
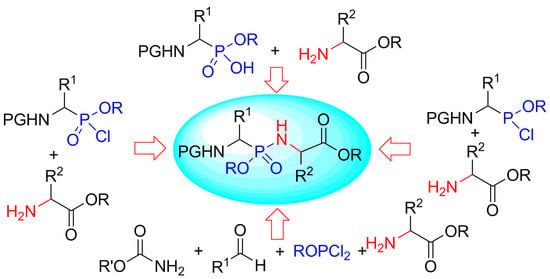
Synthetic Methods of Phosphonopeptides


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Synthesis of Phosphonopeptides via Phosphonochloridates
2.1. Chlorination of Dialkyl Phosphonates with Phosphorus Pentachloride
2.2. Chlorination of Dialkyl Phosphonates with Phosphorus Oxychloride
2.3. Chlorination of Alkyl Phosphonic Acid Monoesters with Thionyl Chloride
2.4. Chlorination of Alkyl Phosphonic Acid Monoesters with Oxalyl Chloride
2.5. Chlorination of Alkyl Trimethylsilyl Phosphonites or Alkyl Phosphinates with Carbon Tetrachloride
2.6. Bromination of Alkyl Phosphinates with Bromine
3. Synthesis of Phosphonopeptides with Coupling Reagents
4. Synthesis of Phosphonopeptides Catalyzed by Enzyme
5. Synthesis of Phosphonopeptides via Phosphonochloridite Followed by Oxidation
6. Synthesis of Phosphonopeptides via Pseudo Four-Component Condensation Reaction
7. Synthesis by Nucleophilic Addition
8. Conclusions
Funding
Conflicts of Interest
References
- Kafarski, P.; Lejczak, B. Synthesis of Phosphono- and Phosphinopeptides in Aminophosphonic and Aminophosphinic Acids; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: West Sussex, UK, 2000; pp. 173–203. [Google Scholar]
- Kafarski, P.; Lejczak, B. The Biological Activity of Phosphono- and Phosphinopeptides in Aminophosphonic and Aminophosphinic Acids; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: West Sussex, UK, 2000; pp. 407–442. [Google Scholar]
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef]
- Bartlett, P.A.; Marlowe, C.K. Evaluation of intrinsic binding energy from a hydrogen bonding group in an enzyme inhibitor. Science 1987, 235, 569–571. [Google Scholar] [CrossRef]
- Krimmer, S.G.; Cramer, J.; Betz, M.; Fridh, V.; Karlsson, R.; Heine, A.; Klebe, G. Rational design of thermodynamic and kinetic binding profiles by optimizing surface water networks coating protein-bound ligands. J. Med. Chem. 2016, 59, 10530–10548. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Yang, K.W.; Cheng, X.; Zhao, C.; Liu, C.C.; Jia, C.; Feng, L.; Xiao, J.M.; Zhou, L.S.; Gao, H.Z.; Yang, X.; et al. Synthesis and activity study of phosphonamidate dipeptides as potential inhibitors of VanX. Bioorg. Med. Chem. Lett. 2011, 21, 7224–7227. [Google Scholar] [CrossRef]
- Demange, L.; Moutiez, M.; Dugave, C. Synthesis and evaluation of Glyψ(PO2R-N)Pro-containing pseudopeptides as novel inhibitors of the human cyclophilin hCyp-18. J. Med. Chem. 2002, 45, 3928–3933. [Google Scholar] [CrossRef]
- Schultz, P.G.; Lerner, R.A. Antibody catalysis of difficult chemical transformations. Acc. Chem. Res. 1993, 26, 392–395. [Google Scholar] [CrossRef]
- Jacobsen, J.R.; Schultz, P.G. Antibody catalysis of peptide-bond formation. Proc. Nat. Acad. Sci. USA 1994, 91, 5888–5892. [Google Scholar] [CrossRef]
- Xie, G.Y.; Chen, J.H.; Lin, S.S.; Jin, S. The synthesis of a novel phosphorus containing antigen. Chin. Chem. Lett. 2001, 12, 497–498. [Google Scholar]
- Xie, G.Y.; Chen, L.M.; Chen, J.H. Synthesis of a novel antigen containing phosphorus. Chem. J. Chin. Univ. 2003, 24, 1037–1039. [Google Scholar]
- Hariharan, M.; Chaberek, S.; Martell, A.E. Synthesis of phosphonopeptide derivatives. Synth. Commun. 1973, 3, 375–379. [Google Scholar] [CrossRef]
- Jackson, D.S.; Fraser, S.A.; Ni, L.-M.; Kam, C.-M.; Winkler, U.; Johnson, D.A.; Froelich, C.J.; Hudig, D.; Powers, J.C. Synthesis and evaluation of diphenyl phosphonate esters as inhibitors of the trypsin-like granzymes A and K and mast cell tryptase. J. Med. Chem. 1998, 41, 2289–2301. [Google Scholar] [CrossRef]
- Cervera-Villanueva, J.M., Jr.; Viveros-Ceballos, J.L.; Linzaga-Elizalde, I.; Ordonez, M. Practical synthesis of novel phosphonopeptides containing AibP. J. Peptide Sci. 2016, 22, 70–75. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Depczynski, R.; Kudzin, M.H.; Drabowicz, J. 1-(N-chloroacetylamino)-alkylphosphonic acids—synthetic precursors of phosphonopeptides. Amino Acids 2008, 34, 163–168. [Google Scholar] [CrossRef]
- Sun, B.Y.; Xu, J.X. Convenient synthesis of phosphonodipeptides containing C-terminal α-aminoalkylphosphonic acids. J. Pept. Sci. 2015, 21, 615–619. [Google Scholar] [CrossRef]
- Sun, B.Y.; Xu, J.X. Convenient and facile synthesis of hybrid sulfonophosphonodipeptides. Synthesis 2015, 47, 1479–1487. [Google Scholar]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Elliott, R.L.; Marks, N.; Berg, M.J.; Portoghese, P.S. Synthesis and biological evaluation of phosphonamidate peptide inhibitors of enkephalinase and angiotensin-converting enzyme. J. Med. Chem. 1985, 28, 1208–1216. [Google Scholar] [CrossRef]
- Xu, J.X.; Xia, C.F.; Yu, L.; Zhou, Q.Z. Synthesis of phosphonopeptides containing 1-aminoalkylphosphonic acid. Phosphorus Sulfur Silicon Relat. Elem. 1999, 152, 35–44. [Google Scholar] [CrossRef]
- Vo Quang, Y.; Gravey, A.M.; Simonneau, R.; Vo Quang, L.; Lacoste, A.M.; Le Goffic, F. Towards new inhibitors of D-alanine:D-alanine ligase: The synthesis of 3-aminobutenylphosphonic and aminophosphonamidic acids. Tetrahedron Lett. 1987, 28, 6167–6170. [Google Scholar] [CrossRef]
- Lacoste, A.M.; Chollet-Gravey, A.M.; Vo Quang, L.; Vo Quang, Y.; Le Goffic, F. Time-dependent inhibition of Streptococcus faecalis D-alanine:D-alanine ligase by α-aminophosphonamidic acids. Eur. J. Med. Chem. 1991, 26, 255–260. [Google Scholar] [CrossRef]
- Musiol, H.-J.; Grams, F.; Rudolph-Boehner, S.; Moroder, L. The chemistry of phosphapeptides: Formation of functionalized phosphonochloridates under mild conditions and their reaction with alcohols and amines. On the synthesis of phosphonamidate peptides. J. Org. Chem. 1994, 59, 6144–6146. [Google Scholar] [CrossRef]
- Gobec, S.; Urleb, U. Synthesis of new lipophilic phosphonate and phosphonamidate analogs of N-acetylmuramyl-D-isoglutamine related to LK 423. Molecules 2002, 7, 394–404. [Google Scholar] [CrossRef]
- Sampson, N.S.; Bartlett, P.A. Synthesis of phosphonic acid derivatives by oxidative activation of phosphinate esters. J. Org. Chem. 1988, 53, 4500–4503. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Plenat, F.; Cristau, H.-J. The preparation of phosphono peptides containing a phosphonamidate bond. Tetrahedron 1994, 50, 12743–12754. [Google Scholar] [CrossRef]
- Yao, Q.L.; Yuan, C.Y. Stereospecific transformation of protected P−H group into P−O or P−N group in one-pot reaction. J. Org. Chem. 2012, 77, 10985–10990. [Google Scholar] [CrossRef]
- Horiguchi, M.; Kandatsu, M. Isolation of 2-aminoethane phosphonic acid from Rumen Protozoa. Nature 1959, 184, 901–902. [Google Scholar] [CrossRef]
- Kittredge, J.S.; Roberts, E.; Simonsen, D.G. The occurrence of free 2-aminoethylphosphonic acid in the sea anemone, Anthopleura elegantissima. Biochemistry 1962, 1, 624–628. [Google Scholar] [CrossRef]
- Yuan, C.Y.; Wang, G. H Studies on organophosphorus compounds. 65. A facile synthetic route to phosphonopeptides. Phosphorus Sulfur Silicon Relat. Elem. 1992, 71, 207–212. [Google Scholar] [CrossRef]
- Yang, K.-W.; Brandt, J.J.; Chatwood, L.L.; Crowder, M.W. Phosphonamidate and phosphothioate dipeptides as potential inhibitors of VanX. Bioorg. Med. Chem. Lett. 2000, 10, 1085–1087. [Google Scholar] [CrossRef]
- Slater, M.J.; Laws, A.P.; Page, M.I. The relative catalytic efficiency of β-lactamase catalyzed acyl and phosphyl transfer. Bioorg. Chem. 2001, 29, 77–95. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P. Transesterification of monophenyl phosphonamidates-chemical modeling of serine protease inhibition. Tetrahedron 2002, 58, 5855–5863. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. Structure-based design and synthesis of dipeptide analogs as new inhibitors of leucine aminopeptidase. Phosphorus Sulfur Silicon Relat. Elem. 2002, 177, 1739–1743. [Google Scholar] [CrossRef]
- Mucha, A.; Grembecka, J.; Cierpicki, T.; Kafarski, P. Hydrolysis of the phosphonamidate bond in phosphono dipeptide analogs—the influence of the nature of the N-terminal functional group. Eur. J. Org. Chem. 2003, 4797–4803. [Google Scholar] [CrossRef]
- Dumy, P.; Escale, R.; Girard, J.P.; Parello, J.; Vidal, J.P. A convenient synthetic approach to new α-(9-fluorenylmethoxycarbonylamino)alkylphosphonic acid derivatives. Synthesis 1992, 1226–1228. [Google Scholar] [CrossRef]
- Cramer, J.; Klebe, G. An allyl protection and improved purification strategy enables the synthesis of functionalized phosphonamidate peptides. Synthesis 2017, 49, 1857–1866. [Google Scholar]
- Mucha, A.; Kunert, A.; Grembecka, J.; Pawelczak, M.; Kafarski, P. A phosphonamidate containing aromatic N-terminal amino group as inhibitor of leucine aminopeptidase-design, synthesis and stability. Eur. J. Med. Chem. 2006, 41, 768–772. [Google Scholar] [CrossRef]
- Malachowski, W.P.; Coward, J.K. The chemistry of phosphapeptides: Formation of functionalized phosphonochloridates under mild conditions and their reaction with alcohols and amines. J. Org. Chem. 1994, 59, 7616–7624. [Google Scholar] [CrossRef]
- Urleb, U.; Gobec, S. Synthesis of new adamantane substituted acyclic MDP analogs related to LK 415. Acta Pharm. 2000, 50, 173–184. [Google Scholar]
- Malachowski, W.P.; Coward, J.K. The chemistry of phosphapeptides: Investigations on the synthesis of phosphonamidate, phosphonate, and phosphinate analogs of glutamyl-γ-glutamate. J. Org. Chem. 1994, 59, 7625–7634. [Google Scholar] [CrossRef]
- Sikora, D.; Nonas, T.; Gajda, T. O-Ethyl 1-azidoalkylphosphonic acids-versatile reagents for the synthesis of protected phosphonamidate peptides. Tetrahedron 2001, 57, 1619–1625. [Google Scholar] [CrossRef]
- Artyushin, O.I.; Sharova, E.V.; Yarkevich, A.N.; Genkina, G.K.; Vinogradova, N.V.; Brel, V.K. Design of phosphonate analogs of short peptides by “Click” chemistry. Russ. Chem. Bull. 2015, 64, 2172–2177. [Google Scholar] [CrossRef]
- Natchev, I. Glycine phospha C-peptides. Izvestiya po Khimiya 1988, 21, 40–45. [Google Scholar]
- Ishibashi, Y.; Miyata, K.; Kitamura, M. (9H-Fluoren-9-yl)methanesulfonyl (Fms): An amino protecting group complementary to Fmoc. Eur. J. Org. Chem. 2010, 4201–4204. [Google Scholar] [CrossRef]
- Nasief, N.N.; Tan, H.W.; Kong, J.; Hangauer, D. Water mediated ligand functional group cooperativity: The contribution of a methyl group to binding affinity is enhanced by a COO− group through changes in the structure and thermodynamics of the hydration waters of ligand-thermolysin complexes. J. Med. Chem. 2012, 55, 8283–8302. [Google Scholar] [CrossRef]
- Nasief, N.N.; Hangauer, D. Influence of neighboring groups on the thermodynamics of hydrophobic binding: An added complex facet to the hydrophobic effect. J. Med. Chem. 2014, 57, 2315–2333. [Google Scholar] [CrossRef]
- Nasief, N.N.; Hangauer, D. Additivity or cooperativity: Which model can predict the influence of simultaneous incorporation of two or more functionalities in a ligand molecule? Eur. J. Med. Chem. 2015, 90, 897–915. [Google Scholar] [CrossRef]
- Natchev, I.A. Organophosphorus analogs and derivatives of the natural L-aminocarboxylic acid and peptides. VII. Enzyme synthesis of phospha-C peptides. Tetrahedron 1991, 47, 1239–1248. [Google Scholar] [CrossRef]
- De Fatima Fernandez, M.; Vlaar, C.P.; Fan, H.; Liu, Y.-H.; Fronczek, F.R.; Hammer, R.P. Synthesis of phosphonate and thiophosphonate esters and amides from hydrogen-phosphinates by a novel one-pot activation-coupling-oxidation procedure. J. Org. Chem. 1996, 60, 7390–7391. [Google Scholar] [CrossRef]
- Rushing, S.D.; Hammer, R.P. Synthesis of phosphonamide and thiophosphonamide dipeptides. J. Am. Chem. Soc. 2001, 123, 4861–4862. [Google Scholar] [CrossRef]
- Xu, J.X.; Fu, N.Y. A facile synthesis of N-protected 1-aminoalkylphosphonamidate derivatives. Synth. Commun. 2000, 30, 4137–4145. [Google Scholar] [CrossRef]
- Xu, J.X.; Fu, N.Y. A novel and convenient method for synthesizing unsymmetrical N-benzyloxycarbonyl-protected 1-amino-1-aryl-alkylphosphonate mixed diesters. J. Chem. Soc. Perkin. Trans. 1 2001, 1223–1226. [Google Scholar] [CrossRef]
- Xu, J.X.; Wei, M. A convenient method for the synthesis of N-protected 1-aminoalkyl-phosphonate mixed monothioesters and dithioesters. Synth. Commun. 2001, 31, 1489–1497. [Google Scholar] [CrossRef]
- Xu, J.X. Convergent synthesis of phosphonopeptides and phosphonodepsipeptides. Sci. Sin. Chim. 2013, 43, 995–1004. [Google Scholar] [CrossRef]
- Xu, J.X. Convergent synthesis of phosphonopeptides via phospha-Mannich reactions: Rationale on reactivity and mechanistic insights. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 487–492. [Google Scholar] [CrossRef]
- Xu, J.X.; Gao, Y.H. Straightforward synthesis of depsiphosphonopeptides via Mannich-type multiple component condensation. Synthesis 2006, 783–788. [Google Scholar] [CrossRef]
- Liu, H.; Cai, S.Z.; Xu, J.X. Asymmetric synthesis of N-protected chiral 1-aminoalkylphosphonic acids and synthesis of side-chain functionalized depsiphosphonopeptides. J. Peptide Sci. 2006, 12, 337–340. [Google Scholar] [CrossRef]
- Fu, N.Y.; Zhang, Q.H.; Duan, L.F.; Xu, J.X. Facile synthesis of phosphonamidate and phosphonate-linked phosphonopeptides. J. Peptide Sci. 2006, 12, 303–309. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Nesterova, L.I. N-Hydrophosphoryl derivatives of N-alkylaminoacetic acid in the synthesis of 1,4,2-diazaphosphorinan-5-ones. Zhurnal Obshchei Khimii 1982, 52, 2465–2467. [Google Scholar]































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J. Synthetic Methods of Phosphonopeptides. Molecules 2020, 25, 5894. https://doi.org/10.3390/molecules25245894
Xu J. Synthetic Methods of Phosphonopeptides. Molecules. 2020; 25(24):5894. https://doi.org/10.3390/molecules25245894
Chicago/Turabian StyleXu, Jiaxi. 2020. "Synthetic Methods of Phosphonopeptides" Molecules 25, no. 24: 5894. https://doi.org/10.3390/molecules25245894
APA StyleXu, J. (2020). Synthetic Methods of Phosphonopeptides. Molecules, 25(24), 5894. https://doi.org/10.3390/molecules25245894