
Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7

 ,
,  ,
, 
 ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Results
2.1. Structure-Based Virtual Screening (SBVS)
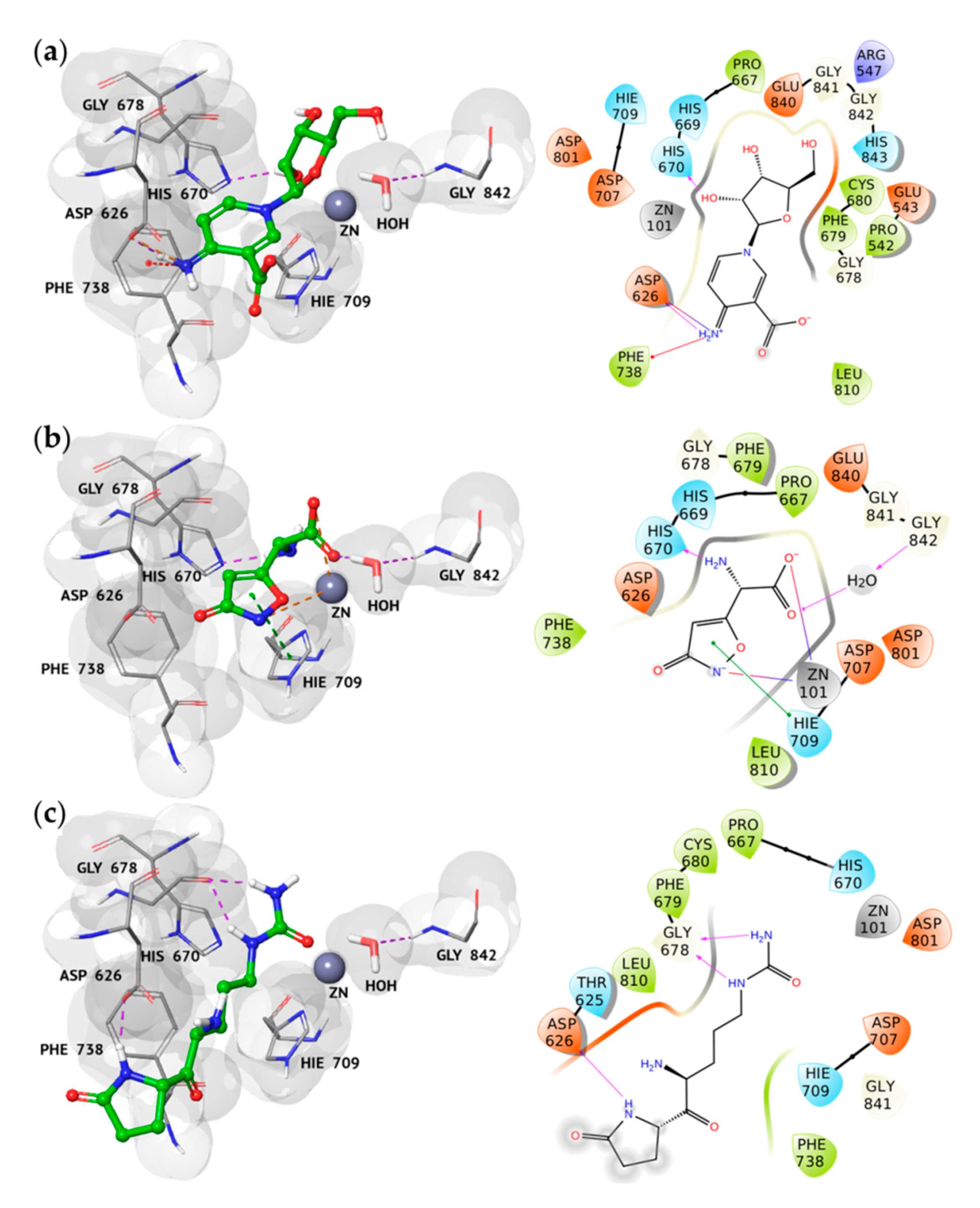
2.2. Docking Analysis of the Best Hits
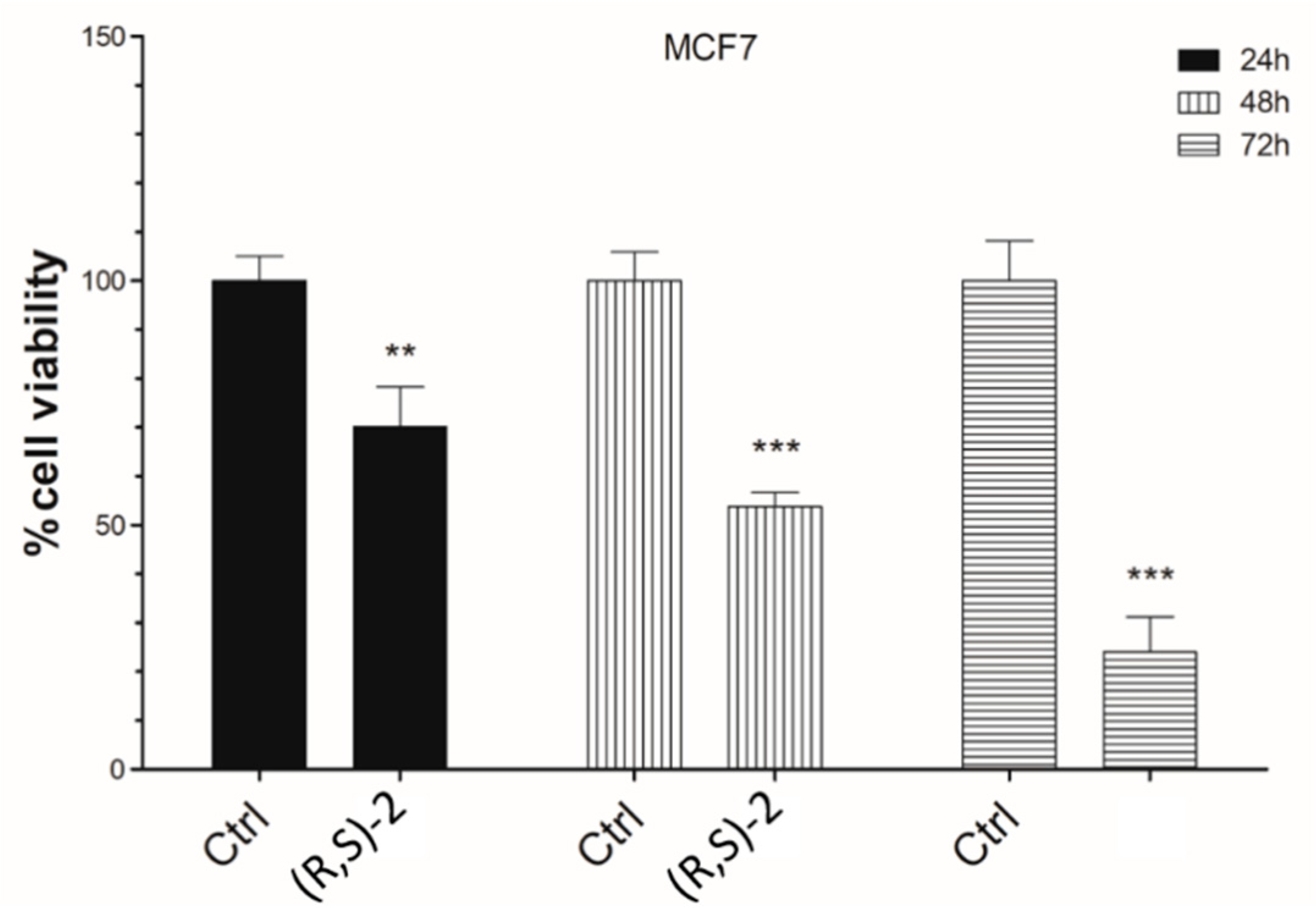
2.3. Cell Viability Assay
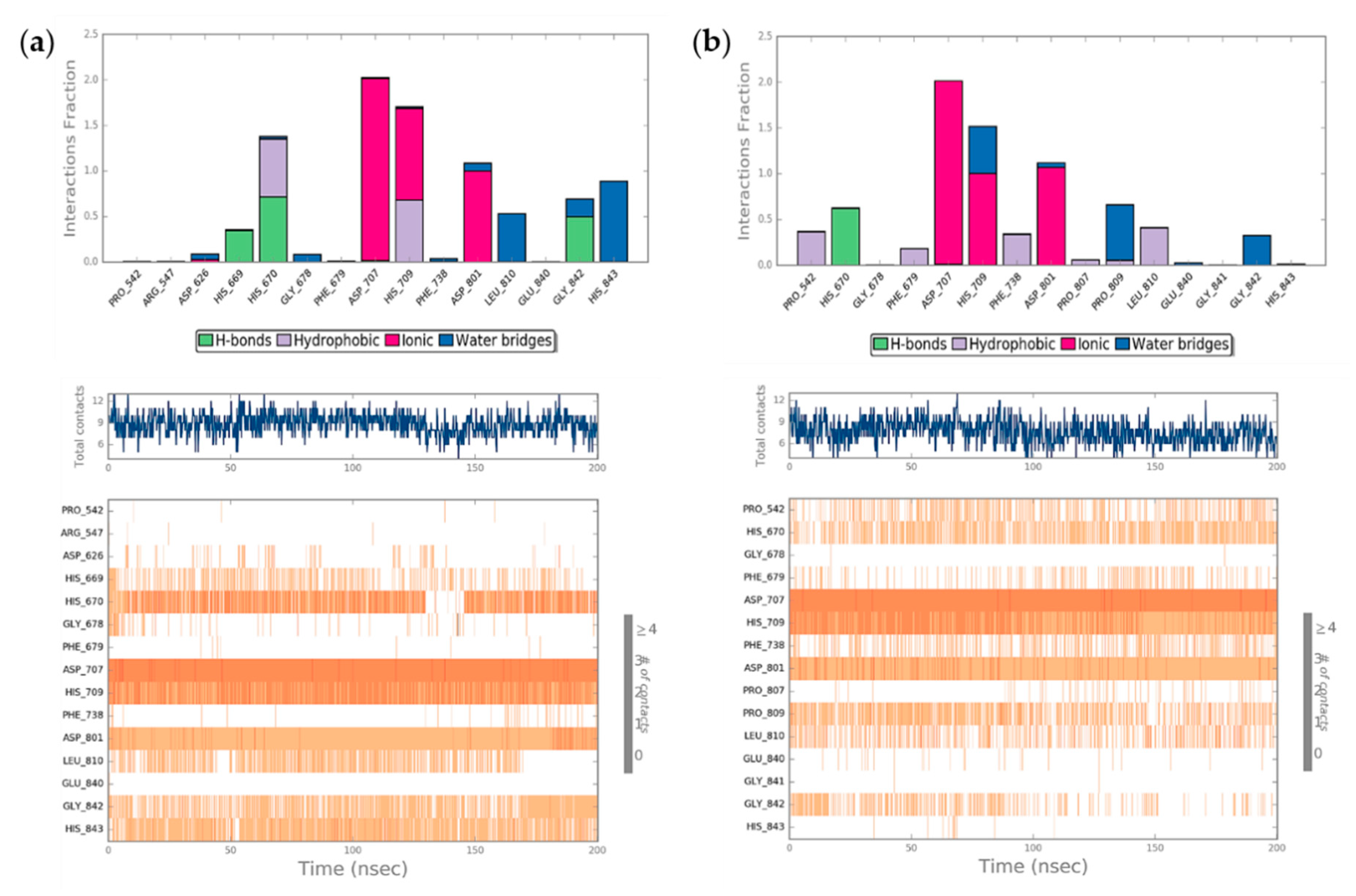
2.4. Molecular Dynamics Simulation (MDs)
3. Discussion
4. Materials and Methods
4.1. Database Preparation
4.2. Receptor Preparation
4.3. Docking Simulations
4.4. Molecular Dynamics Simulations (MDs)
4.5. Cell Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, L.; Yang, X.B. Harnessing the HDAC-histone deacetylase enzymes, inhibitors and how these can be utilised in tissue engineering. Int. J. Oral Sci. 2019, 11, 20. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef]
- Ververis, K.; Karagiannis, T.C. Overview of the Classical Histone Deacetylase Enzymes and Histone Deacetylase Inhibitors. ISRN Cell Biol. 2012, 2012, 130360. [Google Scholar] [CrossRef]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. Epigenetic dysregulation in cancer. Am. J. Pathol. 2009, 175, 1353–1361. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran Biomed. J. 2016, 20, 246–258. [Google Scholar] [PubMed]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The Search for Potent, Small-Molecule HDACIs in Cancer Treatment: A Decade After Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef]
- Zhao, C.; Dong, H.; Xu, Q.; Zhang, Y. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017-present). Expert Opin. Ther. Pat. 2020, 30, 263–274. [Google Scholar] [CrossRef]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef]
- Yeung, A.; Bhargava, R.K.; Ahn, R.; Bahna, S.; Kang, N.H.; Lacoul, A.; Niles, L.P. HDAC inhibitor M344 suppresses MCF-7 breast cancer cell proliferation. Biomed. Pharmacother. 2012, 66, 232–236. [Google Scholar] [CrossRef]
- Chiu, C.F.; Chin, H.K.; Huang, W.J.; Bai, L.Y.; Huang, H.Y.; Weng, J.R. Induction of Apoptosis and Autophagy in Breast Cancer Cells by a Novel HDAC8 Inhibitor. Biomolecules 2019, 9, 824. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, M.Y.; Kim, T.H.; Yoon, S.; Kang, K.W.; Lee, J.; Moon, H.R.; Jung, J.H.; Chung, H.Y.; Kim, H.S. A new synthetic HDAC inhibitor, MHY218, induces apoptosis or autophagy-related cell death in tamoxifen-resistant MCF-7 breast cancer cells. Investig. New Drugs 2012, 30, 1887–1898. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Pellati, F.; Pinzi, L.; Gamberini, M.C. Repositioning Natural Products in Drug Discovery. Molecules 2020, 25, 1154. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, S.H.; Barton, C.L. Repurposing Strategies for Therapeutics. Pharm. Med. 2010, 24, 151–159. [Google Scholar] [CrossRef]
- Maruca, A.; Catalano, R.; Bagetta, D.; Mesiti, F.; Ambrosio, F.A.; Romeo, I.; Moraca, F.; Rocca, R.; Ortuso, F.; Artese, A.; et al. The Mediterranean Diet as source of bioactive compounds with multi-targeting anti-cancer profile. Eur. J. Med. Chem. 2019, 181, 111579. [Google Scholar] [CrossRef] [PubMed]
- Bagetta, D.; Maruca, A.; Lupia, A.; Mesiti, F.; Catalano, R.; Romeo, I.; Moraca, F.; Ambrosio, F.A.; Costa, G.; Artese, A.; et al. Mediterranean products as promising source of multi-target agents in the treatment of metabolic syndrome. Eur. J. Med. Chem. 2020, 186, 111903. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Byun, M.R.; Lee, D.H.; Jang, Y.P.; Lee, H.S.; Choi, J.W.; Lee, S.K. Repurposing natural products as novel HDAC inhibitors by comparative analysis of gene expression profiles. Phytomedicine 2019, 59, 152900. [Google Scholar] [CrossRef]
- Abdalla, M.A. Medicinal significance of naturally occurring cyclotetrapeptides. J. Nat. Med. 2016, 70, 708–720. [Google Scholar] [CrossRef]
- Catalano, R.; Rocca, R.; Juli, G.; Costa, G.; Maruca, A.; Artese, A.; Caracciolo, D.; Tagliaferri, P.; Alcaro, S.; Tassone, P.; et al. A drug repurposing screening reveals a novel epigenetic activity of hydroxychloroquine. Eur. J. Med. Chem. 2019, 183, 111715. [Google Scholar] [CrossRef] [PubMed]
- Maruca, A.; Ambrosio, F.A.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Costa, G.; et al. Computer-based techniques for lead identification and optimization I: Basics. Phys. Sci. Rev. 2019, 4, 113. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shah, S. Structure-Based Virtual Screening. Methods Mol. Biol. 2017, 1558, 111–124. [Google Scholar] [PubMed]
- Lupia, A.; Moraca, F.; Bagetta, D.; Maruca, A.; Ambrosio, F.; Rocca, R.; Catalano, R.; Romeo, I.; Talarico, C.; Ortuso, F.; et al. Computer-based techniques for lead identification and optimization II: Advanced search methods. Phys. Sci. Rev. 2020, 5, 20180114. [Google Scholar]
- Costa, G.; Maruca, A.; Rocca, R.; Ambrosio, F.A.; Berrino, E.; Carta, F.; Mesiti, F.; Salatino, A.; Lanzillotta, D.; Trapasso, F.; et al. In Silico Identification and Biological Evaluation of Antioxidant Food Components Endowed with IX and XII hCA Inhibition. Antioxidants 2020, 9, 775. [Google Scholar] [CrossRef]
- Maruca, A.; Lanzillotta, D.; Rocca, R.; Lupia, A.; Costa, G.; Catalano, R.; Moraca, F.; Gaudio, E.; Ortuso, F.; Artese, A.; et al. Multi-Targeting Bioactive Compounds Extracted from Essential Oils as Kinase Inhibitors. Molecules 2020, 25, 2174. [Google Scholar] [CrossRef]
- Costa, G.; Carta, F.; Ambrosio, F.A.; Artese, A.; Ortuso, F.; Moraca, F.; Rocca, R.; Romeo, I.; Lupia, A.; Maruca, A.; et al. A computer-assisted discovery of novel potential anti-obesity compounds as selective carbonic anhydrase VA inhibitors. Eur. J. Med. Chem. 2019, 181, 111565. [Google Scholar] [CrossRef]
- Catalano, R.; Moraca, F.; Amato, J.; Cristofari, C.; Rigo, R.; Via, L.D.; Rocca, R.; Lupia, A.; Maruca, A.; Costa, G.; et al. Targeting multiple G-quadruplex-forming DNA sequences: Design, biophysical and biological evaluations of indolo-naphthyridine scaffold derivatives. Eur. J. Med. Chem. 2019, 182, 111627. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Talarico, C.; Ortuso, F.; Da Ros, S.; Nicoletto, G.; Sissi, C.; Alcaro, S.; Artese, A. In Silico Identification of Piperidinyl-amine Derivatives as Novel Dual Binders of Oncogene c-myc/c-Kit G-quadruplexes. ACS Med. Chem. Lett. 2018, 9, 848–853. [Google Scholar] [CrossRef]
- Rocca, R.; Talarico, C.; Moraca, F.; Costa, G.; Romeo, I.; Ortuso, F.; Alcaro, S.; Artese, A. Molecular recognition of a carboxy pyridostatin toward G-quadruplex structures: Why does it prefer RNA? Chem. Biol. Drug Des. 2017, 90, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Rocca, R.; Moraca, F.; Costa, G.; Nadai, M.; Scalabrin, M.; Talarico, C.; Distinto, S.; Maccioni, E.; Ortuso, F.; Artese, A.; et al. Identification of G-quadruplex DNA/RNA binders: Structure-based virtual screening and biophysical characterization. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Rocca, R.; Moraca, F.; Talarico, C.; Romeo, I.; Ortuso, F.; Alcaro, S.; Artese, A. A Comparative Docking Strategy to Identify Polyphenolic Derivatives as Promising Antineoplastic Binders of G-quadruplex DNA c-myc and bcl-2 Sequences. Mol. Inform. 2016, 35, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Rocca, R.; Costa, G.; Artese, A.; Parrotta, L.; Ortuso, F.; Maccioni, E.; Pinato, O.; Greco, M.L.; Sissi, C.; Alcaro, S.; et al. Hit Identification of a Novel Dual Binder for h-telo/c-myc G-Quadruplex by a Combination of Pharmacophore Structure-Based Virtual Screening and Docking Refinement. ChemMedChem 2016, 11, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Gagliardi, M.G.; Ambrosio, F.A.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Ambrosio, F.A.; Maleddu, R.; Costa, G.; Rocca, R.; Maccioni, E.; Catalano, R.; Romeo, I.; Eleftheriou, P.; Karia, D.C.; et al. Chromenone derivatives as a versatile scaffold with dual mode of inhibition of HIV-1 reverse transcriptase-associated Ribonuclease H function and integrase activity. Eur. J. Med. Chem. 2019, 182, 111617. [Google Scholar] [CrossRef]
- Maruca, A.; Moraca, F.; Rocca, R.; Molisani, F.; Alcaro, F.; Gidaro, M.C.; Alcaro, S.; Costa, G.; Ortuso, F. Chemoinformatic Database Building and in Silico Hit-Identification of Potential Multi-Targeting Bioactive Compounds Extracted from Mushroom Species. Molecules 2017, 22, 1571. [Google Scholar] [CrossRef]
- Schuetz, A.; Min, J.; Allali-Hassani, A.; Schapira, M.; Shuen, M.; Loppnau, P.; Mazitschek, R.; Kwiatkowski, N.P.; Lewis, T.A.; Maglathin, R.L.; et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2018, 283, 11355–11363. [Google Scholar] [CrossRef]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Aronson, J.K. Old drugs—New uses. Br. J. Clin. Pharmacol. 2007, 64, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Update 2020, 53, 100721. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, S.L. Drug repurposing strategies for COVID-19. Future Drug Discov. 2020, 2, FDD40. [Google Scholar] [CrossRef]
- Breene, W.M. Nutritional and Medicinal Value of Specialty Mushrooms. J. Food Prot. 1990, 53, 883–894. [Google Scholar] [CrossRef]
- Kosanić, M.; Ranković, B.; Rancic, A.; Stanojkovic, T. Evaluation of metal contents and bioactivity of two edible mushrooms Agaricus campestris and Boletus edulis. Emir. J. Food Agric. 2017, 29, 98–103. [Google Scholar] [CrossRef]
- Li, S.; Chen, G.; Bi, Y. Studies on antioxidative and Antitumor Activities for Two Wild Edible Fungi. Edible Fungi China 2005, 3, 58–63. [Google Scholar]
- Zhang, J.J.; Li, Y.; Zhou, T.; Xu, D.P.; Zhang, P.; Li, S.; Li, H.B. Bioactivities and Health Benefits of Mushrooms Mainly from China. Molecules 2016, 21, 938. [Google Scholar] [CrossRef]
- Li, C.; Oberlies, N.H. The most widely recognized mushroom: Chemistry of the genus Amanita. Life Sci. 2005, 78, 532–538. [Google Scholar] [CrossRef]
- Lee, M.R.; Dukan, E.; Milne, I. Amanita muscaria (fly agaric): From a shamanistic hallucinogen to the search for acetylcholine. J. R. Coll. Physicians Edinb. 2018, 48, 85–91. [Google Scholar] [CrossRef]
- Jarrard, L.E. On the use of ibotenic acid to lesion selectively different components of the hippocampal formation. J. Neurosci. Methods 1989, 29, 251–259. [Google Scholar] [CrossRef]
- Ito, Y.; Ito, M.; Takagi, N.; Saito, H.; Ishige, K. Neurotoxicity induced by amyloid β-peptide and ibotenic acid in organotypic hippocampal cultures: Protection by S-allyl-l-cysteine, a garlic compound. Brain Res. 2003, 985, 98–107. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Maranhao, R.C.; Vital, C.G.; Tavoni, T.M.; Graziani, S.R. Clinical experience with drug delivery systems as tools to decrease the toxicity of anticancer chemotherapeutic agents. Expert Opin. Drug Deliv. 2017, 14, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 24 August 2020).
- Available online: https://www.ebi.ac.uk/chembl/ (accessed on 24 August 2020).
- Available online: http://www.chemspider.com/ (accessed on 24 August 2020).
- Available online: https://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 24 August 2020).
- Schrödinger, LigPrep, Schrödinger; LLC: New York, NY, USA, 2018.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Available online: http://zinc15.docking.org/ (accessed on 24 August 2020).
- Available online: www.rcsb.org (accessed on 24 August 2020).
- Schrödinger, Epik, Schrödinger; LLC: New York, NY, USA, 2016.
- Schrödinger, Protein Preparation Wizard, Schrödinger; LLC: New York, NY, USA, 2018.
- Schrödinger, Impact, Schrödinger; LLC: New York, NY, USA, 2016.
- Schrödinger-Glide; LLC: New York, NY, USA, 2018.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, SC ‘06. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Hit | Name | 2D Structure | G-Score (kcal/mol) |
|---|---|---|---|
| 1 | Clitidine |  | −8.73 |
| (S)-2 | (S)-Ibotenic acid |  | −8.60 |
| 3 | Pyroglutamylcitrulline |  | −8.91 |
| 4 | Trichostatin A |  | −8.41 |
Sample Availability: Samples of the compounds not are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maruca, A.; Rocca, R.; Catalano, R.; Mesiti, F.; Costa, G.; Lanzillotta, D.; Salatino, A.; Ortuso, F.; Trapasso, F.; Alcaro, S.; et al. Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7. Molecules 2020, 25, 5524. https://doi.org/10.3390/molecules25235524
Maruca A, Rocca R, Catalano R, Mesiti F, Costa G, Lanzillotta D, Salatino A, Ortuso F, Trapasso F, Alcaro S, et al. Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7. Molecules. 2020; 25(23):5524. https://doi.org/10.3390/molecules25235524
Chicago/Turabian StyleMaruca, Annalisa, Roberta Rocca, Raffaella Catalano, Francesco Mesiti, Giosuè Costa, Delia Lanzillotta, Alessandro Salatino, Francesco Ortuso, Francesco Trapasso, Stefano Alcaro, and et al. 2020. "Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7" Molecules 25, no. 23: 5524. https://doi.org/10.3390/molecules25235524
APA StyleMaruca, A., Rocca, R., Catalano, R., Mesiti, F., Costa, G., Lanzillotta, D., Salatino, A., Ortuso, F., Trapasso, F., Alcaro, S., & Artese, A. (2020). Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7. Molecules, 25(23), 5524. https://doi.org/10.3390/molecules25235524