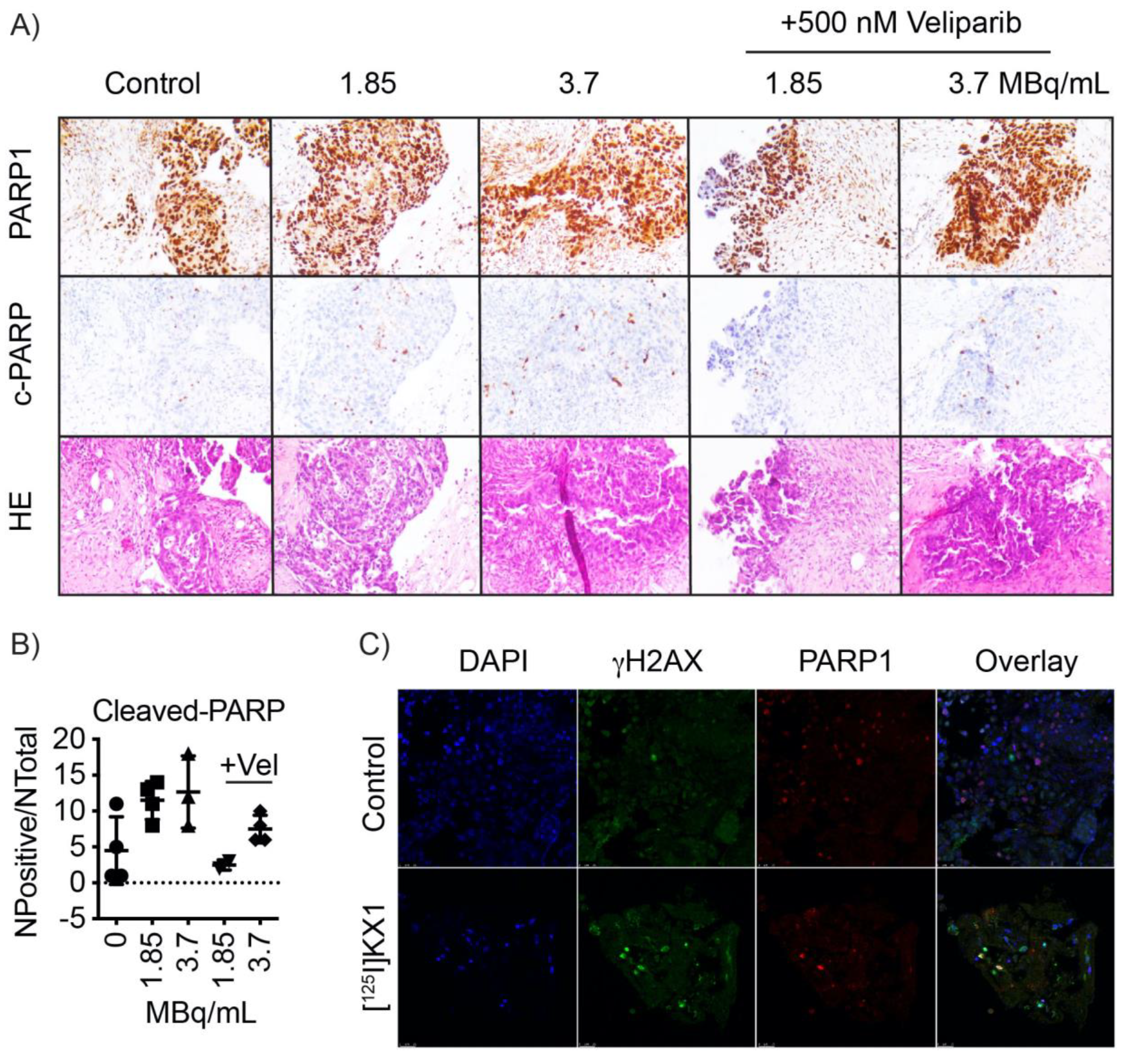
https://www.med.upenn.edu/OCRCBioTrust/). Tissues were embedded in low melting point agarose and viably sectioned at 300 µm using a VF-310-0Z vibrating microtome (Precisionary Instruments, Greenville, NC, USA). Tumor slices were cultured in complete media (DMEM/F12 + 10% FBS + 1% penicillin/streptomycin) overnight. Media was replaced and cells were treated with 25, 50, or 3.7 MBq/mL [
125I]KX1 with or without the addition of 500 nM veliparib. Tissue slices were fixed and stained with primary and secondary antibodies as described above and mounted on glass slides for confocal microscopy.
4.10. Histology
Murine tumors were fixed in 10% neutral buffered formalin for 48 h followed by washing with PBS and dehydrating in a 20% glucose solution for 24 h. After dehydration, tumors were washed with PBS, embedded in OCT, and were sectioned on a cryostat at a thickness of 5 µm. Adjacent tissue sections were stained for hematoxylin and eosin and RPA (dilution 1:1000, NA19L anti-replication protein A (Ab-3) mouse mAb RPA34-20, Millipore) with DAPI mounting medium. Tissue sections were imaged on a Zeiss Observer microscope with motorized stage at 10×. Images were then reconstructed into whole section images using Zeiss Zen software.
Patient tumor slices were fixed in 10% neutral buffered formalin for 48 h followed by washing with PBS then embedded in paraffin wax. Tissues were dehydrated in graded ethanol solutions, cleared in xylene, then embedded in paraffin. Blocks were cut into 5 μm sections and stained using the DAKO CoverStainer for H & E (Agilent, Santa Clara, CA, USA). Immunohistochemistry was performed using the Leica Bond-IIITM with the Bond Polymer Refine Detection System. Then, the tissue was dehydrated, and antigen retrieval was optimized using sodium citrate, pH 6.0 or EDTA, pH 9.0. Primary antibodies used were cleaved-PARP and PARP (Cell Signaling, 46D11). Images were acquired using a Leica DM 2000 microscope. For quantification of tumor-specific cleaved-PARP+, the average number of pixels was calculated from 3 to 4 40× images using Aperio ImageScope software.
4.11. Statistics
When appropriate, results were reported as mean ± standard deviation (SD) or mean ± standard error (SE) unless denoted otherwise. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparisons post-hoc test, or two-way ANOVA with Tukey’s multiple comparisons post-hoc test, or unpaired Student’s T-test. Statistical significance was set at * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001 (GraphPad Prism, La Jolla, CA, USA). In-vitro experiments were repeated at least three times with two or more replicates per experiment.
4.12. Radioligand Binding Assays
OVCAR8 wt and PARP1 KO cells were plated at 50,000 cells/well in 96-well plates 24 h before the assay in complete growth medium. For saturation curves, cells were incubated with increasing concentrations of [125I]KX1 (0.2–50 nM), and allowed to incubate until equilibrium was reached at 1 h. Then, 10 μM veliparib was used to determine non-specific binding. For competitive inhibition curves, 0–500 nM veliparib was co-incubated with [125I]KX1 at its Kd, and allowed to incubate until equilibrium was reached at 1 h. Following incubation, media was aspirated, cells were washed with PBS, and radioactivity was measured on a Perkin Elmer Wizard gamma counter. Competition and saturation curves were plotted using GraphPad Prism version 6.0 and competitive inhibition constants (Ki) and saturation dissociation constants (Kd) were calculated.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




