
Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study

 ,
,  , ,
, , 
Abstract
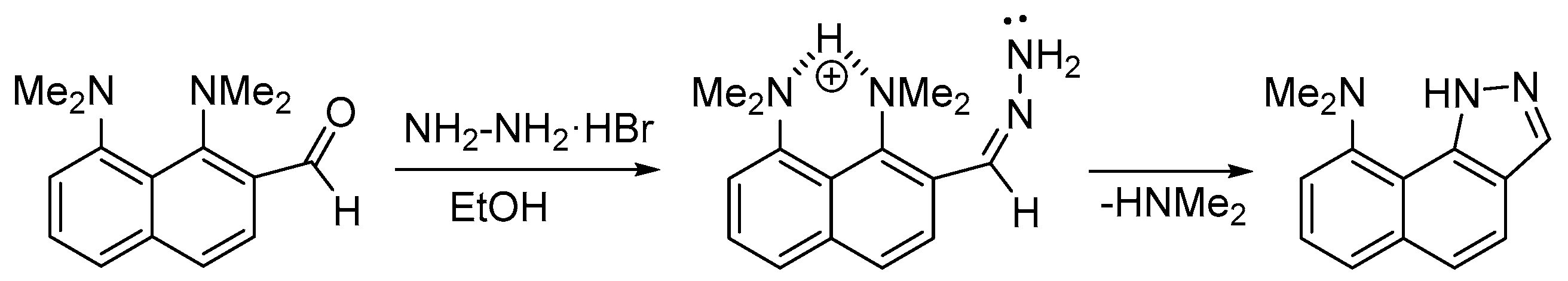
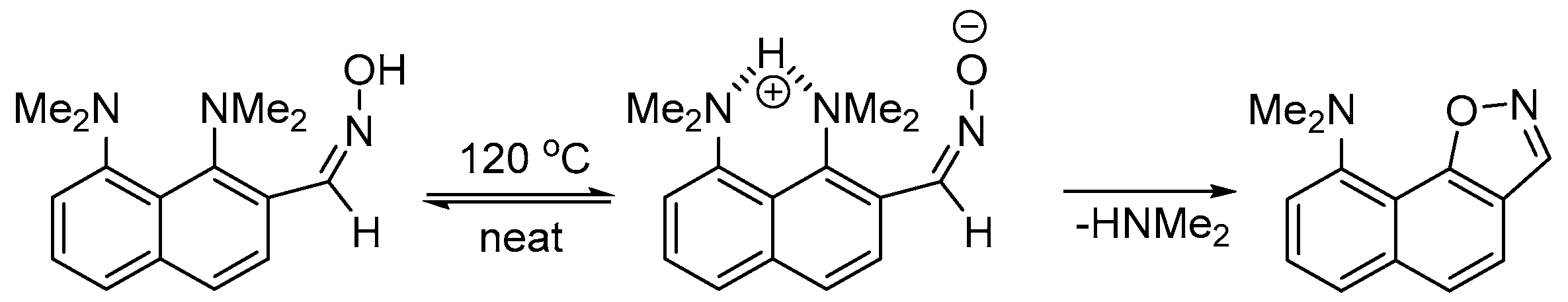
1. Introduction
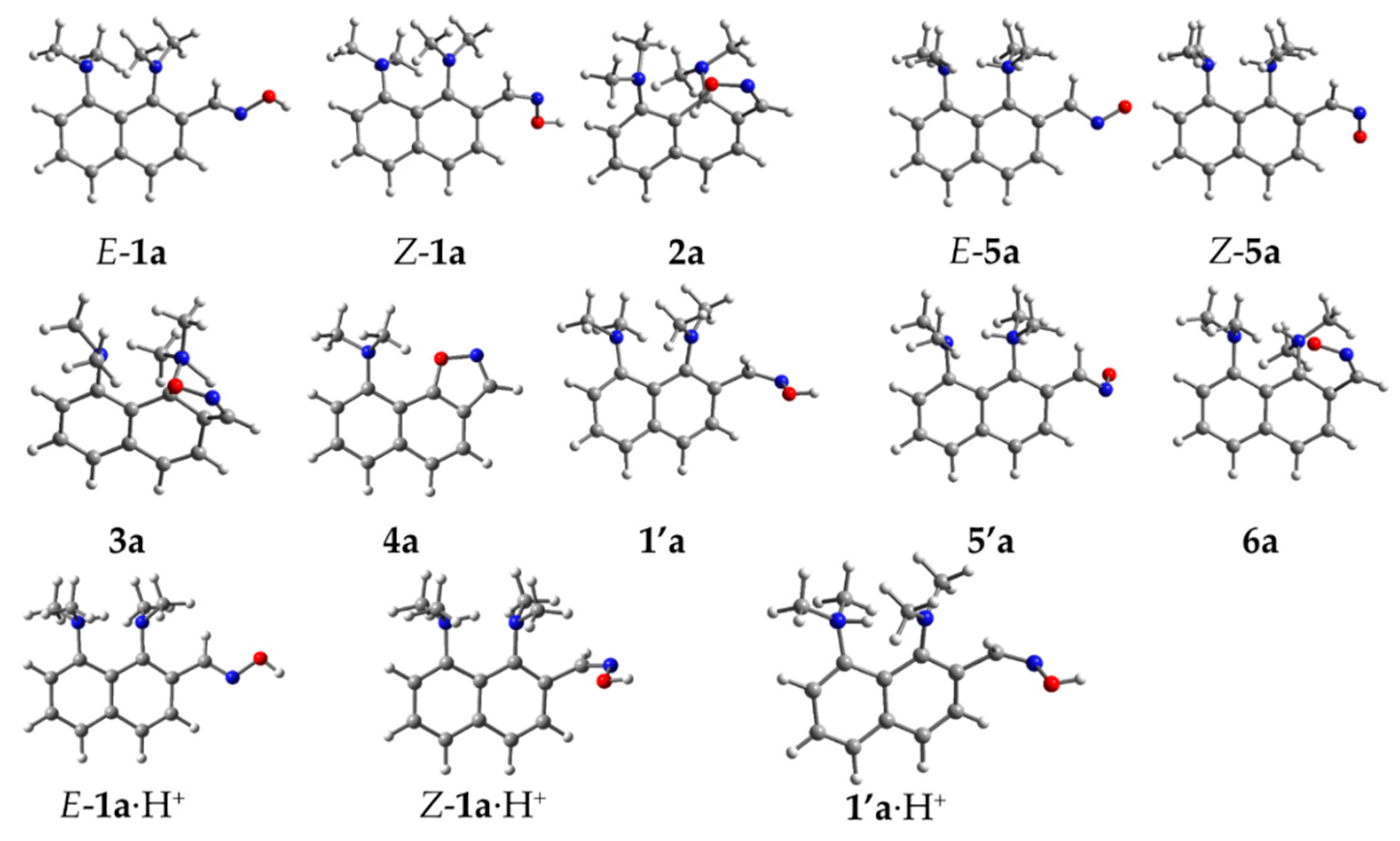
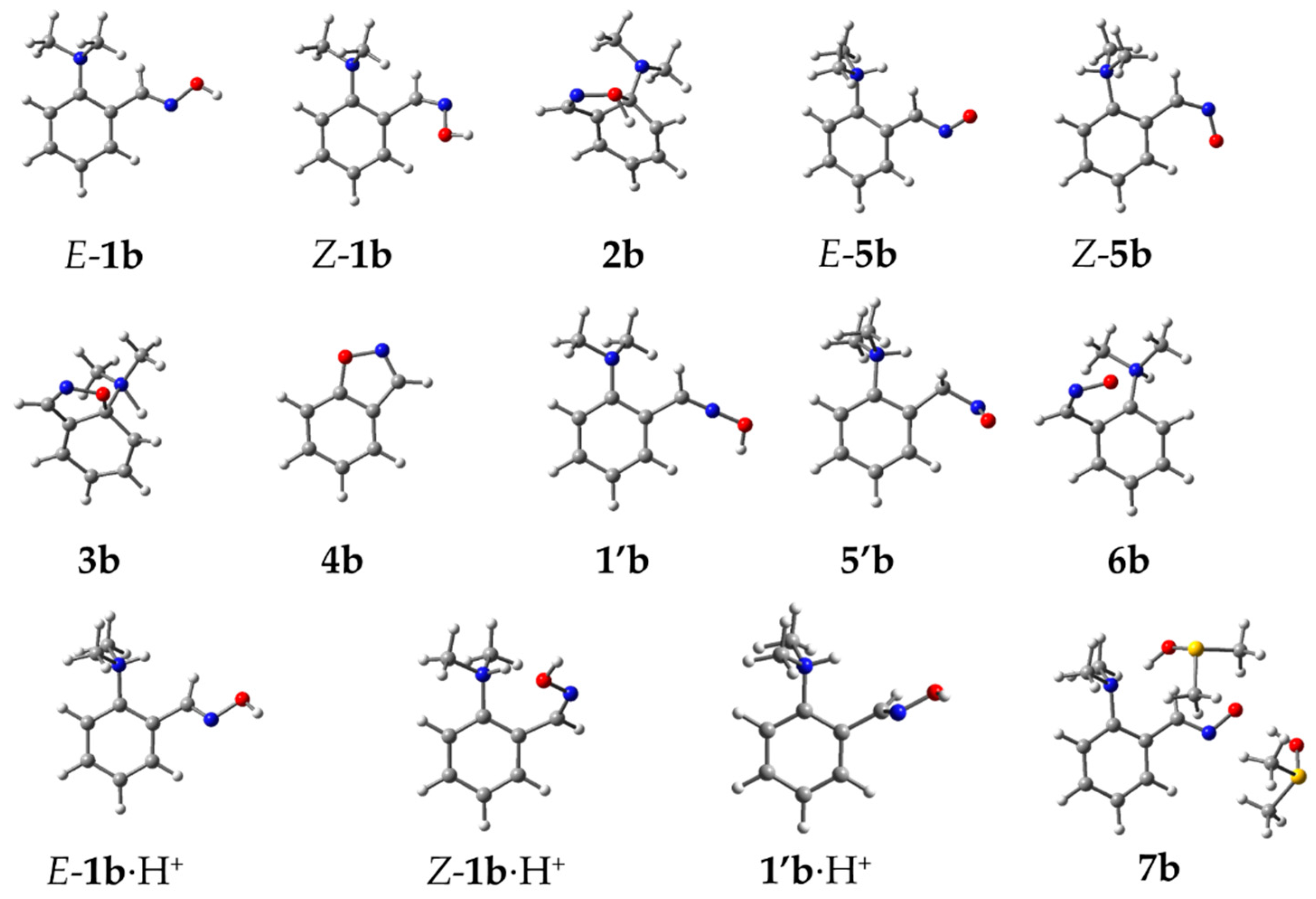
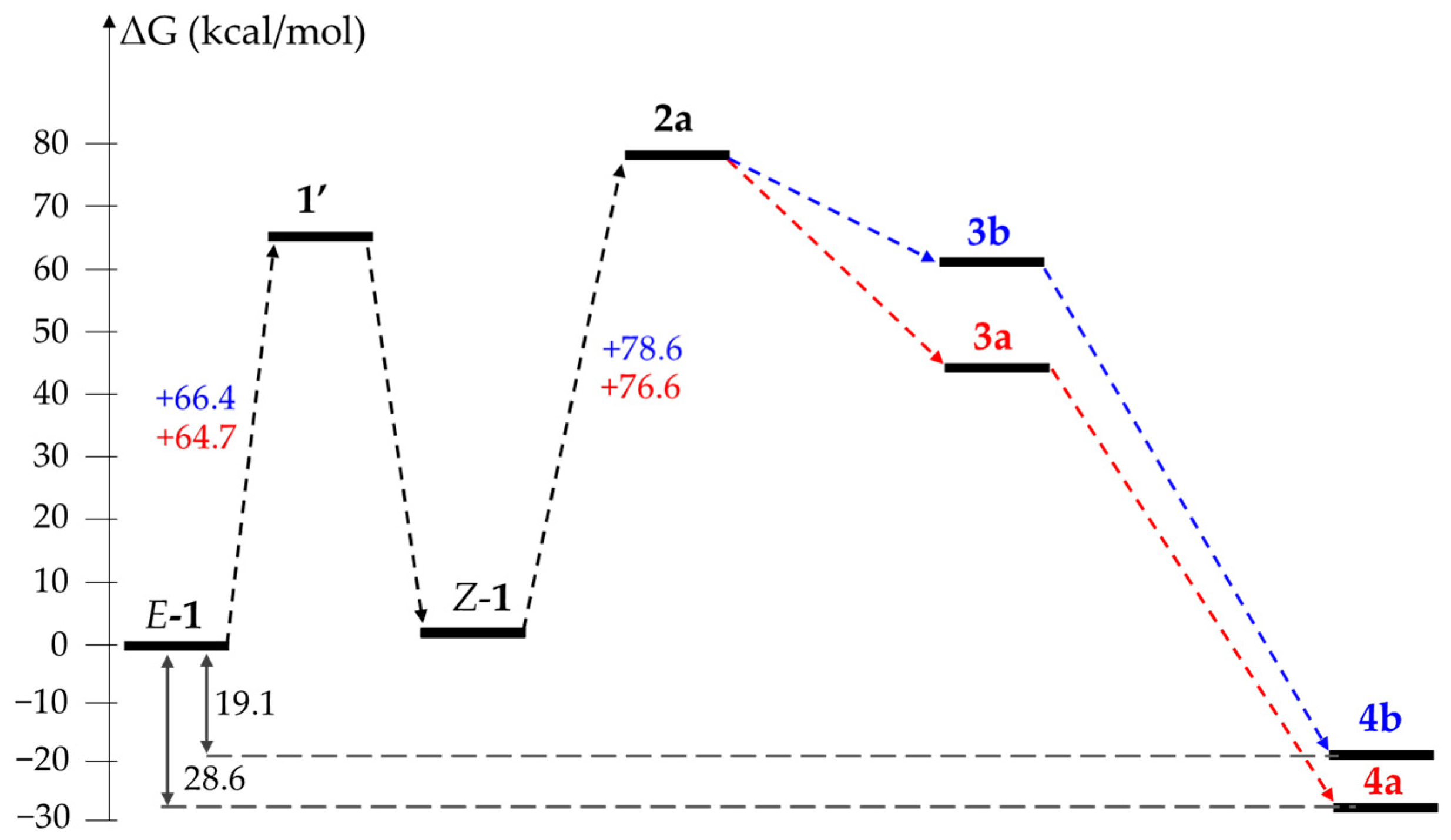
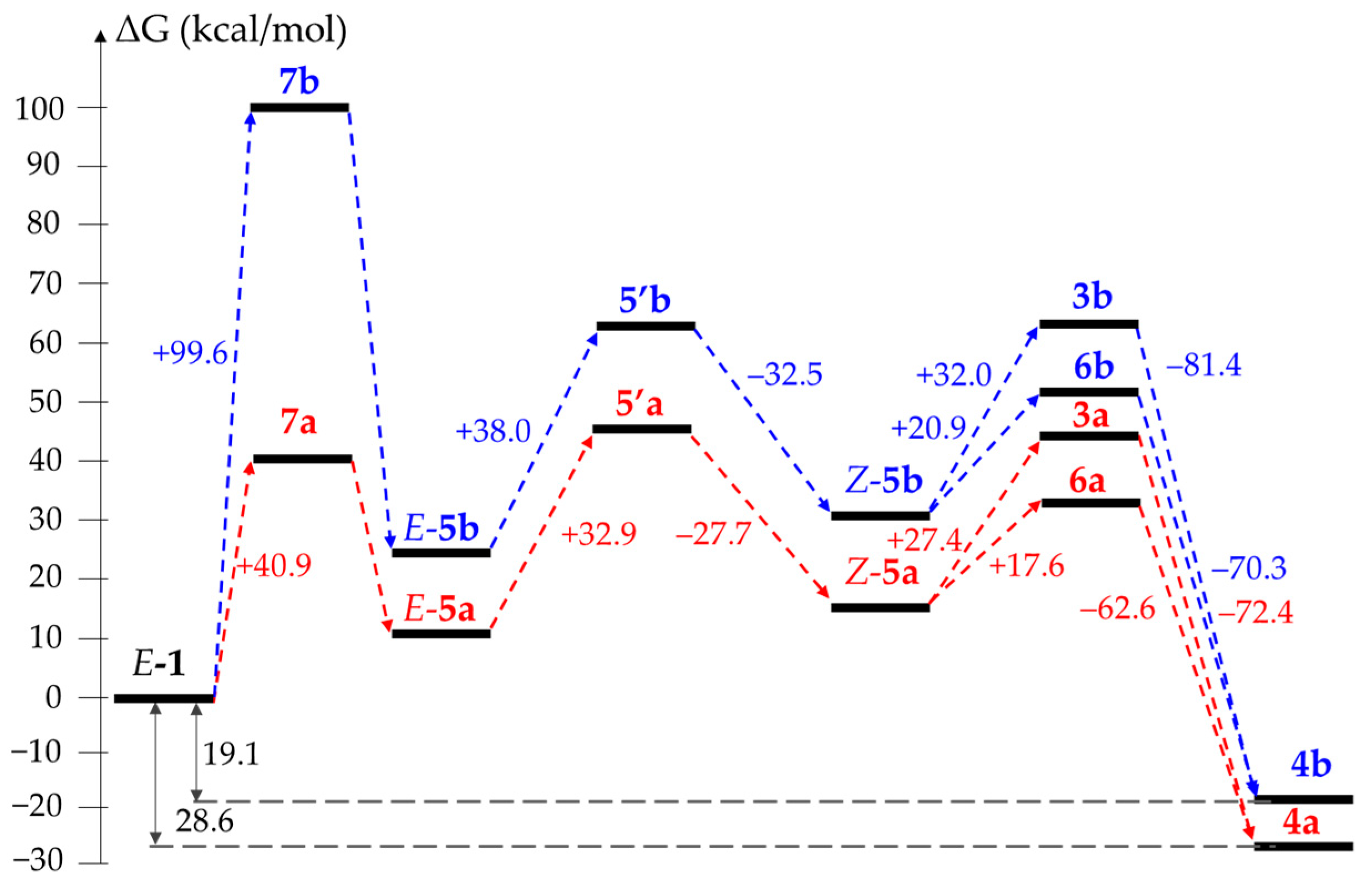
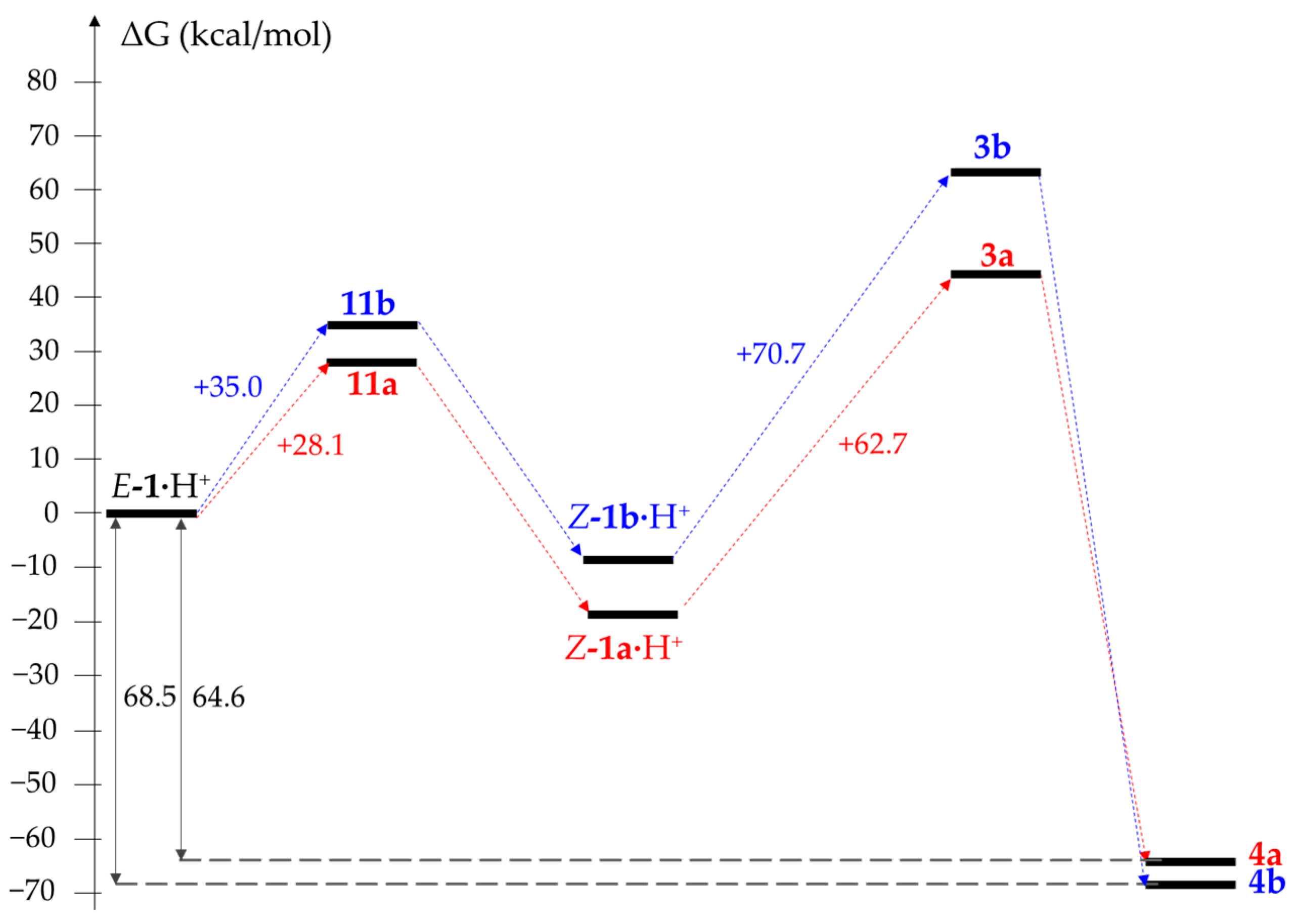
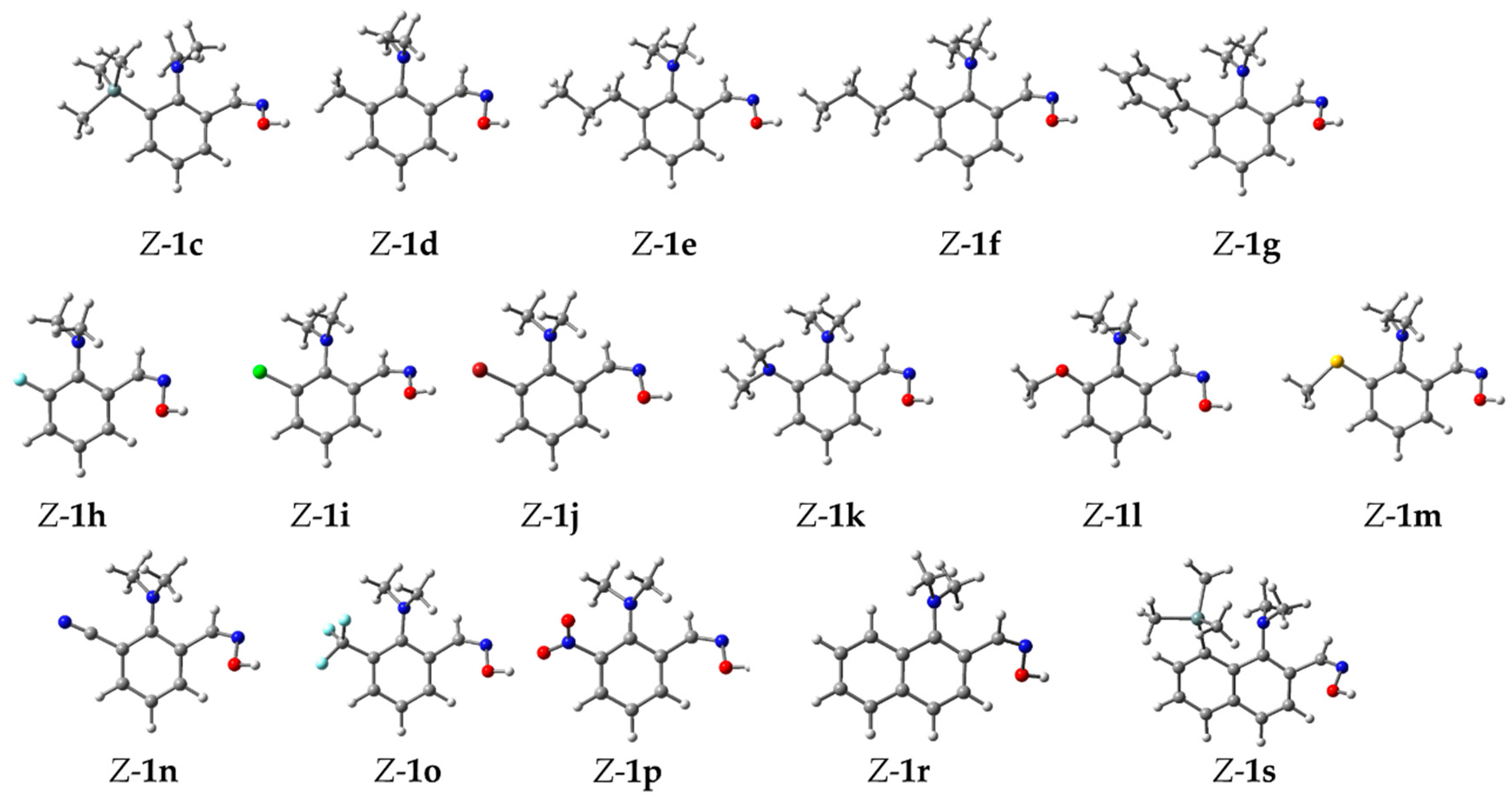
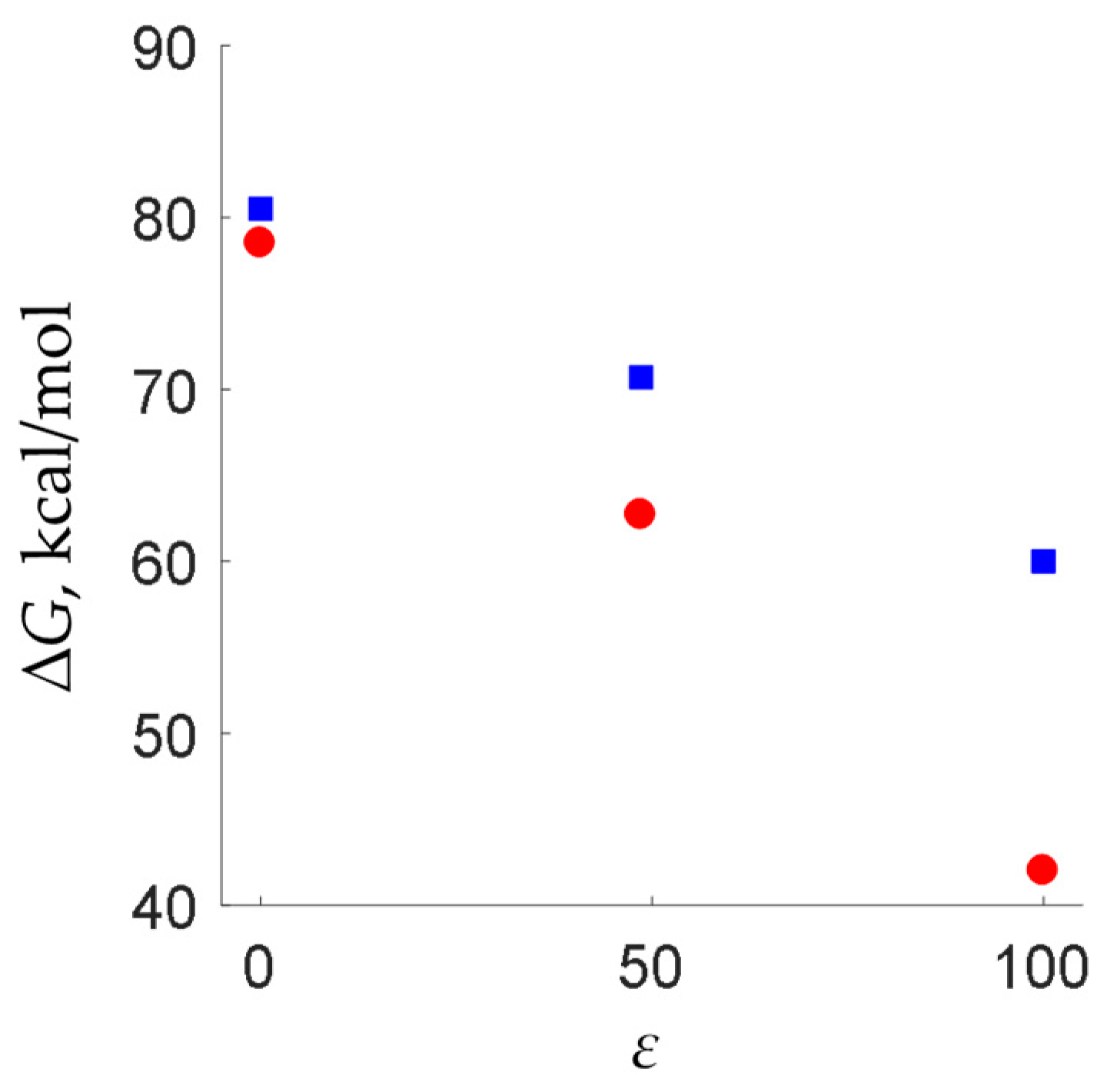
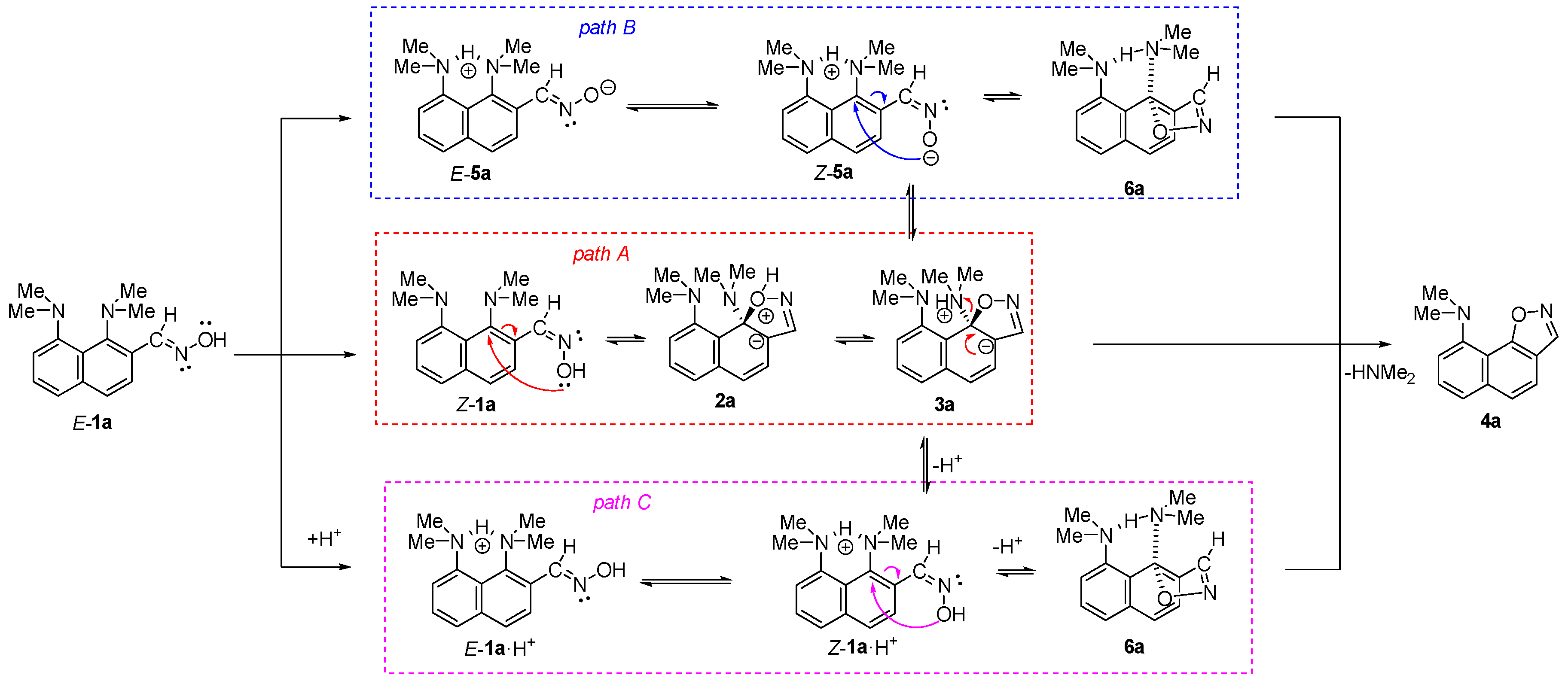
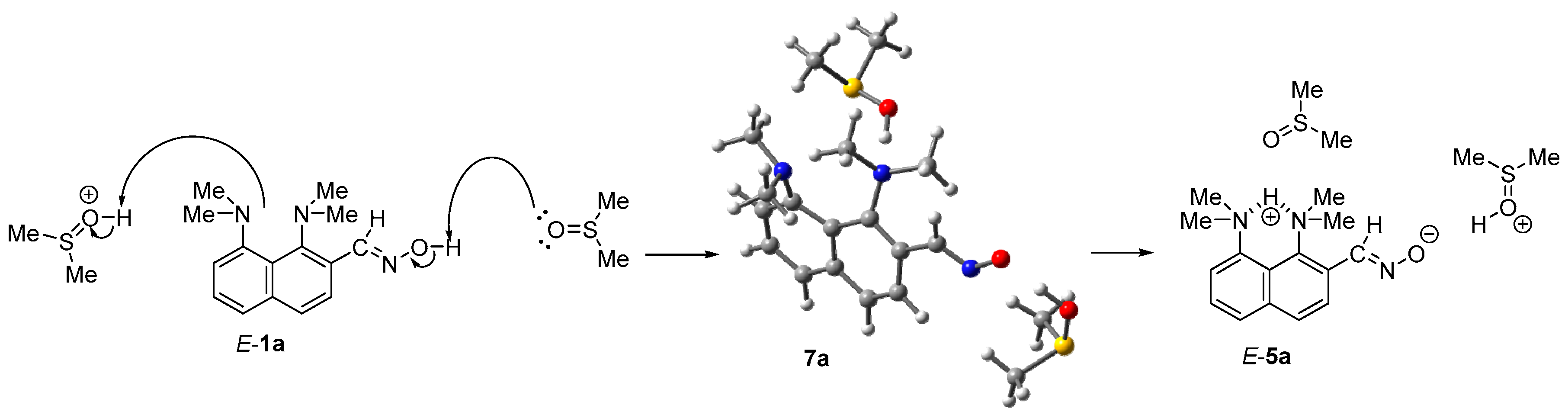
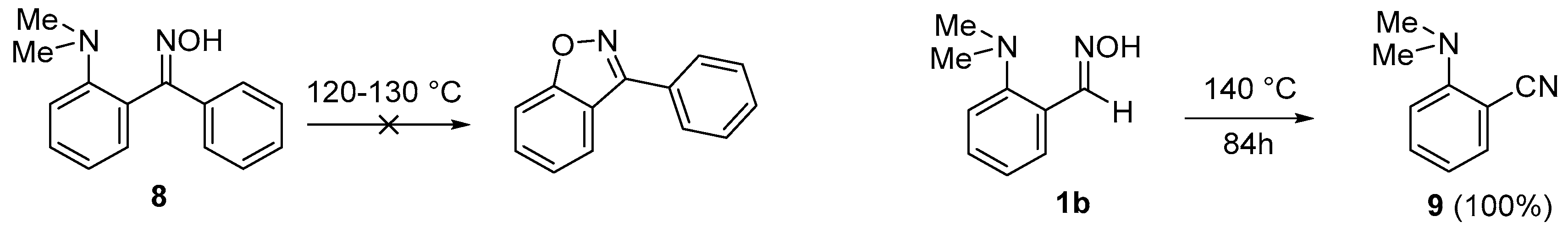
2. Results
3. Materials and Methods
3.1. Synthesis of 1b
3.2. Thermolysis of 1b
3.3. Thermolysis of 1b·MsOH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Hagihara, S.; Itami, K. Making Dimethylamino a Transformable Directing Group by Nickel-Catalyzed C-N Borylation. Chem. A Eur. J. 2015, 21, 16796–16800. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, F.; Wang, L.; Yuan, H.; Feng, L.; Lu, H.; Gao, H. Transition-metal-free aerobic C–O bond formation via C–N bond cleavage. Org. Chem. Front. 2020, 7, 1077–1081. [Google Scholar] [CrossRef]
- Paras, N.A.; Simmons, B.; Macmillan, D.W.C. A process for the rapid removal of dialkylamino-substituents from aromatic rings. Application to the expedient synthesis of (R)-tolterodine. Tetrahedron 2009, 65, 3232–3238. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Yang, Z.K.; Wang, C.; Zhang, A.; Uchiyama, M. From Anilines to Aryl Ethers: A Facile, Efficient, and Versatile Synthetic Method Employing Mild Conditions. Angew. Chem. Int. Ed. 2018, 57, 3641–3645. [Google Scholar] [CrossRef]
- Hojo, M.; Masuda, R.; Okada, E.; Miya, H. Aromatic Nucleophilic Nitrogen-Nitrogen Exchange Reaction of N,N-Dimethyl-2,4-bis(trifluoroacetyl)-1-naphthylamine with Amino Acid Derivatives: A Facile Synthesis of Fluorine-Containing 1H-Benzo[g]indolines and 1H-Benzo[g]indoles. Synthesis (Stuttg.) 1989, 1989, 550–552. [Google Scholar] [CrossRef]
- Hojo, M.; Okada, E.; Masuda, R.; Tomifuji, T. A Facile and Convenient Synthesis of Fluorine-containing Naphth[1,2-d][1,3]oxazines by Novel Cyclization of N,N-Dialkyl-2,4-bis(trifluoroacetyl)-1-naphthylamines. Heterocycles 1993, 36, 845. [Google Scholar] [CrossRef]
- Okada, E.; Otsuki, Y.; Shinohara, M.; Médebielle, M.; Shimizu, Y.; Takeuchi, H. Synthesis and aromatic nucleophilic N–N, N–S and N–O exchange reactions of N,N-dimethyl-2-trifluoroacetyl-1-naphthylamine. Tetrahedron Lett. 2003, 44, 741–743. [Google Scholar] [CrossRef]
- Sekiguchi, S.; Horie, T.; Suzuki, T. Aromatic nucleophilic substitution reactions of 1-dialkylamino-2,4-dinitronaphthalene with primary or secondary amines in organic solvents: Facile amine–amine exchange. J. Chem. Soc. Chem. Commun. 1988, 698–700. [Google Scholar] [CrossRef]
- Sekiguchi, S.; Hosokawa, M.; Suzuki, T.; Sato, M. Kinetic studies of the reaction of 1-dialkylamino-2,4-dinitronaphthalenes with butylamine in dimethyl sulfoxide. J. Chem. Soc. Perkin Trans. 1993, 2, 1111–1118. [Google Scholar] [CrossRef]
- Yang, Z.-K.; Xu, N.-X.; Takita, R.; Muranaka, A.; Wang, C.; Uchiyama, M. Cross-coupling polycondensation via C-O or C-N bond cleavage. Nat. Commun. 2018, 9, 1587. [Google Scholar] [CrossRef]
- Mikshiev, V.Y.; Antonov, A.S.; Pozharskii, A.F. Tandem Synthesis of 10-Dimethylaminobenzo[h]quinazolines from 2-Ketimino-1,8-bis(dimethylamino)naphthalenes via Nucleophilic Replacement of the Unactivated Aromatic NMe2 Group. Org. Lett. 2016, 18, 2872–2875. [Google Scholar] [CrossRef] [PubMed]
- Povalyakhina, M.A.; Antonov, A.S.; Dyablo, O.V.; Ozeryanskii, V.A.; Pozharskii, A.F. H-Bond-Assisted Intramolecular Nucleophilic Displacement of the 1-NMe 2 Group in 1,8-Bis(dimethylamino)naphthalenes as a Route to Multinuclear Heterocyclic Compounds and Strained Naphthalene Derivatives. J. Org. Chem. 2011, 76, 7157–7166. [Google Scholar] [CrossRef] [PubMed]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKaUnits: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Benoit, R.L.; Lefebvre, D.; Fréchette, M. Basicity of 1,8-bis(dimethylamino)naphthalene and 1,4-diazabicyclo[2.2.2]octane in water and dimethylsulfoxide. Can. J. Chem. 1987, 65, 996–1001. [Google Scholar] [CrossRef]
- Kaljurand, I.; Lilleorg, R.; Murumaa, A.; Mishima, M.; Burk, P.; Koppel, I.; Koppel, I.A.; Leito, I. The basicity of substituted N,N-dimethylanilines in solution and in the gas phase. J. Phys. Org. Chem. 2013, 26, 171–181. [Google Scholar] [CrossRef]
- Filatova, E.A.; Pozharskii, A.F.; Gulevskaya, A.V.; Ozeryanskii, V.A. Multiple Transformations of 2-Alkynyl-1,8-bis(dimethylamino)naphthalenes into Benzo[g]indoles. Pd/Cu-Dependent Switching of the Electrophilic and Nucleophilic Sites in Acetylenic Bond and a Puzzle of Porcelain Catalysis. J. Org. Chem. 2015, 80, 872–881. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kachalkina, S.G.; Pozharskii, A.F.; Borodkin, G.S.; Filarowski, A. Reaction of 2-trifluoroacetyl-1,8-Bis(dimethylamino)naphthalene with strong organic bases: Deprotonation of 1-NMe 2 group resulting in the formation of Benzo[g]indole derivatives versus nucleophilic addition to C=O group. Tetrahedron 2017, 73, 3452–3457. [Google Scholar] [CrossRef]
- Kachalkina, S.G.; Borodkin, G.S.; Pozharskii, A.F.; Antonov, A.S.; Borodkina, I.G.; Maltsev, Y.F.; Filatova, E.A.; Filarowski, A.; Ozeryanskii, V.A. Base-promoted transformation of 2-C(O)R-1,8-bis(dimethylamino)naphthalenes into benzo[g]indole derivatives. Mendeleev Commun. 2015, 25, 182–184. [Google Scholar] [CrossRef]
- Ji Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles. In The Chemistry of Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 149–478. [Google Scholar]
- Bordwell, F.G.; Ji, G.Z. Equilibrium acidities and homolytic bond dissociation energies of the H-O bonds in oximes and amidoximes. J. Org. Chem. 1992, 57, 3019–3025. [Google Scholar] [CrossRef]
- Horning, E.C.; Stromberg, V.L. Beckmann Rearrangements. Aldoximes. J. Am. Chem. Soc. 1952, 74, 5151–5152. [Google Scholar] [CrossRef]
- Varvounis, G.; Supsana, P.; Liaskopoulos, T.; Tsoungas, P.G. DMF-Catalysed Thermal Dehydration of Aldoximes: A Convenient Access to Functionalized Aliphatic and Aromatic Nitriles. Synlett 2007, 2007, 2671–2674. [Google Scholar] [CrossRef]
- Blanco, F.; Alkorta, I.; Elguero, J. Barriers about Double Carbon-Nitrogen Bond in Imine Derivatives. Croat. Chem. Acta 2009, 82, 173–183. [Google Scholar]
- Armstrong, D.R.; Walker, G.T. The electronic structure of CH2NCH3, CH2NNH2, CH2NOH and CH2NF. J. Mol. Struct. THEOCHEM 1987, 149, 369–389. [Google Scholar] [CrossRef]
- Nagy, P.I.; Kökösi, J.; Gergely, A.; Rácz, Á. Theoretical Conformational Analysis for Codeinone-6-oximes in Gas Phase and in Solution. J. Phys. Chem. A 2003, 107, 7861–7868. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef]
- Antonov, A.S.; Bardakov, V.G.; Mulloyarova, V.V. Sterically facilitated meta-lithiation of arenes, containing electron-donating groups. J. Organomet. Chem. 2020, 906, 121068. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Torigoe, T.; Ohmura, T.; Suginome, M. Asymmetric Cycloisomerization of o-Alkenyl-N-Methylanilines to Indolines by Iridium-Catalyzed C(sp3)−H Addition to Carbon-Carbon Double Bonds. Angew. Chem. Int. Ed. 2017, 56, 14272–14276. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Starting Material | R | Activation Barriers, kcal/mol | ||||
|---|---|---|---|---|---|---|
| Z-1 → 2 | Z-5 → 3 | Z-5 → 6 | Z-1·H+ → 3 | Z-1·H+ → 6 | ||
| 1a | NMe2 | 76.7 | 27.4 | 17.6 | 62.7 | 52.9 |
| 1b | H | 78.6 | 32.0 | 20.7 | 70.7 | 59.4 |
| 1c | SiMe3 | 72.6 | 23.5 | 19.8 | 56.9 | 53.2 |
| 1d | Me | 78.9 | 32.0 | 22.9 | 67.6 | 58.5 |
| 1e | iPr | 78.4 | 31.1 | 23.0 | 66.6 | 58.5 |
| 1f | tBu | 78.3 | 32.0 | 23.1 | 67.0 | 58.1 |
| 1g | Ph | 80.1 | 33.9 | 23.2 | 67.8 | 57.2 |
| 1h | F | 83.4 | 36.5 | 21.9 | 69.6 | 54.9 |
| 1i | Cl | 78.6 | 31.4 | 21.5 | 64.2 | 54.3 |
| 1j | Br | 77.2 | 29.5 | 20.6 | 62.3 | 53.4 |
| 1k | NMe2 | 82.2 | 41.5 | 23.7 | 75.5 | 57.7 |
| 1l | OMe | 80.2 | 40.9 | 25.6 | 75.1 | 59.9 |
| 1m | SMe | 83.4 | 31.1 | 17.4 | 62.3 | 51.6 |
| 1n | CN | 86.0 | 23.7 | 13.7 | 55.4 | 45.4 |
| 1o | CF3 | 85.8 | 22.8 | 17.9 | 56.0 | 51.2 |
| 1p | NO2 | 83.8 | 24.3 | 14.4 | 55.7 | 45.8 |
| 1r | H | 77.2 | 36.4 | 15.6 | 70.0 | 49.2 |
| 1s | SiMe3 | 72.3 | 8.7 | 7.7 | 45.3 | 44.3 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonov, A.S.; Tupikina, E.Y.; Karpov, V.V.; Mulloyarova, V.V.; Bardakov, V.G. Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study. Molecules 2020, 25, 5977. https://doi.org/10.3390/molecules25245977
Antonov AS, Tupikina EY, Karpov VV, Mulloyarova VV, Bardakov VG. Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study. Molecules. 2020; 25(24):5977. https://doi.org/10.3390/molecules25245977
Chicago/Turabian StyleAntonov, Alexander S., Elena Yu Tupikina, Valerii V. Karpov, Valeriia V. Mulloyarova, and Victor G. Bardakov. 2020. "Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study" Molecules 25, no. 24: 5977. https://doi.org/10.3390/molecules25245977
APA StyleAntonov, A. S., Tupikina, E. Y., Karpov, V. V., Mulloyarova, V. V., & Bardakov, V. G. (2020). Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study. Molecules, 25(24), 5977. https://doi.org/10.3390/molecules25245977