
Antitumor Drugs and Their Targets


Abstract
1. Introduction
2. Development of Cancer
3. Cancer Stem Cells
4. Historical Perspective of Anticancer Therapies
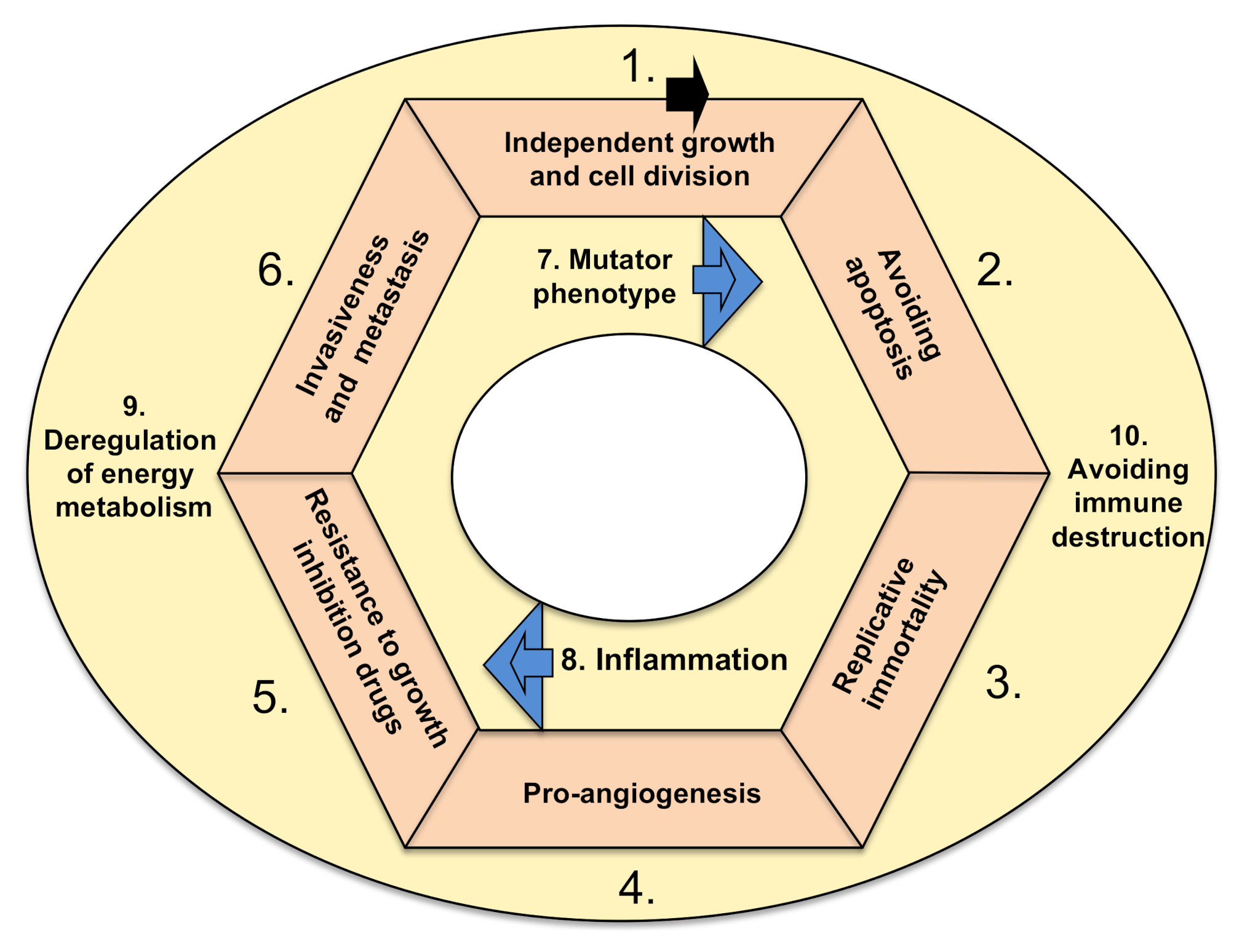
5. Therapies Targeting Cancer Hallmarks
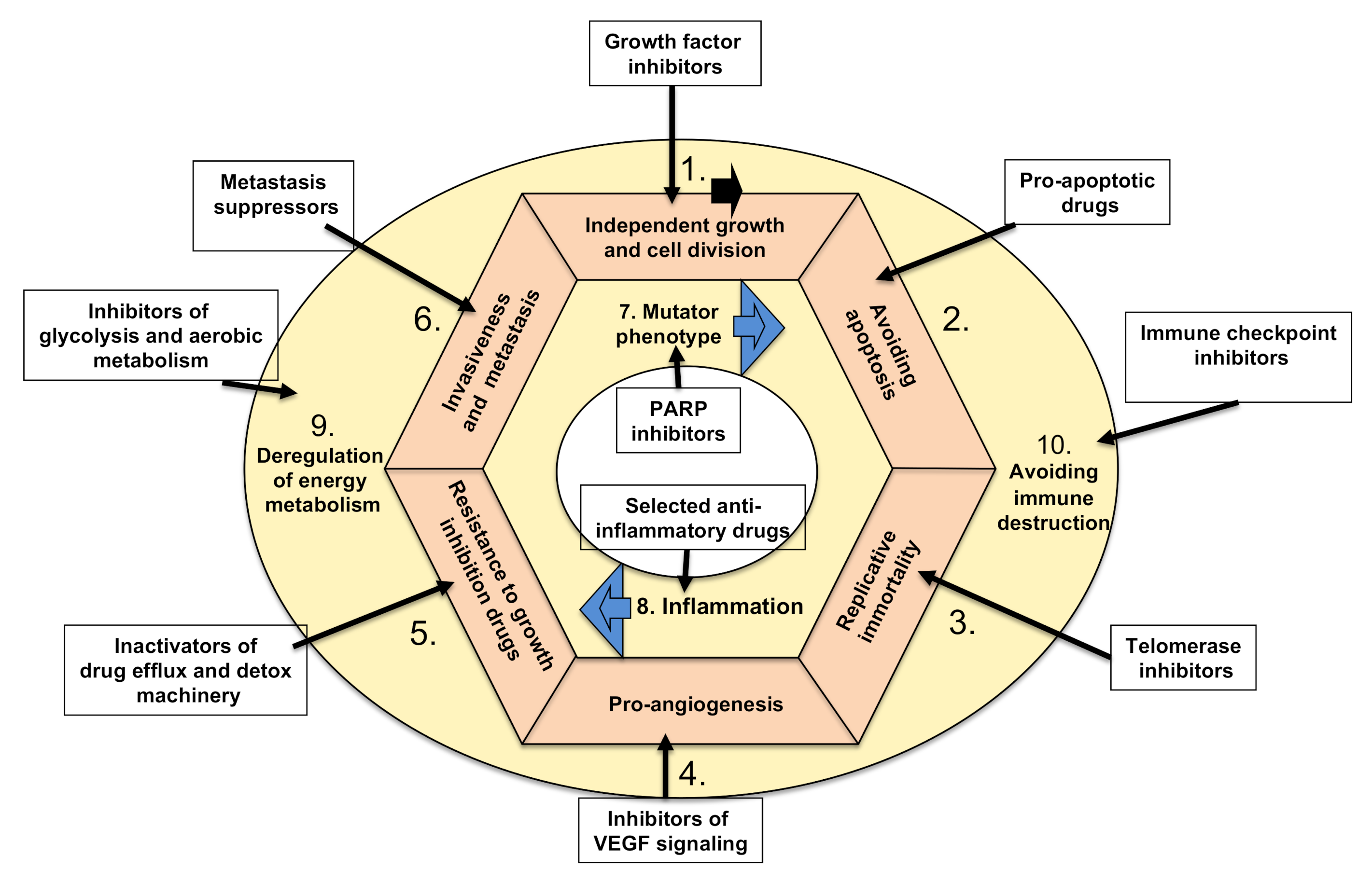
5.1. Targeting Tumors’ Growth Independence
5.1.1. Inhibiting Cancer Proliferation
5.1.2. Targeting Tumor Growth Suppressors
5.2. Inhibiting Tumor Ability to Resist Programmed Cell Death (Apoptosis)
5.3. Targeting Tumors’ Ability to Divide Indefinitely by Prolonging Telomeres
5.4. Targeting Tumors’ Ability to Induce Angiogenesis
5.5. Targeting Growth Suppressor (Drug) Resistance of Cancer Cells
- DNA repair endonucleases (xeroderma pigmentosum group F, XPF and excision repair cross-complementing protein group 1, ERCC1) involved in the nucleotide excision repair (NER) pathway, are central for the effective repair of DNA damage induced by platinum-based agents [171]. Cancer can become resistant to, for example, cisplatin by overexpressing both XPF and ERCC1 proteins [172]. Compounds like E-X PPI2 and E-X AS7 were identified as XPF-ERCC1 pathway inhibitors [173] and are a viable option for treating such cancers. However, Pt-DNA lesions depend, besides the NER pathway, also on the homologous recombination (HR) pathway. Moreover, there are mismatch repair (MMR) [174] and interstrand crosslink (ICL) repair [175] mechanisms which might be involved too. Thus, together with the latter two, Replication protein A (RPA) inhibitors [176], ataxia teleangiectasia-related (ATR) kinase inhibitors [177], DNA-PKcs inhibitors [178], HR inhibitors [179] and translesion synthesis (TLS) inhibitors [180,181] are all potential drugs for restraining multidrug resistance (MDR) in cancer that depends on overactive DNA repair machinery.
- Enhanced efflux of drugs is exemplified by the P-glycoprotein (P-gp) family that binds ATP (ATP-binding cassette [ABC] transporters) [182]. Overexpression of P-gp, multidrug-resistance-associated protein and breast cancer resistance protein (BCRP) that are present in the cell membrane are responsible for a major portion of multidrug resistance in cancer [183,184,185]. This is because they regulate the absorption, excretion and distribution of many drugs used as chemotherapy. They decrease the toxic high intracellular concentration of the administered chemotherapeutic drug, diminishing its bioavailability and therapeutic effect. P-gp, as a multidrug membrane transporter, normally pumps chloride out of the cells and can bind to a variety of chemotherapy agents, including doxorubicin, vinblastine and taxol [183]. Finding a remedy for MDR has been a long-standing challenge in cancer therapy. There is a number of P-gp inhibitors that show significant anticancer effects in various experimental studies, but unfortunately, none have entered clinical trials except sitravatinib [186], which is a receptor tyrosine kinase inhibitor that could reverse multidrug resistance in vitro. Sitravatinib is an orally available, potent small-molecule inhibitor of a closely related spectrum of receptor tyrosine kinases (RTKs) including MET, Axl, MERTK, VEGFR family, PDGFR family, KIT, FLT3, Trk family, RET, DDR2 and selected Eph family members. Sitravatinib is currently in clinical trials with nivolumab (which targets hallmark #10) for metastatic lung cancer (NSCLC, phase 2) [186] and also for renal cell carcinoma (phase 3) [187].
- Elevated metabolism of xenobiotics. Isoforms of cytochrome (CYP) are important for the drug degradation and detoxification of xenobiotic [188] and endogenous compounds like estrogen and testosterone [189,190]. Overexpression of CYP1B1 has been observed in various cancer types, especially those which are estrogen responsive, and is linked with enhanced resistance to a variety of drug therapies, including docetaxel [191]. Glutathione (GSH) maintains cellular redox homeostasis. Cancer cells show higher reactive oxygen species production than normal cells. Due to the increased requirement for energy for proliferation, cancer produces an antioxidant defense system to balance out the elevated oxidant state. Many cancer types show overexpression of GSH. Furthermore, GSH detoxifies extracellular toxins, drugs and biotics and can increase cancer multidrug resistance [192,193]. The inhibition of the GSH antioxidant defense system (flavonoids and chalcone derivatives) [194] could prevent the resistance of cancer to the used chemotherapeutics and is a reasonable strategy to avoid multidrug resistance of cancer.
- Genetic mutations in genes involved in MDR have been common reasons for cancers resisting selected drug therapy. Amplifications of genes that are being repressed by the chemotherapy—for example, dihydrofolate reductase (DHFR) in methotrexate chemotherapy—have been seen in many cancers, including leukemias. Recently, epigenetic modifications were found that can also increase MDR in cancers. There are various miRNAs that influence the sensitivity of cancer cells against anticancer agents which target genes related to cell proliferation, cell cycle and apoptosis (reviewed in [169]). Furthermore, histone deacetylase inhibitors (like mocetinostat) or histone kinase inhibitors (i.e., CUDC-101, -907) that alter the expression of various genes whose products are involved in diverse mechanisms of chemoresistance of cancer could be useful for anticancer resistance prevention, perhaps best tested in combinatorial treatments. Likewise, mocetinostat has been undergoing clinical trials since 2016 to evaluate the clinical activity in NSCLC (phase 2, ends in 2021) and RCC (phase 3, ending in 2024), in combination with three separate investigational agents, one being the immune checkpoint inhibitor nivolumab, and comparing its efficacy against sitravatinib (a multi-targeted RTK inhibitor) or glesatinib (also a multi-targeted tyrosine kinase inhibitor) [186,187].
- As additional manifestation of chemoresistance, cancers can upregulate the autocrine production of various cytokines including growth factors such as IL-1, Il-4, Il-6 and IL-8. Biologics like IL-6 (siltuximab, Sylvant) or IL-6R inhibitors (tocilizumab, Actemra; sarilumab, Kevzara) have been suggested as therapy to counteract cancer MDR, both of which are being tested for the treatment or prevention of SARS-CoV-2 severe pneumonia [195]. Furthermore, FGFs present in solid tumors not derived from cancer cells but from surrounding accompanying cells in a tumor mass can provide resistance to chemotherapy, such as 5-FU, DOX and paclitaxel, which have different mechanisms of action. Suramin, an inhibitor of FGFs, could reverse this resistance in vitro. Small-molecule inhibitors of FGFs are currently in trial for the treatment of glioblastoma [166].
5.6. Targeting Cancers’ Invasive Spreading with Metastasis
5.7. Targeting Tumors’ Mutator Phenotype by Counteracting Their Ability to Increase Genetic/Epigenetic Instability
5.8. Targeting Tumor-Promoting Inflammation by Anti-Inflammatory Drugs
5.9. Targeting Tumors’ Metabolic Shift from Oxidative to Anaerobic Glycolysis
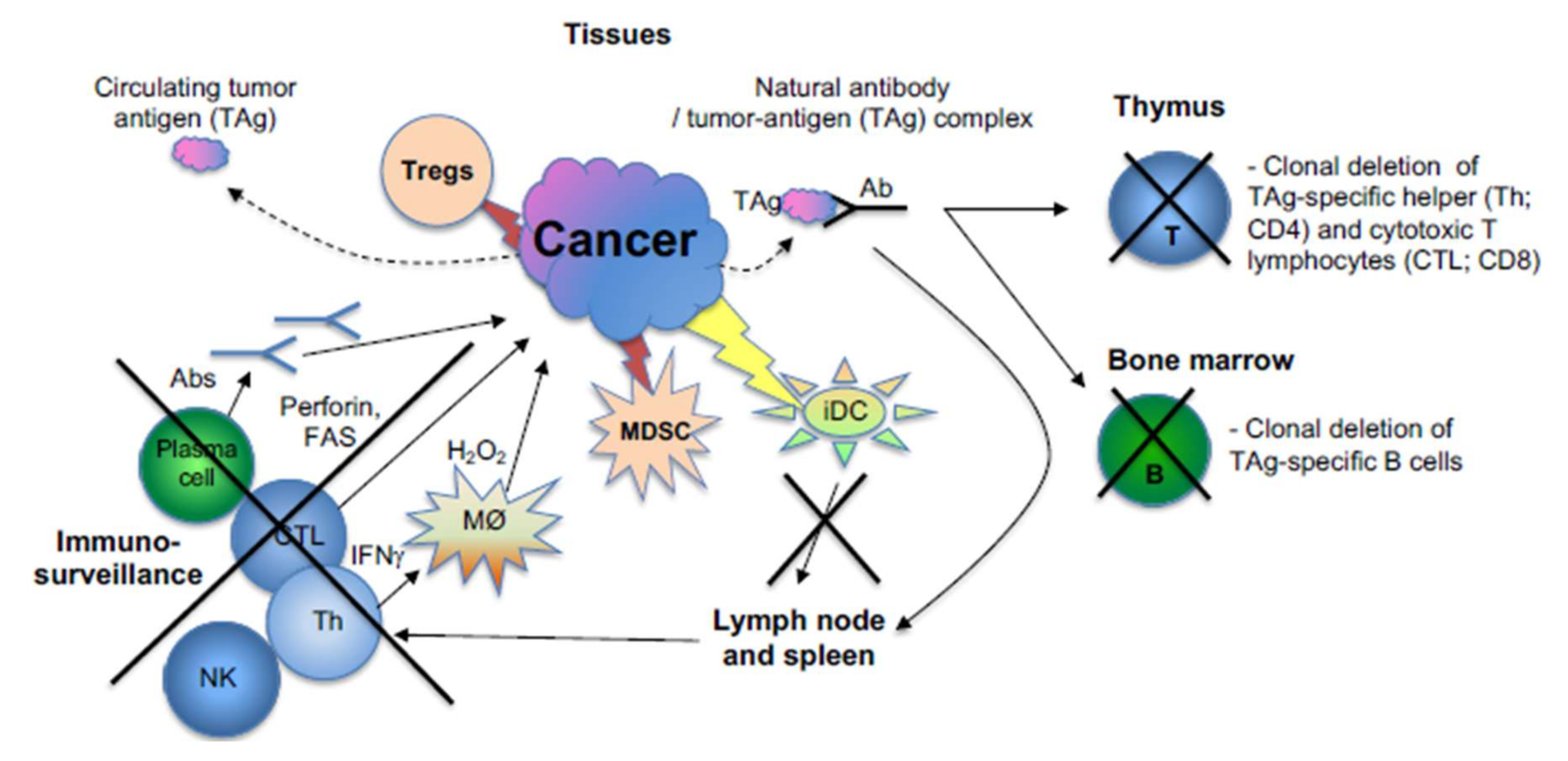
5.10. Targeting Cancers’ Escape from Destruction by the Immune System Using Various Immunotherapies (Biological and Cell-Based)
- Cancer can directly (or indirectly, by secreting a putative factor) diminish the function of either activating or effector T (or B) cells by engaging at least one of the following molecules: CTLA-4, PD1, VISTA, BTLA, CD160, CD244, LAG-3 and TIM-3 [227].
- Deletion of T cell clones (potential antitumor) in the thymus (migrating tumor cells or antigen-presenting cells which have acquired tumor antigens elsewhere) [228].
- Deletion of B cell clones (potential antitumor) in the bone marrow (by presenting tumor antigens to developing B cells) [229].
- Tregs: Cancer can activate or induce suppressor T cells (Tregs) that suppress antitumor T, B and NK immunosurveillance cells [230]. The development of regulatory T cells in the thymus and periphery of the immune system [226] is known, and their role was suggested to be of paramount importance for the immunosurveillance of cancer [225].
- Myeloid-derived suppressor cells (MDSC) are cells that can inhibit the adhesion of other leukocytes onto the endothelial surface in the tumor [233].
6. Therapies That Target Epigenetic Changes in Cancer
7. Concluding Remarks
Funding
Conflicts of Interest
References
- ClinicalTrials.gov. Clinical Studies for Neoplasms (phase 2 and 3). Available online: https://www.clinicaltrials.gov/ct2/results?cond=Neoplasms&recrs=d&age_v=&gndr=&type=Intr&rslt=&phase=1&phase=2&Search=Apply (accessed on 3 November 2020).
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Cho, R.W.; Clarke, M.F. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev. 2008, 18, 48–53. [Google Scholar] [CrossRef]
- Cho, R.W.; Wang, X.; Diehn, M.; Shedden, K.; Chen, G.Y.; Sherlock, G.; Gurney, A.; Lewicki, J.; Clarke, M.F. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells 2008, 26, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Hegar, A. Castration der Frauen; Breitkopf & Hartel: Wiesbaden, Germany, 1878. [Google Scholar]
- Battey, R. Normal ovariotomy. Atlanta Med Surg. J. 1872, 10, 321–329. [Google Scholar]
- Pusey, W.A. Roentgen rays in the treatment of skin diseases and for the removal of hair. J. Cutan. Dis. Incl. Syph. 1900, 18, 302–318. [Google Scholar] [CrossRef]
- Grubbe, E.H. News of Science: Emil H. Grubbe. Science 1957, 125, 18–22. [Google Scholar]
- Grubbe, E.H. X-rays in the treatment of cancer and other malignant diseases. Med Rec. 1902, 62, 692–695. [Google Scholar]
- Nakayama, D.K.; Bonasso, P.C. The history of multimodal treatment of wilms’ tumor. Am. Surg. 2016, 82, 487–492. [Google Scholar] [CrossRef]
- Curie, P.; Curie, M. Sur la radioactivité provoquée par les rayons de Bécquere. Comp. Rendus Acad. Sci. 1899, 129, 714–716. [Google Scholar]
- Kułakowski, A. The contribution of Marie Skłodowska-Curie to the development of modern oncology. Anal. Bioanal. Chem. 2011, 400, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Kassabian, M.K. Roentgen Rays and Electro-Therapeutics; Lippincott Company: Philadelphia, PA, USA, 1907; p. 670. [Google Scholar]
- Coutard, H. The results and methods of treatment of cancer by radiation. Ann. Surg. 1937, 106, 584–598. [Google Scholar] [CrossRef]
- Deloch, L.; Derer, A.; Hartmann, J.; Frey, B.; Fietkau, R.; Gaipl, U.S. Modern radiotherapy concepts and the impact of radiation on immune activation. Front. Oncol. 2016, 6, 141. [Google Scholar] [CrossRef]
- Moulder, J.E.; Seymour, C. Radiation fractionation: The search for isoeffect relationships and mechanisms. Int. J. Radiat. Biol. 2018, 94, 743–751. [Google Scholar] [CrossRef]
- Gilman, A. Therapeutic applications of chemical warfare agents. Fed. Proc. 1946, 5, 285–292. [Google Scholar]
- Golomb, F.M. Agents used in cancer chemotherapy. Am. J. Surg. 1963, 105, 579–590. [Google Scholar] [CrossRef]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- FDA U.S. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 3 November 2020).
- Hirsch, J. An anniversary for cancer chemotherapy. J. Am. Med. Assoc. 2006, 296, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Mattes, W.B.; Hartley, J.A.; Kohn, K.W. DNA sequence selectivity of guanine-N7 alkylation by nitrogen mustards. Nucleic Acids Res. 1986, 14, 2971–2987. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, D.; Xu, Z.; Song, J.; Qian, X.; Lv, X.; Luan, J. Anti-tumor drug targets analysis: Current insight and future prospect. Curr. Drug Targets 2019, 20, 1180–1202. [Google Scholar] [CrossRef]
- Jayashree, B.S.; Nigam, S.; Pai, A.; Patel, H.K.; Reddy, N.D.; Kumar, N.; Rao, C.M. Targets in anticancer research—A review. Indian J. Exp. Biol. 2015, 53, 489–507. [Google Scholar]
- Olgen, S. Overview on anticancer drug design and development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef]
- Kumar, B.; Singh, S.; Skvortsova, I.; Kumar, V. Promising targets in anti-cancer drug development: Recent updates. Curr. Med. Chem. 2017, 24, 4729–4752. [Google Scholar] [CrossRef]
- Sneader, W. Drug Discovery: A History; John Wiley & Sons: Hoboken, NJ, USA, 2005; p. 251. [Google Scholar]
- Ravina, E. The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs; Wiley-VCH: Weinheim, Germany, 2011; p. 157. [Google Scholar]
- Fischer, J.; Ganellin, C.R. Analogue-Based Drug Discovery; John Wiley & Sons: Hoboken, NJ, USA, 2006; p. 511. [Google Scholar]
- Melphalan Monograph for Professionals. Available online: https://www.drugs.com/monograph/melphalan.html (accessed on 3 November 2020).
- Phelinun EMA Marketing Authorisation. Available online: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/phelinun (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Doxorubicin Doxorubicin Hydrochloride. Available online: https://www.drugs.com/monograph/doxorubicin-hydrochloride.html (accessed on 3 November 2020).
- Shealy, Y.F.; Krauth, C.A.; Montgomery, J.A. Imidazole I. Coupling reactions of 5-diazoimidazole-4-carboxamide. J. Org. Chem. 1962, 27, 2150–2154. [Google Scholar] [CrossRef]
- Leukemia & Lyphoma Society. Hodgkin Lymphoma. Available online: https://www.lls.org/lymphoma/hodgkin-lymphoma/treatment/chemotherapy-and-drug-therapy (accessed on 3 November 2020).
- Elias, A.; Ryan, L.; Aisner, J.; Antman, K.H. Mesna, doxorubicin, ifosfamide, dacarbazine (MAID) regimen for adults with advanced sarcoma. Semin. Oncol. 1990, 17, 41–49. [Google Scholar] [PubMed]
- Ewend, M.G.; Brem, S.; Gilbert, M.; Goodkin, R.; Penar, P.L.; Varia, M.; Cush, S.; Carey, L.A. Treatment of single brain metastasis with resection, intracavity carmustine polymer wafers, and radiation therapy is safe and provides excellent local control. Clin. Cancer Res. 2007, 13, 3637–3641. [Google Scholar] [CrossRef] [PubMed]
- Gliadel Carmustine, FDA approval. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/96/020637Orig1s000.pdf (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Ifosfamide. Available online: https://www.drugs.com/mtm/ifosfamide.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Carboplatin. Available online: https://www.drugs.com/monograph/carboplatin.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Paclitaxel. Available online: https://www.drugs.com/monograph/paclitaxel.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Tretinoin. Available online: https://www.drugs.com/monograph/tretinoin.html (accessed on 3 November 2020).
- Topotecan FDA Approves Ovarian Cancer Drug. Available online: https://web.archive.org/web/20090119141150/https://www.fda.gov/bbs/topics/NEWS/NEW00537.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Irinotecan Hydrochloride. Available online: https://www.drugs.com/monograph/irinotecan-hydrochloride.html (accessed on 3 November 2020).
- Raymond, E.; Chaney, S.G.; Taamma, A.; Cvitkovic, E. Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- The American Society of Health-System Pharmacists. Letrozole Femara. Available online: https://www.drugs.com/monograph/letrozole.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Rituximab. Available online: https://www.drugs.com/monograph/rituximab.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Trastuzumab. Available online: https://www.drugs.com/monograph/trastuzumab.html (accessed on 3 November 2020).
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef]
- The American Society of Health-System Pharmacists. Alemtuzumab. Available online: https://www.drugs.com/monograph/alemtuzumab.html (accessed on 3 November 2020).
- Grillo-López, A.J. Zevalin: The first radioimmunotherapy approved for the treatment of lymphoma. Expert Rev. Anticancer Ther. 2002, 2, 485–493. [Google Scholar] [CrossRef]
- FDA. Tositumumab Approval. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2003/tosicor062703L.htm (accessed on 3 November 2020).
- Tositumumab, Iodine (I)-131 RadioImmunoTherapy, FDA Approval. 2013. Available online: https://web.archive.org/web/20180127032045/https://www.fda.gov/ohrms/dockets/ac/02/briefing/3916B1_02_D-FDA%20-%20Product%20Description.htm (accessed on 3 November 2020).
- Milenic, D.E.; Brady, E.D.; Brechbiel, M.W. Antibody-targeted radiation cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 488–499. [Google Scholar] [CrossRef]
- BEXXAR. FDA Withdrawal of Approval of the Indication for Treatment of Patients With Relapsed or Refractory, Low Grade, Follicular, or Transformed CD20 Positive Non-Hodgkin’s Lymphoma Who Have Not Received Prior Rituximab. Available online: https://www.federalregister.gov/documents/2013/10/23/2013-24840/glaxosmithkline-llc-withdrawal-of-approval-of-the-indication-for-treatment-of-patients-with-relapsed (accessed on 3 November 2020).
- EMA. Withdrawal of tositumumab (iodine131) orphan designation on the request of the sponsor (GlaxoSmithKline LLC). Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu303136 (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Cetuximab Erbitux. Available online: https://www.drugs.com/mtm/cetuximab.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Bevacizumab Avastin. Available online: https://www.drugs.com/monograph/bevacizumab.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Anastrozole Arimidex. Available online: https://www.drugs.com/monograph/anastrozole.html (accessed on 3 November 2020).
- Dukes, M. The Relevance of preclinical models to the treatment of postmenopausal breast cancer. Oncology 1997, 54, 6–10. [Google Scholar] [CrossRef]
- Coombes, R.C.; Kilburn, L.S.; Snowdon, C.F.; Paridaens, R.; Coleman, R.E.; Jones, S.E.; Jassem, J.; Van de Velde, C.J.; Delozier, T.; Alvarez, I.; et al. Survival and safety of exemestane versus tamoxifen after 2–3 years’ tamoxifen treatment (Intergroup Exemestane Study): A randomised controlled trial. Lancet 2007, 369, 559–570. [Google Scholar] [CrossRef]
- The American Society of Health-System Pharmacists. Thalidomide. Available online: https://www.drugs.com/monograph/thalidomide.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Lenalidomide. Available online: https://www.drugs.com/monograph/lenalidomide.html (accessed on 3 November 2020).
- Panitumumab (Vectibix) FDA Aproval. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/125147LTR.pdf (accessed on 3 November 2020).
- Itoh, K.; Yamada, A.; Mine, T.; Noguchi, M. Recent advances in cancer vaccines: An overview. Jpn. J. Clin. Oncol. 2008, 39, 73–80. [Google Scholar] [CrossRef]
- MMWR FDA Licensure of Bivalent Human Papillomavirus Vaccine (HPV2, Cervarix) for Use in Females and Updated HPV Vaccination Recommendations from the Advisory Committee on Immunization Practices (ACIP). Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5920a4.htm (accessed on 3 November 2020).
- Plosker, G.L. Sipuleucel-T. Drugs 2011, 71, 101–108. [Google Scholar] [CrossRef] [PubMed]
- EPAR (EMA). Yervoy Autorisation for use in European Union. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yervoy (accessed on 3 November 2020).
- FDA. Ipilimumab (Yervoy) Drug Aproval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125377Orig1s000TOC.cfm (accessed on 3 November 2020).
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- The American Society of Health-System Pharmacists. Peginterferon-Alfa-2b. Available online: https://www.drugs.com/ppa/peginterferon-alfa-2b.html (accessed on 3 November 2020).
- Adcetris Seattle Genetics’ Antibody-Drug Conjugate Receives FDA Okay to Treat Lymphomas. Available online: https://www.genengnews.com/topics/drug-discovery/seattle-genetics-antibody-drug-conjugate-receives-fda-okay-to-treat-lymphomas/ (accessed on 3 November 2020).
- ADC Review. Monomethyl auristatin E (MMAE). J. Antibody-Drug Conjug. 2014. Available online: https://www.adcreview.com/knowledge-center/monomethyl-auristatin-e-mmae/ (accessed on 3 November 2020).
- EMA Adcetris EPAR. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/adcetris (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Blincyto Blinatumomab. Available online: https://www.drugs.com/ppa/blinatumomab.html (accessed on 3 November 2020).
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for patients with B-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- The American Society of Health-System Pharmacists. Pembrolizumab Keytruda. Available online: https://www.drugs.com/monograph/pembrolizumab.html (accessed on 3 November 2020).
- The American Society of Health-System Pharmacists. Nivolumab Opdivo. Available online: https://www.drugs.com/monograph/nivolumab.html (accessed on 3 November 2020).
- EMA. Tisagenlecleucel Kymriah EPAR. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah (accessed on 3 November 2020).
- Kymriah FDA Approval Brings First Gene Therapy to the United States. Available online: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed on 3 November 2020).
- FDA. FDA Grants Accelerated Approval to New Treatment for Advanced Soft Tissue Sarcoma (to Lartrurvo). Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-new-treatment-advanced-soft-tissue-sarcoma (accessed on 3 November 2020).
- Eli Lilly and Company. Lilly Lilly Reports Results of Phase 3 Soft Tissue Sarcoma Study of LARTRUVO®. Available online: https://www.oncnursingnews.com/publications/oncology-nurse/2019/april-2019/fda-advises-against-starting-olaratumab-for-soft-tissue-sarcoma (accessed on 3 November 2020).
- Lartrurvo FDA. EMA Say Olaratumab Should Not Be Given to New Patients With Soft Tissue Sarcoma. Available online: https://www.targetedonc.com/view/fda-ema-say-olaratumab-should-not-be-given-to-new-patients-with-soft-tissue-sarcoma (accessed on 3 November 2020).
- Durvalumab FDA Granted Accelerated Approval to Durvalumab (IMFINZI, AstraZeneca UK Limited). Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/durvalumab-imfinzi (accessed on 3 November 2020).
- Avelumab FDA Approves First Treatment for Rare Form of Skin Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-rare-form-skin-cancer (accessed on 3 November 2020).
- Atezolizumab FDA Approved Atezolizumab (TECENTRIQ, Genentech Oncology) for the Treatment of Patients with Metastatic Non-Small Cell Lung Cancer (NSCLC). Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/atezolizumab-tecentriq (accessed on 3 November 2020).
- Yescarta FDA Approval of YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 3 November 2020).
- Mylotarg FDA Approves Gemtuzumab Ozogamicin for CD33-Positive AML. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-gemtuzumab-ozogamicin-cd33-positive-aml (accessed on 3 November 2020).
- Libtayo FDA Approves First Treatment for Advanced Form of the Second Most Common Skin Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-advanced-form-second-most-common-skin-cancer (accessed on 3 November 2020).
- Libtayo Libtayo: The Australian Prescription Medicine Decision Summary. Available online: https://www.tga.gov.au/apm-summary/libtayo (accessed on 3 November 2020).
- Turalio Pexidartinib Hydrochloride. Available online: https://www.drugs.com/monograph/pexidartinib-hydrochloride.html (accessed on 3 November 2020).
- Roberts, A.W.; Huang, D. Targeting BCL2 with BH3 mimetics: Basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef]
- Venclexta Venetoclax: Drugs.com. Available online: https://www.drugs.com/ppa/venetoclax.html (accessed on 3 November 2020).
- Tecartus FDA Approves Brexucabtagene Autoleucel for Relapsed Or Refractory Mantle Cell Lymphoma. Available online: https://www.fda.gov/drugs/fda-approves-brexucabtagene-autoleucel-relapsed-or-refractory-mantle-cell-lymphoma (accessed on 3 November 2020).
- Gardasil FDA Approves Expanded Uses for Gardasil to Include Preventing Certain Vulvar and Vaginal Cancers. Available online: https://web.archive.org/web/20100306073734/https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116945.htm (accessed on 3 November 2020).
- EMA. Gardasil 9: Human Papillomavirus 9-Valent Vaccine (Recombinant, Adsorbed) EPAR. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/gardasil-9 (accessed on 3 November 2020).
- Dowd, F.J.; Johnson, B.; Mariotti, A.S. Pharmacology and Therapeutics for Dentistry, 7th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Hine, R. A Dictionary of Biology, 6th ed.; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Hall, J.E. Guyton and Hall Textbook of Medical Physiology, 14th ed.; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Bio.l 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Blackhall, F.H. Direct ras G12C inhibitors: Crossing the rubicon. Br. J. Cancer 2019, 121, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Murphree, A.L.; Benedict, W.F. Retinoblastoma: Clues to human oncogenesis. Science 1984, 223, 1028–1033. [Google Scholar] [CrossRef]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets Ther. 2013, 7, 57–68. [Google Scholar]
- Toufektchan, E.; Toledo, F. The guardian of the genome revisited: p53 downregulates genes required for telomere maintenance, DNA repair, and centromere structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Goodrich, D.W.; Wang, N.P.; Qian, Y.W.; Lee, E.Y.; Lee, W.H. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 1991, 67, 293–302. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; McCormack, D.; McDonald, D.; McFadden, D. Pterostilbene induces mitochondrially derived apoptosis in breast cancer cells in vitro. J. Surg. Res. 2013, 180, 208–215. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Rosenfeld, N.; Itzkovitz, S.; Milo, R.; Sigal, A.; Dekel, E.; Yarnitzky, T.; Liron, Y.; Polak, P.; Lahav, G.; et al. Oscillations and variability in the p53 system. Mol. Syst. Biol. 2006, 2, 1–13. [Google Scholar] [CrossRef]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Busson, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The p53 isoform Delta133p53beta promotes cancer stem cell potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Seimiya, H. Revisiting telomere shortening in cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Kaneko, S. Telomerase-targeted cancer immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Middleton, G.; Greenhalf, W.; Costello, E.; Shaw, V.; Cox, T.; Ghaneh, P.; Palmer, D.H.; Neoptolemos, J.P. Immunobiological effects of gemcitabine and capecitabine combination chemotherapy in advanced pancreatic ductal adenocarcinoma. Br. J. Cancer 2016, 114, 510–518. [Google Scholar] [CrossRef]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef]
- Seimiya, H.; Muramatsu, Y.; Ohishi, T.; Tsuruo, T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005, 7, 25–37. [Google Scholar] [CrossRef]
- Crees, Z.; Girard, J.; Rios, Z.; Botting, G.M.; Harrington, K.; Shearrow, C.; Wojdyla, L.; Stone, A.L.; Uppada, S.B.; Devito, J.T.; et al. Oligonucleotides and G-quadruplex stabilizers: Targeting telomeres and telomerase in cancer therapy. Curr. Pharm. Des. 2014, 20, 6422–6437. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Epel, E.S.; Lin, J.; Dhabhar, F.S.; Wolkowitz, O.M.; Puterman, E.; Karan, L.; Blackburn, E.H. Dynamics of telomerase activity in response to acute psychological stress. Brain Behav. Immun. 2010, 24, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Chen, B.; Liu, X. Telomere and telomerase as targets for cancer therapy. Appl. Biochem. Biotechnol. 2010, 160, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Eckhardt, S.G. Development of matrix metalloproteinase inhibitors in cancer therapy. J. Natl. Cancer Inst. 2001, 93, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Yamamoto, H.; Adachi, Y.; Itoh, F.; Iku, S.; Matsuno, K.; Kusano, M.; Arimura, Y.; Endo, T.; Hinoda, Y.; Hosokawa, M.; et al. Association of matrilysin expression with recurrence and poor prognosis in human esophageal squamous cell carcinoma. Cancer Res. 1999, 59, 3313–3316. [Google Scholar]
- Murray, G.I.; Duncan, M.E.; O’Neil, P.; Melvin, W.T.; Fothergill, J.E. Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat. Med. 1996, 2, 461–462. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Qulali, M.; Goulet, R.; Bone, E.A.; Fife, R. Effect of matrix metalloproteinase inhibitor batimastat on breast cancer regrowth and metastasis in athymic mice. J. Natl. Cancer Inst. 1995, 87, 1546–1550. [Google Scholar] [CrossRef]
- Zucker, S.; Cao, J.; Molloy, C.J. Role of Matrix metalloproteinases and plasminogen activators in cancer invasion and metastasis: Therapeutic strategies. Anticancer Drug Dev. 2002, 6, 91–122. [Google Scholar]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern cooperative oncology group trial E2196. J. Clin. Oncol. 2004, 22, 4683–4690. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Oxygen sensing and metabolic homeostasis. Mol. Cell Endocrinol. 2014, 397, 51–58. [Google Scholar] [CrossRef]
- Sohn, S.H.; Kim, B.; Sul, H.J.; Choi, B.Y.; Kim, H.S.; Zang, D.Y. Foretinib inhibits cancer stemness and gastric cancer cell proliferation by decreasing CD44 and c-MET signaling. OncoTargets Ther. 2020, 13, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Clinical Study on the Treatment of Recurrent Glioblastoma With Anlotinib. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04004975?term=FGF&cond=Glioblastoma&draw=2&rank=1 (accessed on 3 November 2020).
- Kachalaki, S.; Ebrahimi, M.; Mohamed Khosroshahi, L.; Mohammadinejad, S.; Baradaran, B. Cancer chemoresistance; biochemical and molecular aspects: A brief overview. Eur. J. Pharm. Sci. 2016, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kushwaha, P.P.; Gupta, S. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019, 2, 161–177. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Kirschner, K.; Melton, D.W. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010, 30, 3223–3232. [Google Scholar]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19–28. [Google Scholar] [CrossRef]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Bodenmiller, D.; Fitch, R.W.; Jalal, S.I.; Turchi, J.J. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol. Cancer Ther. 2011, 10, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef]
- Albarakati, N.; Abdel-Fatah, T.M.; Doherty, R.; Russell, R.; Agarwal, D.; Moseley, P.; Perry, C.; Arora, A.; Alsubhi, N.; Seedhouse, C.; et al. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol. Oncol. 2015, 9, 204–217. [Google Scholar] [CrossRef]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A Small-molecule inhibitor of RAD51 reduces homologous recombination and sensitizes multiple myeloma cells to doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef]
- Sail, V.; Rizzo, A.A.; Chatterjee, N.; Dash, R.C.; Ozen, Z.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Identification of small molecule translesion synthesis inhibitors that target the Rev1-CT/RIR protein-protein interaction. ACS Chem. Biol. 2017, 12, 1903–1912. [Google Scholar] [CrossRef]
- Inoue, A.; Kikuchi, S.; Hishiki, A.; Shao, Y.; Heath, R.; Evison, B.J.; Actis, M.; Canman, C.E.; Hashimoto, H.; Fujii, N. A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem. 2014, 289, 7109–7120. [Google Scholar] [CrossRef]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef][Green Version]
- Di Pietro, A.; Dayan, G.; Conseil, G.; Steinfels, E.; Krell, T.; Trompier, D.; Baubichon-Cortay, H.; Jault, J. P-glycoprotein-mediated resistance to chemotherapy in cancer cells: Using recombinant cytosolic domains to establish structure-function relationships. Braz. J. Med. Biol. Res. 1999, 32, 925–939. [Google Scholar] [CrossRef]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; van Gastelen, M.A.; de Jong, L.A.; Pluim, D.; van Waardenburg, R.C.; Ruevekamp-Helmers, M.C.; Floot, B.G.; Schellens, J.H. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999, 59, 4559–4563. [Google Scholar]
- ClinicalTrials.gov. Sitravatinib Combined with Nivolumab in Non Small Cell Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/ct2/show/NCT02954991 (accessed on 3 November 2020).
- ClinicalTrials.gov. Sitravatinib Combined with Nivolumab in Renal Cell Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03015740 (accessed on 3 November 2020).
- Sutter, T.R.; Tang, Y.M.; Hayes, C.L.; Wo, Y.Y.P.; Jabs, E.W.; Li, X.; Yin, H.; Cody, C.W.; Greenlee, W.F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [Google Scholar] [PubMed]
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Sissung, T.M.; Danesi, R.; Price, D.K.; Steinberg, S.M.; De Wit, R.; Zahid, M.; Gaikwad, N.; Cavalieri, E.; Dahut, W.L.; Sackett, D.L. Association of the CYP1B1* 3 allele with survival in patients with prostate cancer receiving docetaxel. Mol. Cancer Ther. 2008, 7, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.-I.; Kamada, M.; Furumoto, H.; Hirao, T.; Aono, T. Expression of glutathione S-transferase-π in human ovarian cancer as an indicator of resistance to chemotherapy. Gynecol. Oncol. 1994, 52, 313–319. [Google Scholar] [CrossRef]
- Batist, G.; Tulpule, A.; Sinha, B.K.; Katki, A.G.; Myers, C.E.; Cowan, K.H. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J. Biol. Chem. 1986, 261, 15544–15549. [Google Scholar]
- Elliott, A.J.; Scheiber, S.A.; Thomas, C.; Pardini, R.S. Inhibition of glutathione reductase by flavonoids: A structure-activity study. Biochem. Pharmacol. 1992, 44, 1603–1608. [Google Scholar] [CrossRef]
- COVID-19 Regeneron, Sanofi Launch Clinical Trial of Kevzara as Coronavirus Treatment. Available online: https://www.genengnews.com/news/regeneron-sanofi-launch-clinical-trial-of-kevzara-as-coronavirus-treatment/ (accessed on 3 November 2020).
- Meirson, T.; Gil-Henn, H.; Samson, A.O. Invasion and metastasis: The elusive hallmark of cancer. Oncogene 2020, 39, 2024–2026. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Wu, A.T.; Lee, W.H.; Chang, T.C.; Chiou, J.F.; Wang, L.S.; Wu, C.H.; Huang, C.Y.; Shieh, Y.S.; Chao, T.Y.; et al. Pterostilbene, a bioactive component of blueberries, suppresses the generation of breast cancer stem cells within tumor microenvironment and metastasis via modulating NF-kappaB/microRNA 448 circuit. Mol. Nutr. Food Res. 2013, 57, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab. Investig. 2018, 98, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. The multiple roles of exosomes in metastasis. Cancer Genom. Proteom. 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Milholland, B.; Dong, X.; Zhang, L.; Hao, X.; Suh, Y.; Vijg, J. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 2017, 8, 15183. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Loeb, L.A. Human cancers express a mutator phenotype: Hypothesis, origin, and consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef]
- Natali, F.; Rancati, G. The mutator phenotype: Adapting microbial evolution to cancer biology. Front. Genet. 2019, 10, 713. [Google Scholar] [CrossRef]
- Hill, V.K.; Kim, J.S.; Waldman, T. Cohesin mutations in human cancer. Biochim. Biophys. Acta 2016, 1866, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tosti, E.; Edelmann, W. Mouse models of DNA mismatch repair in cancer research. DNA Repair 2016, 38, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Raynes, Y.; Halstead, A.L.; Sniegowski, P.D. The effect of population bottlenecks on mutation rate evolution in asexual populations. J. Evol. Biol. 2014, 27, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Good, B.H.; Desai, M.M. Evolution of mutation rates in rapidly adapting asexual populations. Genetics 2016, 204, 1249–1266. [Google Scholar] [CrossRef]
- Hoong, B.Y.D.; Gan, Y.H.; Liu, H.; Chen, E.S. cGAS-STING pathway in oncogenesis and cancer therapeutics. Oncotarget 2020, 11, 2930–2955. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, J.; Zhu, T.; Zhuo, W.; Yi, Y.; Hu, S.; Yin, J.; Zhang, W.; Zhou, H.; Liu, Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 2020, 19, 133. [Google Scholar] [CrossRef]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-inflammatory drugs as anticancer agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Schölmerich, J.; Oefner, P.J.; et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef]
- Gao, M.; Huang, J.; Jiang, X.; Yuan, Y.; Pang, H.; Luo, S.; Wang, N.; Yao, C.; Lin, Z.; Pu, D.; et al. Regulation of aerobic glycolysis to decelerate tumor proliferation by small molecule inhibitors targeting glucose transporters. Protein Cell 2020, 11, 446–451. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269–67286. [Google Scholar] [CrossRef]
- Dembic, Z. On recognizing ‘shades-of-gray’ (self-nonself discrimination) or ‘colour’ (Integrity model) by the immune system. Scand. J. Immunol. 2013, 78, 325–338. [Google Scholar] [CrossRef]
- Dembic, Z. On integrity in immunity during ontogeny or how thymic regulatory T cells work. Scand. J. Immunol. 2019, 90, e12806. [Google Scholar] [CrossRef]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Bogen, B.; Dembic, Z.; Weiss, S. Clonal deletion of specific thymocytes by an immunoglobulin idiotype. EMBO J. 1993, 12, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.M.; Dembic, Z.; Morahan, G.; Miller, J.; Burki, K.; Nemazee, D. Peripheral deletion of self-reactive B-cells. Nature 1991, 354, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Dembic, Z. Chapter 9—Theories about the function of the immune system. In The Cytokines of the Immune System; Dembic, Z., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 283–302. [Google Scholar]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef]
- Dembic, Z. Chapter 4—The role and regulation of the immune responses. In The Cytokines of the Immune System; Dembic, Z., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 99–122. [Google Scholar]
- Dembic, Z. Chapter 3—Activation of cells of the immune system. In The Cytokines of the Immune System; Dembic, Z., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 57–98. [Google Scholar]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Larkin, J.; Minor, D.; D’Angelo, S.; Neyns, B.; Smylie, M.; Miller Jr, W.H.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: A randomized, controlled, open-label phase III trial. J. Clin. Oncol. 2018, 36, 383. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Belum, V.R.; Benhuri, B.; Postow, M.A.; Hellmann, M.D.; Lesokhin, A.M.; Segal, N.H.; Motzer, R.J.; Wu, S.; Busam, K.J.; Wolchok, J.D.; et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur. J. Cancer 2016, 60, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Rzepecki, A.K.; Cheng, H.; McLellan, B.N. Cutaneous toxicity as a predictive biomarker for clinical outcome in patients receiving anticancer therapy. J. Am. Acad. Dermatol. 2018, 79, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, D.D.; Sharma, S.; You, J.S.; Su, S.F.; Taberlay, P.C.; Kelly, T.K.; Yang, X.; Liang, G.; Jones, P.A. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012, 21, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hambach, L.; Ling, K.W.; Pool, J.; Aghai, Z.; Blokland, E.; Tanke, H.J.; Bruijn, J.A.; Halfwerk, H.; van Boven, H.; Wieles, B.; et al. Hypomethylating drugs convert HA-1-negative solid tumors into targets for stem cell-based immunotherapy. Blood 2009, 113, 2715–2722. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef]
- FDA. U.S. Farydak, Novartis (Panabinostat), Approval. 2015. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process (accessed on 3 November 2020).
- FDA. U.S. Revlimid, Celgene (Lenalidomide), Approval. 2005. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021880s000_Revlimid_PharmR.pdf (accessed on 3 November 2020).
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef]
- Azam, F.; Mehta, S.; Harris, A.L. Mechanisms of resistance to antiangiogenesis therapy. Eur. J. Cancer 2010, 46, 1323–1332. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year of Approval | Drug (Therapy) | Category | Mode of Action | Targeted Hallmark | First Indications (Current) | ||
|---|---|---|---|---|---|---|---|
| Solid Tumors | Blood Borne | Institution or Country | |||||
| 1949 | Nitrogen mustard (Mustine, mechlorethamine) | Chemotherapeutic | Nonspecific DNA alkylating agent; binds and crosslink DNA, prevents cell duplication | 1,2 | Bronchogenic carcinoma | Hodgkin’s disease, lymphosarcoma, chronic myelocytic leukemia [CML], polycythemia vera | USA (FDA) |
| 1953 | Methotrexate; 6-Mercaptopurine | Chemotherapeutic | Blocks cell cycle in S phase | 1,2 | Breast, ovarian, bladder, head and neck cancer, osteosarcoma, choricarcinoma | Acute lymphoblastic leukemia [ALL] | FDA |
| 1959 | Cyclophosphamide | Chemotherapeutic | Nitrogen mustard, DNA alkylating agent; crosslinks DNA, blocks cell cycle | 1,2 | Multiple myeloma | --′′-- | |
| 1961 | Vinblastine | Chemotherapeutic | Blocks cell cycle in M phase | 1,2 | Cancer | --′′-- | |
| 1962 | 5-Fluorouracil (5-FU) | Chemotherapeutic | Blocks cell cycle in S phase | 1,2 | Cancer | --′′-- | |
| 1964 | Melphalan | Chemotherapeutic | Nitrogen mustard, DNA alkylating agent; crosslinks DNA, blocks cell cycle | 1,2 | (Childhood neuroblastoma, ovarian cancer, and mammary adenocarcinoma) | Multiple myeloma (Hodgkin lymphoma, non-Hodgkin lymphoma, ALL and AML) | FDA (EMA in 2020) |
| 1974 | Doxorubicin | Chemotherapeutic | Inhibiting eukaryotic cell growth (anthracydine) | 1,2 | Breast cancer, bladder cancer, Kaposi’s sarcoma | Lymphoma, and ALL | FDA |
| 1975 | Dacarbazine | Chemotherapeutic | Nitrogen mustard, DNA alkylating agent; crosslinks DNA, blocks cell cycle | 1 | Melanoma (sarcoma; MAID * regimen) | Hodgkin lymphoma [a part of ABVD′′ regimen] | --′′-- |
| 1977 | Carmustine | Chemotherapeutic | Blocks cell cycle; nitrosourea, alkylates DNA, action not fully understood | 1,2 | Palliative in glioblastoma, and brain tumors | Multiple myeloma [palliative in refractory Hodgkin or non-Hodgkin tumors] | --′′-- |
| --′′-- | Tamoxifen | Chemotherapeutic | Inhibiting growth (anti-estrogen synthesis), cell cycle in G1 phase | 1 | Breast cancer | --′′-- | |
| 1987 | Ifosfamid | Chemotherapeutic | Nitro mustard, DNA alkylating agent; crosslinking DNA alkylating agent; crosslinking DNA and blocking cell cycle | 1 | (Testicular, ovarian, bladder, cervical, small cell lung cancer, E wing and soft tissue sarcoma, osteosarcoma, thymoma) | Hodgkin, and non-Hodgkin lymphoma | --′′-- |
| 1989 | Carboplatin | Chemotherapeutic | Blocks cell cycle | 1,2 | cancer | --′′-- | |
| 1991 | Paclitaxel | Chemotherapeutic | First oftaxans, antimicrotubule agent, blocks cell cycle in M phase | 1,2 | Advanced ovarian carcinoma (breast, NSCLC, SCLC, opancreatic, es) | --′′-- | |
| 1995 | Anastrozole | Chemotherapeutic | Aromatase inhibitor (inhibits estrogen synthesis) | 1 | Advanced breast cancer [postmenopausal; if progressed on tamoxifen therapy] | UK, --′′-- | |
| --′′-- | Tretinoin | Chemotherapeutic | Vitamin A related | 1 | Acute promyelocytic leukemia | FDA | |
| 1996 | Oxaliplatin | Chemotherapeutic | Blocks cell cycle | 1,2 | Advanced colorectal carcinoma [for the treatment of 5-FU pretreated patients] | France (FDA in 2004) | |
| 1996 | Topotecan; Irinotecan | Chemotherapeutic | DNA-modifying enzyme inhibitors; topoisomerase-1 inhibitors, block cell cycle in S phase | 1,2 | Metastastatic ovarian and colorectal carcinoma (cervical SCLC, pancreatic) | FDA | |
| 1996 | Letrozole | Chemotherapeutic | Aromatase inhibitor (inhibits estrogen synthesis) | 1 | Early stage breast cancer [poswtmenopausal] | France (FDA in 2004) | |
| 1997 | Rituximab | Biologic | Inbihiting proliferation (anti-CD20) | 1 | Non-Hodgkin lymphoma | FDA | |
| 1998 | Trastuzumab (herceptin) | Biologic | Inbihiting proliferation (anti-EGFR2) | 1 | Metast brest cancer | --′′-- | |
| 2001 | Alemtuzumab (Campath1) | Biologic | (anti-CD52) | 1 | Chronic lymphocytic leukemia [CLL] | --′′-- | |
| --′′-- | Imatinib (Gleevec) | Chemotherapeutic/small molecule inhibitor | Inhibit of Bcr-Abl tyrosine kinase | 2 | Gastro intestinal stromal tumor [GIST] | CML, ALL-Philadelphia chromosome positive | --′′-- |
| 2003 | Bortezomib (Velcade) | Chemotherapeutic | Reversible proteasome inhibitor; cell growth arrest, apoptosis | 2 | Relapsed or refractory multiple melanoma | --′′-- | |
| 2003-2 | Ibritumomab tiuxetan (Zevalin) | Radionuclide-linked biologic | (Anti-CD20) | 1 | Non-Hodgkin lymphoma | --′′-- | |
| --′′-- | Tositumomab (Bexxar) | Radionuclide-linked biologic | (Anti-CD19) | 1 | Non-Hodgkin lymphoma [withdrawn in 2014-15] | FDA-EMEA | |
| 2004 | Cetuximab (Erbitux) | Biologic | Inhibiting proliferation (anti-EGFR signaling), inducing apoptosis | 1,2 | Metastatic colorectal carcinoma | FDA | |
| --′′-- | Bevacizumab (Avastin) | Biologic | Inhibiting angiogenesis (anti-VEGF) | 4 | Metastatic colorectal carcinoma (NSCLC, glioblastoma, renal cell carcinoma, breast, ovarian cancer) | --′′-- | |
| 2005 | sorafenib | Chemotherapeutic | Multi-kinase inhibitor of Ras (Raf-MEK-ERK) pathway, anti-angiogenic (anti-VEGFR2,3) | 1,4 | Advanced renal cell carcinomas (from 2007, hepatocellular carcinoma) | --′′-- | |
| --′′-- | Exemestane (and anastrozole) | Chemotherapeutic | Aromatase inhibitor (inhibits estrogen synthesis) | 1 | Early breast cancer [hormone receptor positive] | --′′-- | |
| 2006 | Gardasil | Prophylactic Vaccine | Anti-HPV types 6,11,16 and 18 | 10 | Prevention of cervical carcinoma | --′′-- | |
| --′′-- | Thalidomide (Thalomid) | Chemotherapeutic | An immune omodulatory drug with spectrum of activities notfully charaterized | 1,8 | Relapsed or refractory multiple myeloma [with dexamethasone in combination] | --′′-- | |
| --′′-- | Lemalidomide (Revlimid) | Chemotherapeutic (Thalidomude analogue) | An immunomodulatory drug; inhibits COX2, inhibits angiogenesis, induces apoptosis via G1 arrest | 1,2,4,8 | Relapsed or refractory multiple myeloma [with dexamethasone in combination] | --′′-- | |
| --′′-- | Panitumumab (Vectibix) | Biologic | EGF receptor inhibitor, inhibiting proliferation and inducing apoptosis | 1,2 | Metastatic colorectal cancer [after failing oxalplatin and/or irinotecan regimens] | --′′-- | |
| --′′-- | Vorinostat (Zolinza) | Chemotherapeutic | Histone deacetylase (HDAC) inhibitor, promoting apoptotic cell death and cell cycle arrest in G1, G2/M | 1,2 | Refractory cutaneous T cell lymphoma | --′′-- | |
| Alkylating Agents | Drugs | Mechanism of Anti-Tumor or Action |
|---|---|---|
| Nitrogen mustards: | busulfan, chlorambucil, melplatin | Proliferation block by creating inter- or intra-strand cross links in DNA, or |
| Platinum based: | cisplatin, carboplatin, oxalplatin | causing DNA base mispair, thereby |
| Qxazaphosphorines: | cyclophosphamide, ifosfamide | preventing strand separation during cell cycle progression |
| Hydrazine | ||
| Carmustine | ||
| Antimetabolites | ||
| Purine analogs: | 6-mercaptopurine, azathioprine, cladribine | Proliferation or cell cycle block by: |
| Purine antagonists: | fludarabine | interference with biosynthetic pathways, |
| Pyrimidine antagonists: | cytarabine, 5-fluorouracil (5-FU), gemcitabine, capecitabine | disturbance of DNA/RNA formation, |
| Antifolates | methothrexate, pemetrexed, pralatrexate | causing DNA strand breaks, and |
| Inhibitors of ribonucleotide reductase | hydroxyurea | Incorporation of false analogues. These events ultimately can trigger apoptosis. |
| Mitotic Spindle Poisons (Mitosis Poisons) | ||
| Taxans: | docetaxel, paclitaxel, cabazitaxel | Preventing depolymerization of mitotic spindle by stabilizing GDP-bound tubulin in microtubule. |
| Vinca alkaloids: | vincristine, vinblastine, vinorelbine, vindesine, vinflunine | Preventing mitotic spindle formation by inhibition of tubulin polymerization. |
| Others | ||
| Antibiotics: | bleomycin, actinomycin D, anthracyclines | Intercalates into DNA stopping transcription. |
| Proteasome inhibitors | bortezomib | Apoptotic cell death. |
| Tyrosine kinase inhibitors: | imatinib, erlotinib | Affecting multiple signaling pathways. |
| Enzymes | l-asparaginase | Deregulates normal metabolism. |
| Topoisomerase I; inhibitors: | irinotecan, topotecan | DNA strand breaks during replication and |
| Topoisomerase II; inhibitors: | etoposide, anthracyclines: doxorubicin, | causing cell cycle block, and indirectly apoptosis. |
| Year of Approval | Drug (Therapy) | Category | Mode of Action | Targeted Hallmark | First Indications (Current) | ||
|---|---|---|---|---|---|---|---|
| Solid Tumors | Blood Borne | Institution or Country | |||||
| 2008 | Oncophage | Therapeutic Vaccline | Bolstering anticancer immune response by autologous tumor-deriv heat shock protein gpg6 | 10 | Renal cell carcinoma | Russia | |
| 2009 | Cervarix | Therapeutic Vaccline | Vaccine against two types of HPV (16 and 18) | 10 | Prevention of cervical cancer and other cancers in the reproductivr organs | FDA | |
| 2011 | Sipuleucel-T (Provenge) | Therapeutic Vaccline (autologous cellular immunotherapy) | Bolstering anti prostate cancer adaptive immune response | 10 | Castration resistant prostate cancer | FDA | |
| --′′-- | Lpilimumab (Yervuy) | Immunotherapeutic/Biologic | Immune checkpoint inhibitor of CTLA-4 | 10 | Melanoma [matastatic] | EMA, TGA, FDA | |
| --′′-- | Vemurafenib (Zelboraf) | Chemotherapeutic | Inhibits proliferation without growth factors by inhibiting mutated BRAF serine-threonine kinase | 1 | Advanced melanoma with BRAF V600 mutation | FDA | |
| --′′-- | Brentuximab vedotin (Adcetris) | Drug-kibked biologic | Cytot10oxic ag10ent-linked10 chimer10ic mouse/hum10an anti-huna1n CD301,2 | 1,2 | Hodgkin lymphoma, anaplastic large cell lymphoma; (cutaneous T cell lymphoma, peripheral T cell lymphoma) | FDA (EMA in 2012) | |
| --′′-- | Peginterferon alfa-2b (Sylatron) | Biologic | Cytokine, stimulates killing of tumor cells | 10 | Melanoma | FDA | |
| 2012 | Carfilzomib (Kyprolis) | Chemotherapeutic | Irreversible proteasome inhibitor, cell cycle block, apoptosis | 2 | Relapsed or refractory multiple melanoma | FDA | |
| 2013 | Pomalidomid (Pomalyst) | Chemotherapeutic (Thalidomide analogue) | An immunomoduatory drug, targets, protein cereblon; inhibits COX2, inhibits angiogenesis, induces apoptosis via G1 arrest | 1,2,8 | Relapsed or refractory multiple melanoma | FDA | |
| 2014 | Blinatumomab (Blincyto) | Biologic | moAb, a bispecific T-cell engager (BiTE); CD 19 poditive cancers are killed by cytotoxic T cells | 10 | B cell acute lymphoblastic leukemia [ALL] | FDA | |
| --′′-- | Tisagenlecleucel (Kymriah) | CAR T cell immunotherapy | Targeting the CD 19 receptor on cancer cells | 1,2 | B-ALL, (EMA in 2016, relapsed or refractory diffuse large B cell lymphoma; FDA in 2018) | EMA (FDA in 2017) | |
| --′′-- | Ramucriumab (Cyramaza) | Biologic | moAB that blocks interaction of VEGFR2 with ligands, inhibiting angiogenesis | 4 | Advanced stomach cancer and gastroesophageal junction adenocarcinoma after prior therapy | FDA | |
| --′′-- | Pembrolizumab (Keytruda) and nivolumab (Opdivo) | Immunotherapeutic/Biologic | Immune checkpoint inhibitor of PD-1 | 10 | Not resectable melanoma; with ipilimumab-numerous indications (see in Table 5) | EMA, FDA, TGA and Japan | |
| Year of Approval | Drug (Therapy) | Category | Mode of Action | Targeted Hallmark | First Indications (Current) | ||
|---|---|---|---|---|---|---|---|
| Solid Tumors | Blood Borne | Institution or Country | |||||
| 2015 | Panabinostat (Farydak) | Chemotherapeutic | Histone deacetylase inhibitor, promoting cell death and cell cycle arrest | 2 | Multiple myeloma, relapsed or refractory, in those previously treated with bortezomib and lenalidomide or thalidomide | FDA | |
| --′′-- | Palbociclib (Ibrance) | Chemotherapeutic/small molecules inhibitor | Inhibitor of cyclin-dependent kinase (CDK) 4 and 6 | 1 | With an aromatase inhibitors as initial therapy of postmenopausal, HR-positive, HER2-negative advanced or metastatic breast cancer | FDA | |
| 2017 | Atezolizumab (Tecentriq) | Immunotherapeutic/biologic | Anti-PD-L1 checkponit inhibitor | 10 | Metastatic, chemotherapy-resistant non-small cell lung cancer [NSCLC] | FDA | |
| --′′-- | Olaratumab (Lartruvo) | Immunotherapeutic/biologic | Antibody against the PDGFRα | 1 | Soft tissue sarcoma [STS], provided ineffective surgery and radiation therapy (withdrawn in 2019, EMA and FDA) | EMA, FDA | |
| --′′-- | Gemtuzumab ozogamicin (Mylotarg) | Drug-linked biologic | Anti-CD33 conjugated to toxin | 1,2 | CD33-positive acute myeloid leukermia [AML] | FDA | |
| --′′-- | Durvalumab (Imfinzi) and avelumab (Bavencio) | Immunotherapeutic/biologic | Anti-PD-1/PD-L1 checkpoint inhibitors | 10 | Advanced bladder cancer | FDA | |
| --′′-- | Axicabtagene ciloleucel (Yescarta) | CAR T cell immunotherapy | Targeting the CD19 receptor on cancer cells | 1,2 | Several types non-Hodgkin large B cell lymphomas refractory or twice relapsed | FDA | |
| --′′-- | Ribociclib (Kisqali) | Chemotherapeutic/ small molecules inhibitor | Cyclin dependent kinase inhibitor (CDKi) | 1 | With an aromatase inhibitors as initial therapy of postmenopausal, HR-positive, HER2-negative advanced or metastatic breast cancer | FDA | |
| --′′-- | Abemaciclib (Verzenio) | Chemotherapeutic/ small molecules inhibitor | Inhibitor of cyclin-dependent kinase (CDK) 4 and 6 | 1 | With an aromatase inhibitors as initial therapy of postmenopausal, HR-positive, HER2-negative advanced or metastatic breast cancer | FDA | |
| 2018 | Cemiplimab (Libtayo) | Immunotherapeutic/biologic | moAB, anti-PD-1 checkpoint inhibitor | 10 | Metastatic cutaneous squamous cell carcinoma [CSCC] or lacally advanced CSCC who are not candidates for curative surgery or surative radiation | FDA (EMA in 2019, TGA in 2020) | |
| 2019 | Pexidartinib (Turalio) | Small molecule immunomodulator (chemotherapeutic) | Targeting the cytokine CSF-1 receptor pathway | 1 | Symptomatic tenosynovial giant cell tumor | FDA | |
| --′′-- | Venetoclax (Venclexta, Venclyto) | Chemotherapeutic/ small molecules inhibitor | Targeting Bcl-2 | 2 | CLL, small lymphocytic lymphoma [SLL], AML | FDA (EMA in 2020) | |
| 2020 | Brexucabtagene autoleucel (Tecartus) | CAR T cell immunotherapy | Targeting the CD19 receptor on cancer cells | 1,2 | Relapsed or refractory Mantle cell lymphoma | FDA | |
| --′′-- | Gardasil 9 | Prophylactic vaccine | Anti-HPV (type 6, 11, 16 and 18) | 10 | Head and neck HPV-related cancer prevention | EMA, FDA | |
| Therapy | Mode of Action | Approval | Indications |
|---|---|---|---|
| Ipilimumab (Yervoy) | Inhibitor of CTLA-4 | Since 2011 | Melanoma (metastatic) |
| Nivolumab (Opdivo) | Inhibitor of PD-1 | Since 2014 | (1) surgically inoperative melanoma; |
| (2) relapsed colorectal cancer that is characterized by high microsatellite instability (MSI-hi), | |||
| (3) gastric cancer (The Pharmaceuticals and Medical Devices Agency (PMDA) of Japan), | |||
| (4) advanced liver cancer that has been previously treated with sorafenib; | |||
| Since 2018 | (5) mesothelioma (PMDA); | ||
| Since 2020 | (6) unresectable advanced or recurrent esophageal cancer that has progressed following chemotherapy (PMDA), | ||
| (7) unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma after previous fluoropyrimidine- and platinum-based chemotherapy. | |||
| Pembrolizumab (Keytruda) | Inhibitor of PD-1 | Since 2014 | (1) surgically inoperative melanoma; |
| Since 2017 | (2) advanced non-small cell lung cancer (NSCLC, first line), | ||
| (3) bladder cancer (first line), | |||
| (4) all metastatic solid tumor types classified as MSI-hi (high microsatellite instability) or dMMR (deficient DNA mismatch repair) (second line), | |||
| (5) advanced recurrent cancer of the stomach and gastroesophageal junction; | |||
| Since 2018 | (6) patients with cervical cancer expressing PD-L1 that is metastatic or has recurred after previous chemotherapy treatment, | ||
| (7) adult and pediatric patients with primary mediastinal large B-cell lymphoma (PMBCL) that is refractory or has relapsed after two or more prior systemic treatments, | |||
| (8) advanced, treatment-resistant hepatocellular carcinoma, the most common type of liver cancer; | |||
| Since 2019 | (9) stage III non-small cell lung cancer (NSCLC) that is PD-L1-positive and is not amenable to surgery or chemo-radiation treatment (first-line), | ||
| (10) advanced esophageal squamous cell cancer, | |||
| (11) advanced endometrial carcinoma in patients with disease progression following prior systemic therapy but are ineligible for surgery or radiation, | |||
| Since 2020 | (12) advanced endometrial carcinoma in patients with disease progression following prior systemic therapy but are ineligible for surgery or radiation, | ||
| (13) unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer (first line), | |||
| (14) recurrent or metastatic cutaneous squamous cell carcinoma that is not curable by surgery or radiation, | |||
| (15) unresectable or metastatic tumor mutational burden-high solid tumors, which have progressed and have no satisfactory alternative treatment options. | |||
| Durvalumab (Imfinzi) | anti-PD-L1 inhibitor | Since 2014 | (1) advanced bladder cancer, |
| Since 2018 | (2) unresectable, stage III non-small cell lung cancer (NSCLC) that hasn’t progressed after prior chemo-radiation treatment; | ||
| Since 2020 | (3) extensive-stage small cell lung cancer (ES-SCLC) in combination with standard-of-care chemotherapy (as a first line). | ||
| Avelumab (Bavencio) | a PD-L1 inhibitor | Since 2014 | (1) advanced bladder cancer, |
| Since 2017 | (2) for the treatment of Merkel cell carcinoma (EMA), | ||
| Since 2020 | (3) for maintenance treatment of patients with locally advanced or metastatic urothelial carcinoma that has not progressed with first-line platinum-based chemotherapy. | ||
| Atezolizumab (Tecentriq) | anti-PD-L1 inhibitor | Since 2014 | (1) metastatic, chemotherapy-resistant NSCLC, |
| Since 2019 | (2) unresectable (inoperable) or metastatic triple-negative breast cancer that also expresses PD-L1, (in combination with chemotherapy, as a first line). | ||
| (3) small cell lung cancer-ES-SCLC, (in combination with chemotherapy, as a first line). | |||
| (4) metastatic non-small cell lung cancer-nonsquamous NSCLC without EGFR or ALK molecular aberrations, (in combination with chemotherapy, as a first line). | |||
| Since 2020 | (5) BRAF V600 mutation-positive advanced melanoma (in combination with cobimetinib and vemurafenib). | ||
| Cemiplimab (Libtayo) | anti-PD-1 inhibitor | Since 2018 | cutaneous squamous cell carcinoma, metastatic or locally advanced |
| Nivolumab | inhibitor of PD-1 | Since 2018 | (1) melanoma (PMDA), |
| plus | (2) advanced renal cell carcinoma, the most common form of kidney cancer (FDA, EMA) | ||
| ipilimumab | inhibitor of CTLA-4 | (3) relapsed or refractory colorectal cancer characterized by high microsatellite instability (MSI-hi) or deficient DNA mismatch repair (dMMR) (FDA). | |
| Since 2020 | (4) advanced hepatocellular carcinoma, the most common form of liver cancer, in patients who have previously been treated with sorafenib.(FDA) | ||
| (5) metastatic non-small cell lung cancer (NSCLC) that expresses PD-L1 and does not possess mutations in the EGFR or ALK genes. A triple combination comprising nivolumab, ipilimumab and platinum-doublet chemotherapy was approved (FDA) as a first-line therapy for the same indication including recurrent NSCLC. | |||
| Atezolizumab | anti-PD-L1 inhibitor | Since 2020 | previously untreated hepatocellular carcinoma. |
| plus | |||
| bevacizumab | anti-VEGF Ab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dembic, Z. Antitumor Drugs and Their Targets. Molecules 2020, 25, 5776. https://doi.org/10.3390/molecules25235776
Dembic Z. Antitumor Drugs and Their Targets. Molecules. 2020; 25(23):5776. https://doi.org/10.3390/molecules25235776
Chicago/Turabian StyleDembic, Zlatko. 2020. "Antitumor Drugs and Their Targets" Molecules 25, no. 23: 5776. https://doi.org/10.3390/molecules25235776
APA StyleDembic, Z. (2020). Antitumor Drugs and Their Targets. Molecules, 25(23), 5776. https://doi.org/10.3390/molecules25235776