The Ketamine Antidepressant Story: New Insights

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ketamine as an Antidepressant
3. Emerging Antidepressant Mechanisms
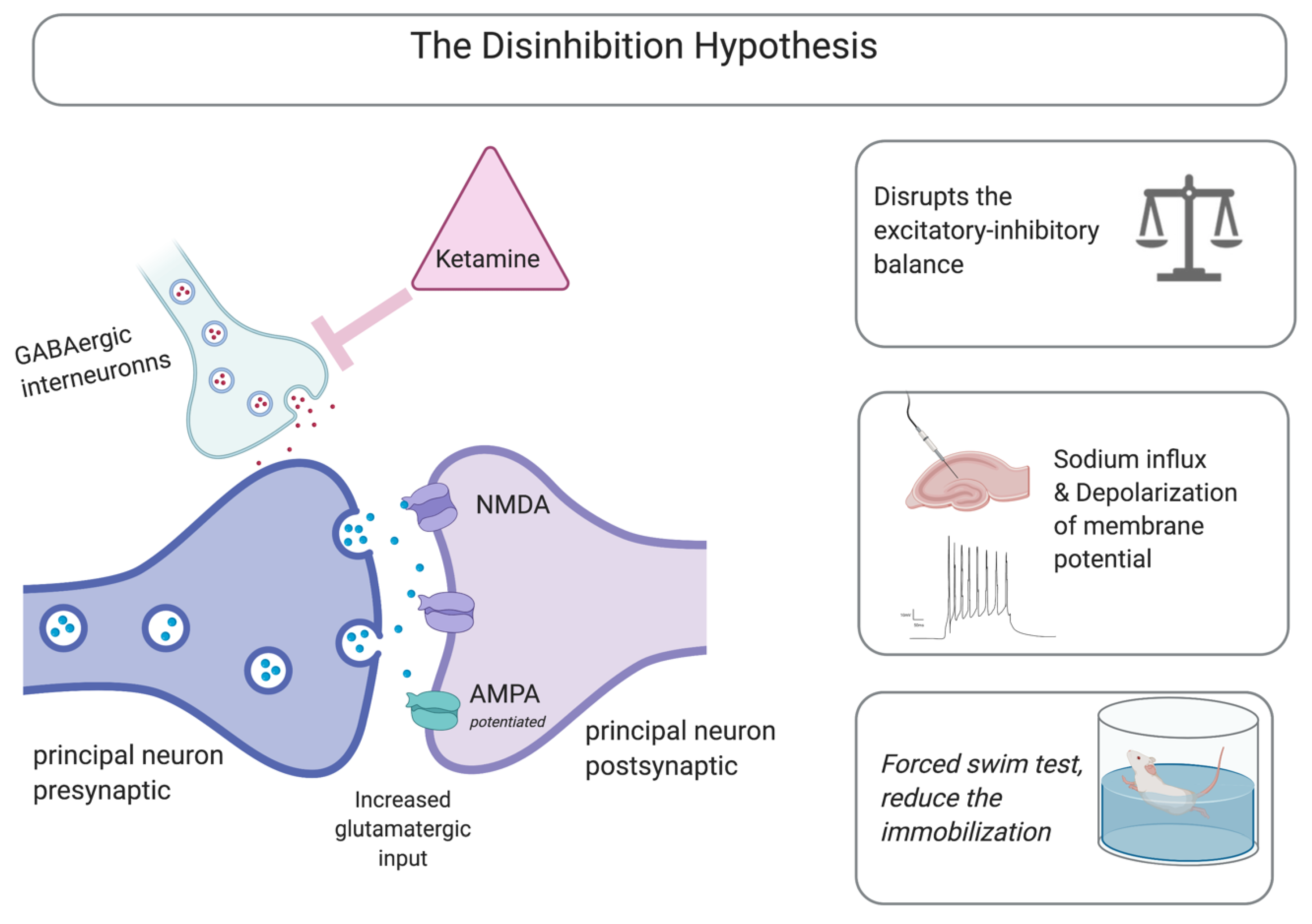
3.1. NMDA Receptors as Mediators
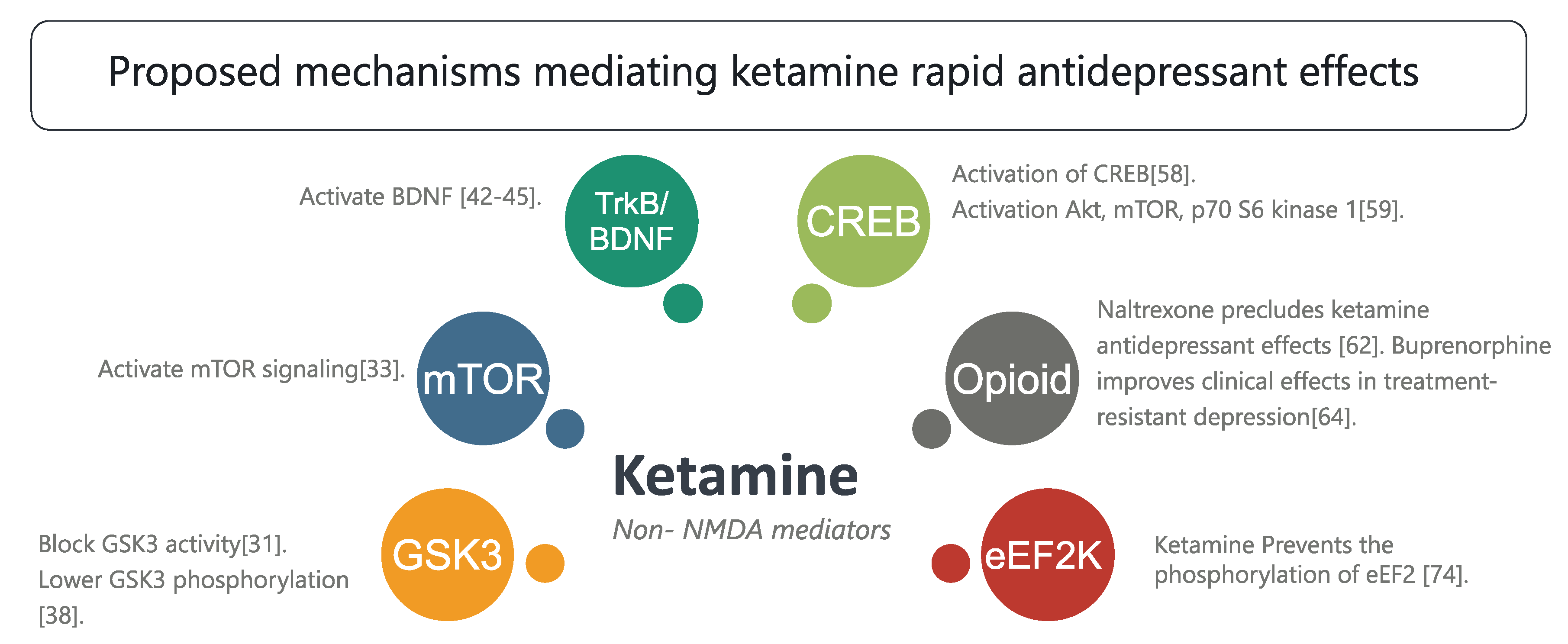
3.2. Non-NMDA Mediators
Neurochemical Cascades and Other Mechanisms
4. Metabolites
5. Conclusions
Funding
Conflicts of Interest
References
- Maddox, V.H.; Godefroi, E.F.; Parcell, R.F. The Synthesis of Phencyclidine and Other 1-Arylcyclohexylamines. J. Med. Chem. 1965, 8, 230–235. [Google Scholar] [CrossRef]
- Domino, E.F.; Luby, E.D. Phencyclidine/schizophrenia: One view toward the past, the other to the future. Schizophr. Bull. 2012, 38, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, D.; Wu, B.; Zhou, W. Ketamine abuse potential and use disorder. Brain Res. Bull. 2016, 126, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Davis, P. Ketamine hydrochloride (Ketalar). Clin. Pharm. Ther. 1970, 11, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Domino, E.F.; Domino, S.E.; Smith, R.E.; Domino, L.E.; Goulet, J.R.; Domino, K.E.; Zsigmond, E.K. Ketamine kinetics in unmedicated and diazepam-premedicated subjects. Clin. Pharm. Ther. 1984, 36, 645–653. [Google Scholar] [CrossRef]
- Corssen, G.; Domino, E.F. Dissociative Anesthesia. Anesth. Analg. 1966, 45, 29–40. [Google Scholar] [CrossRef]
- Pomarol-Clotet, E.; Honey, G.D.; Murray, G.K.; Corlett, P.R.; Absalom, A.R.; Lee, M.; McKenna, P.J.; Bullmore, E.T.; Fletcher, P.C. Psychological effects of ketamine in healthy volunteers. Phenomenological study. Br. J. Psychiatry J. Ment. Sci. 2006, 189, 173–179. [Google Scholar] [CrossRef]
- Wolff, K.; Winstock, A.R. Ketamine. CNS Drugs 2006, 20, 199–218. [Google Scholar] [CrossRef]
- Niesters, M.; Martini, C.; Dahan, A. Ketamine for chronic pain: Risks and benefits. Br. J. Clin. Pharmacol. 2014, 77, 357–367. [Google Scholar] [CrossRef]
- Morgan, C.J.A.; Muetzelfeldt, L.; Curran, H.V. Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: A 1-year longitudinal study. Addiction 2010, 105, 121–133. [Google Scholar] [CrossRef]
- Weiner, A.L.; Vieira, L.; McKay, C.A.; Bayer, M.J. Ketamine abusers presenting to the Emergency Department: A case series. J. Emerg. Med. 2000, 18, 447–451. [Google Scholar] [CrossRef]
- Strayer, R.J.; Nelson, L.S. Adverse events associated with ketamine for procedural sedation in adults. Am. J. Emerg. Med. 2008, 26, 985–1028. [Google Scholar] [CrossRef] [PubMed]
- Van Pelt, L.F. Ketamine and xylazine for surgical anesthesia in rats. J. Am. Vet. Med. Assoc. 1977, 171, 842–844. [Google Scholar] [PubMed]
- Parise, E.M.; Alcantara, L.F.; Warren, B.L.; Wright, K.N.; Hadad, R.; Sial, O.K.; Kroeck, K.G.; Iñiguez, S.D.; Bolaños-Guzmán, C.A. Repeated ketamine exposure induces an enduring resilient phenotype in adolescent and adult rats. Biol. Psychiatry 2013, 74, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Strong, C.E.; Kabbaj, M. On the safety of repeated ketamine infusions for the treatment of depression: Effects of sex and developmental periods. Neurobiol. Stress 2018, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Narendran, R.; Frankle, W.G.; Keefe, R.; Gil, R.; Martinez, D.; Slifstein, M.; Kegeles, L.S.; Talbot, P.S.; Huang, Y.; Hwang, D.R.; et al. Altered prefrontal dopaminergic function in chronic recreational ketamine users. Am. J. Psychiatry 2005, 162, 2352–2359. [Google Scholar] [CrossRef]
- Scott, L.; Kruse, M.S.; Forssberg, H.; Brismar, H.; Greengard, P.; Aperia, A. Selective up-regulation of dopamine D1 receptors in dendritic spines by NMDA receptor activation. Proc. Natl. Acad. Sci. USA 2002, 99, 1661–1664. [Google Scholar] [CrossRef]
- Zimmerman, M.E.; Brickman, A.M.; Paul, R.H.; Grieve, S.M.; Tate, D.F.; Gunstad, J.; Cohen, R.A.; Aloia, M.S.; Williams, L.M.; Clark, C.R.; et al. The Relationship Between Frontal Gray Matter Volume and Cognition Varies Across the Healthy Adult Lifespan. Am. J. Geriatr. Psychiatry 2006, 14, 823–833. [Google Scholar] [CrossRef]
- Liao, Y.; Tang, J.; Corlett, P.R.; Wang, X.; Yang, M.; Chen, H.; Liu, T.; Chen, X.; Hao, W.; Fletcher, P.C. Reduced dorsal prefrontal gray matter after chronic ketamine use. Biol. Psychiatry 2011, 69, 42–48. [Google Scholar] [CrossRef]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Zuccoli, G.S.; Saia-Cereda, V.M.; Nascimento, J.M.; Martins-de-Souza, D. The Energy Metabolism Dysfunction in Psychiatric Disorders Postmortem Brains: Focus on Proteomic Evidence. Front. Neurosci. 2017, 11, 493. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Gorini, A.; Villa, R.F. Affective disorders, antidepressant drugs and brain metabolism. Mol. Psychiatry 2003, 8, 773–785. [Google Scholar] [CrossRef] [PubMed]
- McCormick, L.M.; Boles Ponto, L.L.; Pierson, R.K.; Johnson, H.J.; Magnotta, V.; Brumm, M.C. Metabolic correlates of antidepressant and antipsychotic response in patients with psychotic depression undergoing electroconvulsive therapy. J. ECT 2007, 23, 265–273. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Evans, K.R.; Kruger, S.; Mayberg, H.S.; Meyer, J.H.; McCann, S.; Arifuzzman, A.I.; Houle, S.; Vaccarino, F.J. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am. J. Psychiatry 2001, 158, 899–905. [Google Scholar] [CrossRef]
- Tunc-Ozcan, E.; Peng, C.Y.; Zhu, Y.; Dunlop, S.R.; Contractor, A.; Kessler, J.A. Activating newborn neurons suppresses depression and anxiety-like behaviors. Nat. Commun. 2019, 10, 3768. [Google Scholar] [CrossRef] [PubMed]
- Gass, N.; Becker, R.; Reinwald, J.; Cosa-Linan, A.; Sack, M.; Weber-Fahr, W.; Vollmayr, B.; Sartorius, A. The influence of ketamine’s repeated treatment on brain topology does not suggest an antidepressant efficacy. Transl. Psychiatry 2020, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Ketamine and rapid-acting antidepressants: A new era in the battle against depression and suicide. F1000Research 2018, 7, F1000 Faculty Rev-1659. [Google Scholar] [CrossRef]
- Glasgow, N.G.; Povysheva, N.V.; Azofeifa, A.M.; Johnson, J.W. Memantine and Ketamine Differentially Alter NMDA Receptor Desensitization. J. Neurosci. 2017, 37, 9686–9704. [Google Scholar] [CrossRef]
- Izumi, Y.; Zorumski, C.F. Metaplastic effects of subanesthetic ketamine on CA1 hippocampal function. Neuropharmacology 2014, 86, 273–281. [Google Scholar] [CrossRef]
- Chaki, S. Beyond Ketamine: New Approaches to the Development of Safer Antidepressants. Curr. Neuropharmacol. 2017, 15, 963–976. [Google Scholar] [CrossRef]
- Trullas, R.; Skolnick, P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol. 1990, 185, 1–10. [Google Scholar] [CrossRef]
- Browne, C.A.; Lucki, I. Antidepressant effects of ketamine: Mechanisms underlying fast-acting novel antidepressants. Front. Pharmacol. 2013, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Homayoun, H.; Moghaddam, B. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, D.M.; Pothula, S.; Liu, R.J.; Wu, M.; Li, X.Y.; Girgenti, M.J.; Taylor, S.R.; Duman, C.H.; Delpire, E.; Picciotto, M.; et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J. Clin. Investig. 2020, 130, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, T.K.; Alshammari, M.A.; Nenov, M.N.; Hoxha, E.; Cambiaghi, M.; Marcinno, A.; James, T.F.; Singh, P.; Labate, D.; Li, J.; et al. Genetic deletion of fibroblast growth factor 14 recapitulates phenotypic alterations underlying cognitive impairment associated with schizophrenia. Transl. Psychiatry 2016, 6, e806. [Google Scholar] [CrossRef] [PubMed]
- Di Re, J.; Wadsworth, P.A.; Laezza, F. Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front. Cell Neurosci. 2017, 11, 103. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Widman, A.J.; McMahon, L.L. Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, E3007–E3016. [Google Scholar] [CrossRef]
- Cui, Y.; Hu, S.; Hu, H. Lateral Habenular Burst Firing as a Target of the Rapid Antidepressant Effects of Ketamine. Trends Neurosci. 2019, 42, 179–191. [Google Scholar] [CrossRef]
- Ren, Z.; Pribiag, H.; Jefferson, S.J.; Shorey, M.; Fuchs, T.; Stellwagen, D.; Luscher, B. Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biol. Psychiatry 2016, 80, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Handelman, S.A.; Allyn-Feuer, A.; Ade, A.S.; Burns, J.S.; Omenn, G.S.; Athey, B.D. Ketamine’s pharmacogenomic network in human brain contains sub-networks associated with glutamate neurotransmission and with neuroplasticity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Beurel, E.; Song, L.; Jope, R.S. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol. Psychiatry 2011, 16, 1068–1070. [Google Scholar] [CrossRef]
- Paul, J.R.; DeWoskin, D.; McMeekin, L.J.; Cowell, R.M.; Forger, D.B.; Gamble, K.L. Regulation of persistent sodium currents by glycogen synthase kinase 3 encodes daily rhythms of neuronal excitability. Nat. Commun. 2016, 7, 13470. [Google Scholar] [CrossRef]
- James, T.F.; Nenov, M.N.; Wildburger, N.C.; Lichti, C.F.; Luisi, J.; Vergara, F.; Panova-Electronova, N.I.; Nilsson, C.L.; Rudra, J.S.; Green, T.A.; et al. The Nav1.2 channel is regulated by GSK3. Biochim. Biophys. Acta 2015, 1850, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Scala, F.; Nenov, M.N.; Crofton, E.J.; Singh, A.K.; Folorunso, O.; Zhang, Y.; Chesson, B.C.; Wildburger, N.C.; James, T.F.; Alshammari, M.A.; et al. Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway. Cell Rep. 2018, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Cade, J.F. Lithium salts in the treatment of psychotic excitement. Med. J. Aust. 1949, 2, 349–352. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.T.; Harper, A.D.; Jové, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef]
- Peng, H.; Wang, H.B.; Wang, L.; Zhou, B.; Li, X.Y.; Tan, J. Gsk3β aggravates the depression symptoms in chronic stress mouse model. J. Integr. Neurosci. 2018, 17, 169–175. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Machado-Vieira, R. GSK-3: A key regulatory target for ketamine’s rapid antidepressant effects mediated by enhanced AMPA to NMDA throughput. Bipolar Disord. 2016, 18, 702–705. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Wang, W.; Pan, B.; Liu, X.; Zhang, Z.; Long, J.Z.; Zhang, H.T.; Cravatt, B.F.; Liu, Q.S. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014, 39, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lao, L.; Cui, W.; Rong, J. Potential link between the RagA-mTOR-p70S6K axis and depressive-behaviors during bacterial liposaccharide challenge. J. Neuroinflamm. 2019, 16, 211. [Google Scholar] [CrossRef]
- Yang, C.; Shirayama, Y.; Zhang, J.c.; Ren, Q.; Yao, W.; Ma, M.; Dong, C.; Hashimoto, K. R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 2015, 5, e632. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.-F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Carreno, F.R.; Donegan, J.J.; Boley, A.M.; Shah, A.; DeGuzman, M.; Frazer, A.; Lodge, D.J. Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol. Psychiatry 2016, 21, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Kohtala, S.; Theilmann, W.; Rosenholm, M.; Müller, H.K.; Kiuru, P.; Wegener, G.; Yli-Kauhaluoma, J.; Rantamäki, T. Ketamine-induced regulation of TrkB-GSK3β signaling is accompanied by slow EEG oscillations and sedation but is independent of hydroxynorketamine metabolites. Neuropharmacology 2019, 157, 107684. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Luo, X.; Hua, D.; Wang, Y.; Zhan, G.; Huang, N.; Jiang, R.; Yang, L.; Zhu, B.; Yuan, X.; et al. Ketamine Alleviates Postoperative Depression-Like Symptoms in Susceptible Mice: The Role of BDNF-TrkB Signaling. Front. Pharmacol. 2020, 10, 1702. [Google Scholar] [CrossRef]
- Lepack, A.E.; Bang, E.; Lee, B.; Dwyer, J.M.; Duman, R.S. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 2016, 111, 242–252. [Google Scholar] [CrossRef]
- Ma, Z.; Zang, T.; Birnbaum, S.G.; Wang, Z.; Johnson, J.E.; Zhang, C.-L.; Parada, L.F. TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat. Commun. 2017, 8, 1668. [Google Scholar] [CrossRef]
- Alshammari, M.A.; Alshammari, T.K.; Nenov, M.N.; Scala, F.; Laezza, F. Fibroblast Growth Factor 14 Modulates the Neurogenesis of Granule Neurons in the Adult Dentate Gyrus. Mol. Neurobiol. 2016, 53, 7254–7270. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.S.; Sahay, A.; Hen, R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology 2015, 40, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Abrous, D.N.; Wojtowicz, J.M. Interaction between Neurogenesis and Hippocampal Memory System: New Vistas. Cold Spring Harbor Perspect. Biol. 2015, 7, a018952. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hikosaka, O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 2007, 447, 1111–1115. [Google Scholar] [CrossRef]
- Belujon, P.; Grace, A.A. Dopamine System Dysregulation in Major Depressive Disorders. Int. J. Neuropsychopharmacol. 2017, 20, 1036–1046. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Yin, J.; Fu, B.; Wang, Y.; Yu, T. Effects of ketamine on voltage-gated sodium channels in the barrel cortex and the ventral posteromedial nucleus slices of rats. Neuroreport 2019, 30, 1197–1204. [Google Scholar] [CrossRef]
- Payandeh, J. Progress in understanding slow inactivation speeds up. J. Gen. Physiol. 2018, 150, 1235–1238. [Google Scholar] [CrossRef]
- Yamamura, T.; Okamoto, Y.; Okada, G.; Takaishi, Y.; Takamura, M.; Mantani, A.; Kurata, A.; Otagaki, Y.; Yamashita, H.; Yamawaki, S. Association of thalamic hyperactivity with treatment-resistant depression and poor response in early treatment for major depression: A resting-state fMRI study using fractional amplitude of low-frequency fluctuations. Transl. Psychiatry 2016, 6, e754. [Google Scholar] [CrossRef]
- Wray, N.H.; Schappi, J.M.; Singh, H.; Senese, N.B.; Rasenick, M.M. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 2019, 24, 1833–1843. [Google Scholar] [CrossRef]
- Suzuki, A.; Murakami, K.; Tajima, Y.; Hara, H.; Kunugi, A.; Kimura, H. TAK-137, an AMPA receptor potentiator with little agonistic effect, produces antidepressant-like effect without causing psychotomimetic effects in rats. Pharmacol. Biochem. Behav. 2019, 183, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.M.; Maldonado-Aviles, J.G.; Lepack, A.E.; DiLeone, R.J.; Duman, R.S. Ribosomal protein S6 kinase 1 signaling in prefrontal cortex controls depressive behavior. Proc. Natl. Acad. Sci. USA 2015, 112, 6188–6193. [Google Scholar] [CrossRef] [PubMed]
- Matveychuk, D.; Thomas, R.K.; Swainson, J.; Khullar, A.; MacKay, M.; Baker, G.; Dursun, S. Ketamine as an antidepressant: Overview of its mechanisms of action and potential predictive biomarkers. Ther. Adv. Psychopharmacol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.R.; Heifets, B.D.; Bentzley, B.S.; Blasey, C.; Sudheimer, K.D.; Hawkins, J.; Lyons, D.M.; Schatzberg, A.F. Attenuation of antidepressant and antisuicidal effects of ketamine by opioid receptor antagonism. Mol. Psychiatry 2019, 24, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- McKendrick, G.; Garrett, H.; Jones, H.E.; McDevitt, D.S.; Sharma, S.; Silberman, Y.; Graziane, N.M. Ketamine Blocks Morphine-Induced Conditioned Place Preference and Anxiety-Like Behaviors in Mice. Front. Behav. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Peckham, A.M.; De La Cruz, A.; Dufresne, R.L. Kappa opioid receptor antagonism: Are opioids the answer for treatment resistant depression? Ment. Health Clin. 2018, 8, 175–183. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zhao, J.; Li, L.; Wang, Y.; Zhang, Y.; Li, Y.; Chen, Y.; Liu, W.; Gao, L. Administration of Ketamine Causes Autophagy and Apoptosis in the Rat Fetal Hippocampus and in PC12 Cells. Front. Cell. Neurosci. 2018, 12, 21. [Google Scholar] [CrossRef]
- Gassen, N.C.; Rein, T. Is There a Role of Autophagy in Depression and Antidepressant Action? Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef]
- Bosnjak, Z.J.; Yan, Y.; Canfield, S.; Muravyeva, M.Y.; Kikuchi, C.; Wells, C.W.; Corbett, J.A.; Bai, X. Ketamine induces toxicity in human neurons differentiated from embryonic stem cells via mitochondrial apoptosis pathway. Curr. Drug Saf. 2012, 7, 106–119. [Google Scholar] [CrossRef]
- Lundberg, M.; Curbo, S.; Bohman, H.; Agartz, I.; Ogren, S.O.; Patrone, C.; Mansouri, S. Clozapine protects adult neural stem cells from ketamine-induced cell death in correlation with decreased apoptosis and autophagy. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-L.; Chen, T.-L.; Chen, R.-M. Mechanisms of ketamine-induced immunosuppression. Acta Anaesthesiol. Taiwanica 2012, 50, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.; Duan, M.; Zhou, Z.; Lin, N.; Wang, Z.; Sun, J.; Xu, J. Large dose ketamine inhibits lipopolysaccharide-induced acute lung injury in rats. Inflamm. Res. 2005, 54, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-W.; He, G.-N.; Ma, H.; Wang, J.-K. Ketamine reduces inducible superoxide generation in human neutrophils in vitro by modulating the p38 mitogen-activated protein kinase (MAPK)-mediated pathway. Clin. Exp. Immunol. 2010, 160, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Reus, G.Z.; Vieira, F.G.; Abelaira, H.M.; Michels, M.; Tomaz, D.B.; dos Santos, M.A.; Carlessi, A.S.; Neotti, M.V.; Matias, B.I.; Luz, J.R.; et al. MAPK signaling correlates with the antidepressant effects of ketamine. J. Psychiatr. Res. 2014, 55, 15–21. [Google Scholar] [CrossRef]
- Malemud, C.J.; Miller, A.H. Pro-inflammatory cytokine-induced SAPK/MAPK and JAK/STAT in rheumatoid arthritis and the new anti-depression drugs. Expert. Opin. Ther. Targets 2008, 12, 171–183. [Google Scholar] [CrossRef]
- Bowman, M.A.; Vitela, M.; Clarke, K.M.; Koek, W.; Daws, L.C. Serotonin Transporter and Plasma Membrane Monoamine Transporter Are Necessary for the Antidepressant-Like Effects of Ketamine in Mice. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Gideons, E.; Kavalali, E.T. The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol. Psychiatry 2013, 73, 1199–1203. [Google Scholar] [CrossRef]
- Gideons, E.S.; Kavalali, E.T.; Monteggia, L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA 2014, 111, 8649–8654. [Google Scholar] [CrossRef]
- Czeh, B.; Nagy, S.A. Clinical Findings Documenting Cellular and Molecular Abnormalities of Glia in Depressive Disorders. Front. Mol. Neurosci. 2018, 11, 56. [Google Scholar] [CrossRef]
- Oliveira, J.F.; Gomes, C.A.; Vaz, S.H.; Sousa, N.; Pinto, L. Editorial: Glial Plasticity in Depression. Front. Cell Neurosci. 2016, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.L.; Abdul-Khaliq, H.; Sacher, J.; Steiner, J.; Blasig, I.E.; Mueller, K. Mood disorders are glial disorders: Evidence from in vivo studies. Cardiovasc. Psychiatry Neurol. 2010, 2010, 780645. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Price, J.L.; Simpson, J.R., Jr.; Todd, R.D.; Reich, T.; Vannier, M.; Raichle, M.E. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997, 386, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Gittins, R.A.; Harrison, P.J. A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J. Affect. Disord. 2011, 133, 328–332. [Google Scholar] [CrossRef]
- Altshuler, L.L.; Abulseoud, O.A.; Foland-Ross, L.; Bartzokis, G.; Chang, S.; Mintz, J.; Hellemann, G.; Vinters, H.V. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar. Disord 2010, 12, 541–549. [Google Scholar] [CrossRef]
- Rial, D.; Lemos, C.; Pinheiro, H.; Duarte, J.M.; Gonçalves, F.Q.; Real, J.I.; Prediger, R.D.; Gonçalves, N.; Gomes, C.A.; Canas, P.M.; et al. Depression as a Glial-Based Synaptic Dysfunction. Front. Cell Neurosci. 2016, 9, 521. [Google Scholar] [CrossRef]
- Croft, W.; Dobson, K.L.; Bellamy, T.C. Plasticity of Neuron-Glial Transmission: Equipping Glia for Long-Term Integration of Network Activity. Neural Plast 2015, 2015, 765792. [Google Scholar] [CrossRef]
- Eyo, U.B.; Wu, L.J. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013, 2013, 456857. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Thrane, A.S.; Rangroo Thrane, V.; Zeppenfeld, D.; Lou, N.; Xu, Q.; Nagelhus, E.A.; Nedergaard, M. General anesthesia selectively disrupts astrocyte calcium signaling in the awake mouse cortex. Proc. Natl. Acad. Sci. USA 2012, 109, 18974–18979. [Google Scholar] [CrossRef]
- Stenovec, M.; Božić, M.; Pirnat, S.; Zorec, R. Astroglial Mechanisms of Ketamine Action Include Reduced Mobility of Kir4.1-Carrying Vesicles. Neurochem. Res. 2020, 45, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ren, Q.; Qu, Y.; Zhang, J.C.; Ma, M.; Dong, C.; Hashimoto, K. Mechanistic Target of Rapamycin-Independent Antidepressant Effects of (R)-Ketamine in a Social Defeat Stress Model. Biol. Psychiatry 2018, 83, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Li, S.X.; Hashimoto, K. R (−)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharm. Biochem. Behav. 2014, 116, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, T.K. The Ketamine Antidepressant Story: New Insights. Molecules 2020, 25, 5777. https://doi.org/10.3390/molecules25235777
Alshammari TK. The Ketamine Antidepressant Story: New Insights. Molecules. 2020; 25(23):5777. https://doi.org/10.3390/molecules25235777
Chicago/Turabian StyleAlshammari, Tahani K. 2020. "The Ketamine Antidepressant Story: New Insights" Molecules 25, no. 23: 5777. https://doi.org/10.3390/molecules25235777
APA StyleAlshammari, T. K. (2020). The Ketamine Antidepressant Story: New Insights. Molecules, 25(23), 5777. https://doi.org/10.3390/molecules25235777