
Chagas Disease: Perspectives on the Past and Present and Challenges in Drug Discovery

 ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
1.1. Current Drugs
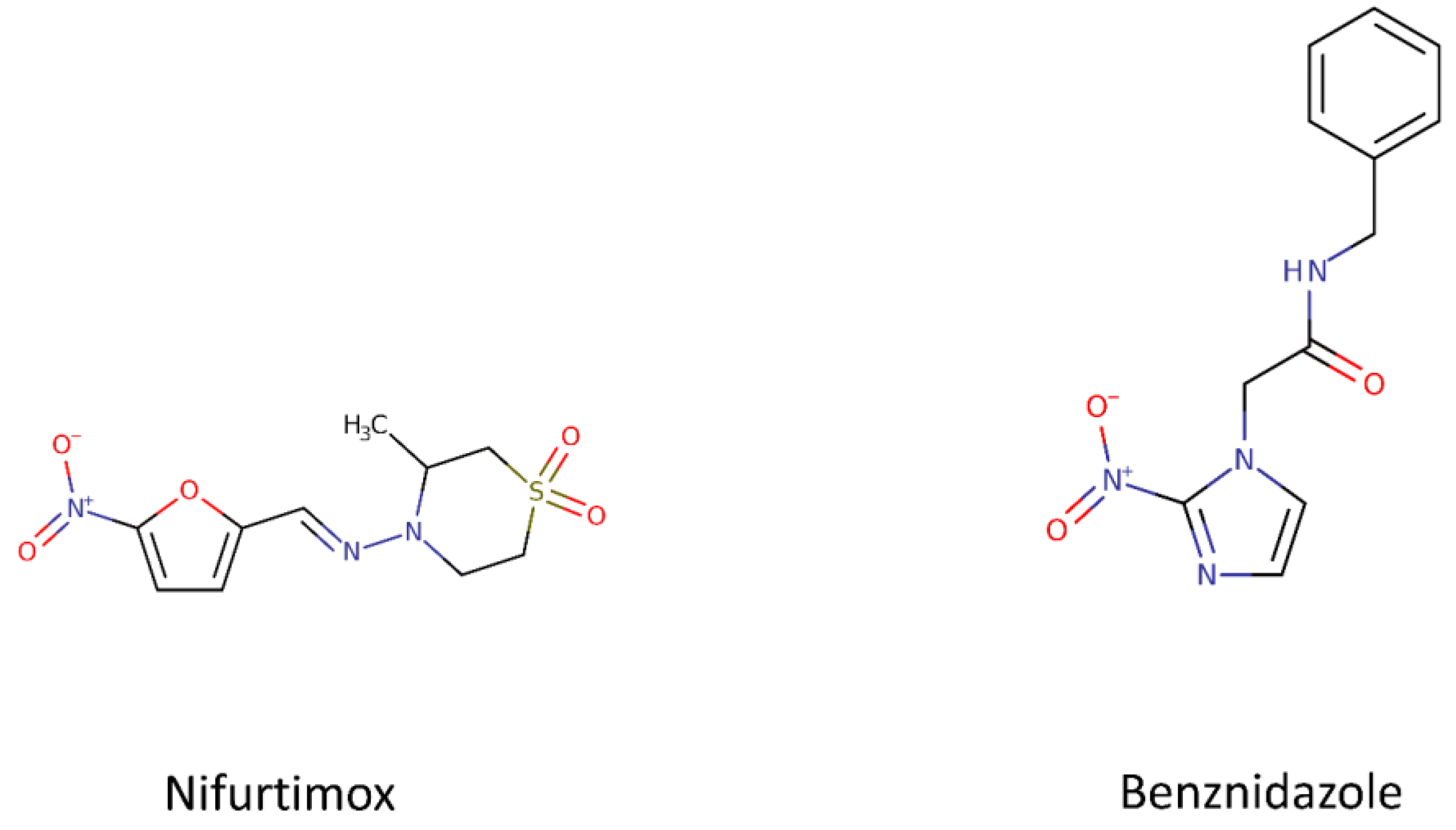
1.1.1. Benznidazole and Nifurtimox
1.1.2. Drug Discovery for Chagas Disease: A Challenge
2. Drugs, Targets and Inhibitors
2.1. Ergosterol Pathway and Inhibitors of CYP51
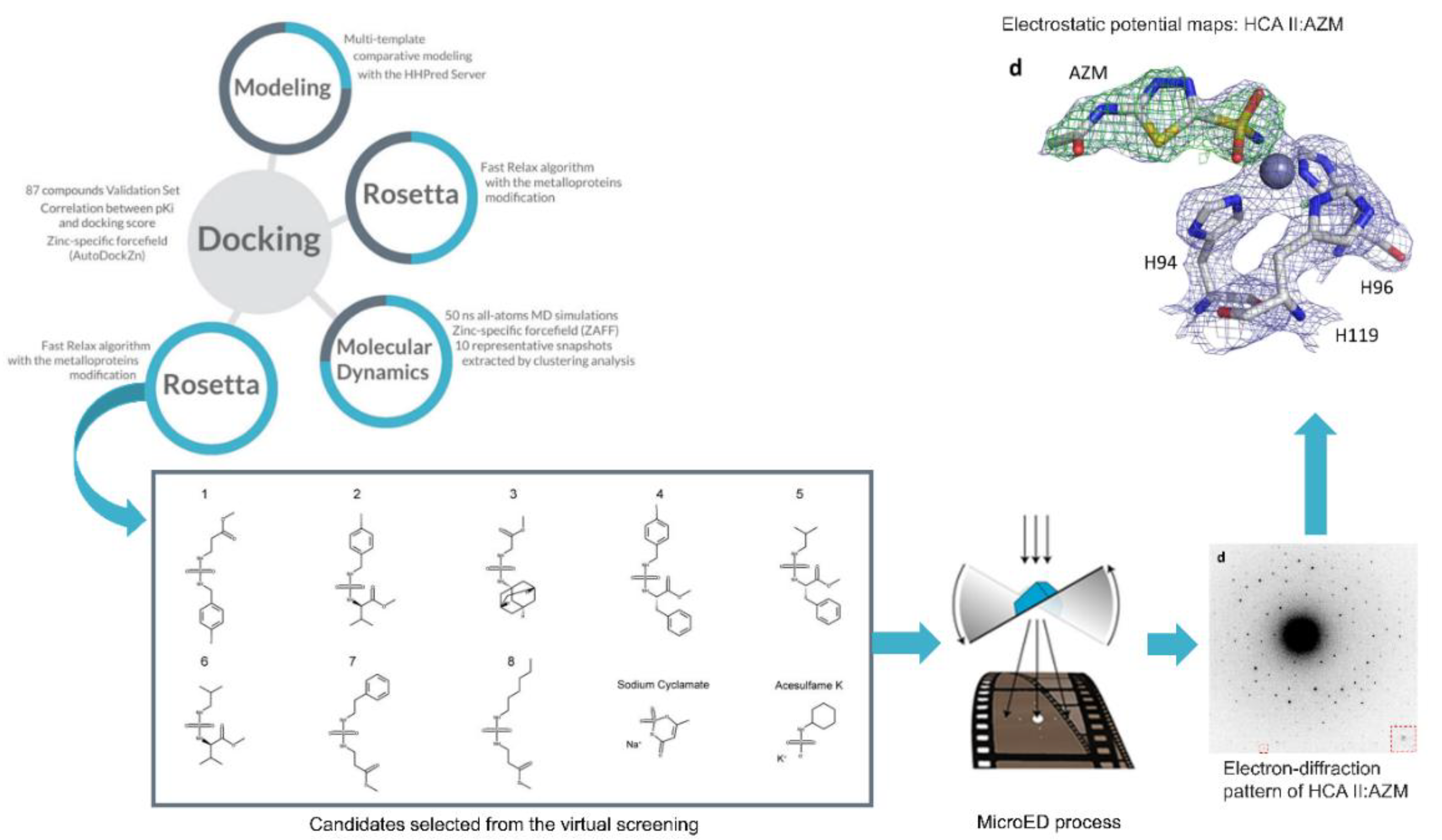
2.2. Carbonic α-Anhydrase and the Inhibitors Sulfonamides, Thiols and Hydroxamates
2.3. Tc80 Proteinase and Peptides
2.4. Cysteine Peptidase and K777
2.5. Proteasome Inhibitors
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Losada Galván, I.; Alonso-Padilla, J.; Cortés-Serra, N.; Alonso-Vega, C.; Gascón, J.; Pinazo, M.J. Benznidazole for the treatment of Chagas disease. Expert Rev. Anti. Infect. Ther. 2020, 1–10. [Google Scholar] [CrossRef]
- DNDI Chagas. Available online: https://dndi.org/diseases/chagas/ (accessed on 10 October 2020).
- Rassi, A.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Bern, C. Chagas’ Disease. N. Engl. J. Med. 2015, 373, 456–466. [Google Scholar] [CrossRef]
- Caldas, I.S.; Santos, E.G.; Novaes, R.D. An evaluation of benznidazole as a Chagas disease therapeutic. Expert Opin. Pharmacother. 2019, 20, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Vermelho, A.B.; Rodrigues, G.C.; Supuran, C.T. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin. Drug Discov. 2020, 15, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; O’Connor, P.D.; Marshall, A.J.; Francisco, A.F.; Kelly, J.M.; Riley, J.; Read, K.D.; Perez, C.J.; Cornwall, S.; Thompson, R.C.A.; et al. Re-evaluating pretomanid analogues for Chagas disease: Hit-to-lead studies reveal both in vitro and in vivo trypanocidal efficacy. Eur. J. Med. Chem. 2020, 207, 112849. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Sunyoto, T. Partnerships for better neglected disease drug discovery and development: How have we fared? Expert Opin. Drug Discov. 2020, 15, 531–537. [Google Scholar] [CrossRef]
- Martínez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.-J.; Gascon, J.; Alonso-Padilla, J. Emerging agents for the treatment of Chagas disease: What is in the preclinical and clinical development pipeline? Expert Opin. Investig. Drugs 2020, 29, 947–959. [Google Scholar] [CrossRef]
- Freyhult, E.; Prusis, P.; Lapinsh, M.; Wikberg, J.E.S.; Moulton, V.; Gustafsson, M.G. Unbiased descriptor and parameter selection confirms the potential of proteochemometric modelling. BMC Bioinform. 2005, 6, 50. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Anuwongcharoen, N.; Phanus-umporn, C.; Sriwanichpoom, N.; Wikberg, J.E.S.; Nantasenamat, C. Proteochemometric Modeling for Drug Repositioning. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 281–302. [Google Scholar]
- Chatelain, E.; Scandale, I. Animal models of Chagas disease and their translational value to drug development. Expert Opin. Drug Discov. 2020, 1–22. [Google Scholar] [CrossRef]
- Sales Junior, P.A.; Molina, I.; Fonseca Murta, S.M.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Carneiro, C.M. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef]
- Villalta, F.; Rachakonda, G. Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin. Drug Discov. 2019, 14, 1161–1174. [Google Scholar] [CrossRef]
- DNDI Screening Chagas disease. Available online: https://dndi.org/research-development/portfolio/screening-chagas/0 (accessed on 10 October 2020).
- DNDI Chagas Hit-to-lead. Available online: https://dndi.org/research-development/portfolio/chagas-h2l/ (accessed on 10 October 2020).
- Yunta, M.J.R.; Dietrich, R.C. Tropical and Subtropical Parasitic Diseases: Targets for a New Approach to Virtual Screening. Mol. Inform. 2019, 38, 1900052. [Google Scholar] [CrossRef]
- Llanos, M.A.; Sbaraglini, M.L.; Villalba, M.L.; Ruiz, M.D.; Carrillo, C.; Alba Soto, C.; Talevi, A.; Angeli, A.; Parkkila, S.; Supuran, C.T.; et al. A structure-based approach towards the identification of novel antichagasic compounds: Trypanosoma cruzi carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 21–30. [Google Scholar] [CrossRef]
- Villalta, F.; Dobish, M.C.; Nde, P.N.; Kleshchenko, Y.Y.; Hargrove, T.Y.; Johnson, C.A.; Waterman, M.R.; Johnston, J.N.; Lepesheva, G.I. VNI Cures Acute and Chronic Experimental Chagas Disease. J. Infect. Dis. 2013, 208, 504–511. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Hargrove, T.Y.; Rachakonda, G.; Wawrzak, Z.; Pomel, S.; Cojean, S.; Nde, P.N.; Nes, W.D.; Locuson, C.W.; Calcutt, M.W.; et al. VFV as a New Effective CYP51 Structure-Derived Drug Candidate for Chagas Disease and Visceral Leishmaniasis. J. Infect. Dis. 2015, 212, 1439–1448. [Google Scholar] [CrossRef]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized Trial of Posaconazole and Benznidazole for Chronic Chagas’ Disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef]
- DNDI Azoles E1224. Available online: https://dndi.org/research-development/portfolio/azoles-e1224/ (accessed on 10 October 2020).
- Moraes, C.B.; Giardini, M.A.; Kim, H.; Franco, C.H.; Araujo-Junior, A.M.; Schenkman, S.; Chatelain, E.; Freitas-Junior, L.H. Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: Implications for Chagas disease drug discovery and development. Sci. Rep. 2015, 4, 4703. [Google Scholar] [CrossRef] [PubMed]
- Guedes-da-Silva, F.H.; Batista, D.G.J.; Da Silva, C.F.; De Araújo, J.S.; Pavão, B.P.; Simões-Silva, M.R.; Batista, M.M.; Demarque, K.C.; Moreira, O.C.; Britto, C.; et al. Antitrypanosomal Activity of Sterol 14α-Demethylase (CYP51) Inhibitors VNI and VFV in the Swiss Mouse Models of Chagas Disease Induced by the Trypanosoma cruzi Y Strain. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- de Soeiro, M.N.C.; de Souza, E.M.; da Silva, C.F.; da Batista, D.G.J.; Batista, M.M.; Pavão, B.P.; Araújo, J.S.; Aiub, C.A.F.; da Silva, P.B.; Lionel, J.; et al. In Vitro and In Vivo Studies of the Antiparasitic Activity of Sterol 14α-Demethylase (CYP51) Inhibitor VNI against Drug-Resistant Strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2013, 57, 4151–4163. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C. Carbonic Anhydrases An Overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: An underexplored therapeutic opportunity. Future Med. Chem. 2016, 8, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Capasso, C. Protozoan Carbonic Anhydrases. In Zinc Enzyme Inhibitors; Springer: Cham, Switzwerland, 2016; pp. 111–133. ISBN 978-3-319-46111-3. [Google Scholar]
- Pan, P.; Vermelho, A.B.; Capaci Rodrigues, G.; Scozzafava, A.; Tolvanen, M.E.E.; Parkkila, S.; Capasso, C.; Supuran, C.T. Cloning, Characterization, and Sulfonamide and Thiol Inhibition Studies of an α-Carbonic Anhydrase from Trypanosoma cruzi, the Causative Agent of Chagas Disease. J. Med. Chem. 2013, 56, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Aspatwar, A.; Barker, H.; Tolvanen, M.; Emameh, R.Z.; Parkkila, S. Carbonic anhydrases from pathogens. In Carbonic Anhydrases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 449–475. [Google Scholar]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019, 14, 1175–1197. [Google Scholar] [CrossRef]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef] [PubMed]
- Clabbers, M.T.B.; Fisher, S.Z.; Coinçon, M.; Zou, X.; Xu, H. Visualizing drug binding interactions using microcrystal electron diffraction. Commun. Biol. 2020, 3, 417. [Google Scholar] [CrossRef] [PubMed]
- Nannenga, B.L. MicroED methodology and development. Struct. Dyn. 2020, 7, 014304. [Google Scholar] [CrossRef] [PubMed]
- Nannenga, B.L.; Gonen, T. The cryo-EM method microcrystal electron diffraction (MicroED). Nat. Methods 2019, 16, 369–379. [Google Scholar] [CrossRef]
- D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Are Carbonic Anhydrases Suitable Targets to Fight Protozoan Parasitic Diseases? Curr. Med. Chem. 2019, 25, 5266–5278. [Google Scholar] [CrossRef]
- Pereira, C.A.; Sayé, M.; Reigada, C.; Silber, A.M.; Labadie, G.R.; Miranda, M.R.; Valera-Vera, E. Computational approaches for drug discovery against trypanosomatid-caused diseases. Parasitology 2020, 147, 611–633. [Google Scholar] [CrossRef]
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 671–686. [Google Scholar] [CrossRef]
- Bourguignon, S.C.; Cavalcanti, D.F.B.; de Souza, A.M.T.; Castro, H.C.; Rodrigues, C.R.; Albuquerque, M.G.; Santos, D.O.; da Silva, G.G.; da Silva, F.C.; Ferreira, V.F.; et al. Trypanosoma cruzi: Insights into naphthoquinone effects on growth and proteinase activity. Exp. Parasitol. 2011, 127, 160–166. [Google Scholar] [CrossRef]
- Bivona, A.E.; Sánchez Alberti, A.; Matos, M.N.; Cerny, N.; Cardoso, A.C.; Morales, C.; González, G.; Cazorla, S.I.; Malchiodi, E.L. Trypanosoma cruzi 80 kDa prolyl oligopeptidase (Tc80) as a novel immunogen for Chagas disease vaccine. PLoS Negl. Trop. Dis. 2018, 12, e0006384. [Google Scholar] [CrossRef]
- Joyeau, R.; Maoulida, C.; Guillet, C.; Frappier, F.; Teixeira, A.R.L.; Schrével, J.; Santana, J.; Grellier, P. Synthesis and activity of pyrrolidinyl- and thiazolidinyl-dipeptide derivatives as inhibitors of the Tc80 prolyl oligopeptidase from Trypanosoma cruzi. Eur. J. Med. Chem. 2000, 35, 257–266. [Google Scholar] [CrossRef]
- Silva, J.V.; da Santos, S.S.; Machini, M.T.; Giarolla, J. Neglected tropical diseases and infectious illnesses: Potential targeted peptides employed as hits compounds in drug design. J. Drug Target. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Salas-Sarduy, E.; Landaburu, L.U.; Karpiak, J.; Madauss, K.P.; Cazzulo, J.J.; Agüero, F.; Alvarez, V.E. Novel scaffolds for inhibition of Cruzipain identified from high-throughput screening of anti-kinetoplastid chemical boxes. Sci. Rep. 2017, 7, 12073. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, A. Antidiabetic agents: Past, present and future. Future Med. Chem. 2013, 5, 411–430. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, F.; Sastry, L.; Shepherd, S.M.; Jones, D.; Scott, A.; Craggs, P.D.; Cortes, A.; Gray, D.W.; Torrie, L.S.; De Rycker, M. Identification of Novel Trypanosoma cruzi Proteasome Inhibitors Using a Luminescence-Based High-Throughput Screening Assay. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Rao, S.P.S.; Lakshminarayana, S.B.; Jiricek, J.; Kaiser, M.; Ritchie, R.; Myburgh, E.; Supek, F.; Tuntland, T.; Nagle, A.; Molteni, V.; et al. Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis. Trop. Med. Infect. Dis. 2020, 5, 28. [Google Scholar] [CrossRef]
- D’Souza, S.; Prema, K.V.; Balaji, S. Machine learning models for drug–target interactions: Current knowledge and future directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- Shen, C.; Ding, J.; Wang, Z.; Cao, D.; Ding, X.; Hou, T. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. WIREs Comput. Mol. Sci. 2020, 10. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminform. 2020, 12, 1–30. [Google Scholar] [CrossRef]
- Reich, S.H.; Webber, S.E. Structure-based drug design (SBDD): Every structure tells a story. Perspect. Drug Discov. Des. 1993, 1, 371–390. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Gaieb, Z.; Amaro, R.E. An Analysis of Proteochemometric and Conformal Prediction Machine Learning Protein-Ligand Binding Affinity Models. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Danishuddin; Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef]
- Qiu, T.; Qiu, J.; Feng, J.; Wu, D.; Yang, Y.; Tang, K.; Cao, Z.; Zhu, R. The recent progress in proteochemometric modelling: Focusing on target descriptors, cross-term descriptors and application scope. Brief. Bioinform. 2017, 18, 125–136. [Google Scholar] [CrossRef][Green Version]
- Lapinsh, M.; Prusis, P.; Gutcaits, A.; Lundstedt, T.; Wikberg, J.E.S. Development of proteo-chemometrics: A novel technology for the analysis of drug-receptor interactions. Biochim. Biophys. Acta Gen. Subj. 2001, 1525, 180–190. [Google Scholar] [CrossRef]
- van Westen, G.J.P.; Wegner, J.K.; IJzerman, A.P.; van Vlijmen, H.W.T.; Bender, A. Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. Medchemcomm 2011, 2, 16–30. [Google Scholar] [CrossRef]
- Playe, B.; Stoven, V. Evaluation of deep and shallow learning methods in chemogenomics for the prediction of drugs specificity. J. Cheminform. 2020, 12, 11. [Google Scholar] [CrossRef]
- Magarinos, M.P.; Carmona, S.J.; Crowther, G.J.; Ralph, S.A.; Roos, D.S.; Shanmugam, D.; Van Voorhis, W.C.; Aguero, F. TDR Targets: A chemogenomics resource for neglected diseases. Nucleic Acids Res. 2012, 40, D1118–D1127. [Google Scholar] [CrossRef] [PubMed]
- Urán Landaburu, L.; Berenstein, A.J.; Videla, S.; Maru, P.; Shanmugam, D.; Chernomoretz, A.; Agüero, F. TDR Targets 6: Driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Valera-Vera, E.A.; Sayé, M.; Reigada, C.; Miranda, M.R.; Pereira, C.A. In silico repositioning of etidronate as a potential inhibitor of the Trypanosoma cruzi enolase. J. Mol. Graph. Model. 2020, 95, 107506. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Alberca, L.N.; Sbaraglini, M.L.; Morales, J.F.; Dietrich, R.; Ruiz, M.D.; Pino Martínez, A.M.; Miranda, C.G.; Fraccaroli, L.; Alba Soto, C.D.; Carrillo, C.; et al. Cascade Ligand- and Structure-Based Virtual Screening to Identify New Trypanocidal Compounds Inhibiting Putrescine Uptake. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Bellera, C.L.; Alberca, L.N.; Sbaraglini, M.L.; Talevi, A. In Silico Drug Repositioning for Chagas Disease. Curr. Med. Chem. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Chatelain, E.; Ioset, J.-R. Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin. Drug Discov. 2018, 13, 141–153. [Google Scholar] [CrossRef]
- Álvarez-Bardón, M.; Pérez-Pertejo, Y.; Ordóñez, C.; Sepúlveda-Crespo, D.; Carballeira, N.M.; Tekwani, B.L.; Murugesan, S.; Martinez-Valladares, M.; García-Estrada, C.; Reguera, R.M.; et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Mar. Drugs 2020, 18, 187. [Google Scholar] [CrossRef]
- Gilbert, I.H. Drug Discovery for Neglected Diseases: Molecular Target-Based and Phenotypic Approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Aulner, N.; Danckaert, A.; Ihm, J.; Shum, D.; Shorte, S.L. Next-Generation Phenotypic Screening in Early Drug Discovery for Infectious Diseases. Trends Parasitol. 2019, 35, 559–570. [Google Scholar] [CrossRef]
- Ekins, S.; Lage de Siqueira-Neto, J.; McCall, L.-I.; Sarker, M.; Yadav, M.; Ponder, E.L.; Kallel, E.A.; Kellar, D.; Chen, S.; Arkin, M.; et al. Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl. Trop. Dis. 2015, 9, e0003878. [Google Scholar] [CrossRef] [PubMed]
- Roquero, I.; Cantizani, J.; Cotillo, I.; Manzano, M.P.; Kessler, A.; Martín, J.J.; McNamara, C.W. Novel chemical starting points for drug discovery in leishmaniasis and Chagas disease. Int. J. Parasitol. Drugs Drug Resist. 2019, 10, 58–68. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NCT Number | Acronym | Status | Interventions | Phases | Estimated Enrollment | Funded By | Locations |
|---|---|---|---|---|---|---|---|
| NCT02625974 | CHICO | Active, not recruiting | Nifurtimox (Lampit, BAYA2502), Placebo | Phase 3 | 330 participants | Industry | Argentina, Bolivia, Colombia |
| NCT03334838 | Completed | Nifurtimox (Lampit, BAYA2502) | Phase 1 | 36 participants | Industry | Argentina | |
| NCT03350295 | Completed | Nifurtimox (Lampit, BAYA2502) | Phase 1 | 48 participants | Industry | Argentina | |
| NCT02606864 | Completed | Nifurtimox (BAYa2502) | Phase 1 | 36 participants | Industry | Argentina | |
| NCT01927224 | Completed | Nifurtimox (BAYa2502) | Phase 1 | 37 participants | Industry | Argentina | |
| NCT03892213 | Completed | Benznidazole, E1224 | Phase 1 | 28 participants | Other | Argentina | |
| NCT03587766 | FEXI12 | Completed | Fexinidazole, Placebo Oral Tablet | Phase 2 | 45 participants | Other | Spain |
| NCT01377480 | STOP CHAGAS | Completed | Posaconazole, Placebo for posaconazole, Benznidazole | Phase 2 | 120 participants | Industry | Argentina, Chile, Colombia, Guatemala, Mexico, Spain |
| NCT01162967 | CHAGASAZOL | Completed | Benznidazole, Posaconazole | Phase 2 | 78 participants | Other | Spain |
| NCT02154269 | Completed | Treatment with G-CSF (Granulocyte colony stimulating factor), Placebo saline | Phase 2 | 70 participants | Other | Brazil | |
| NCT02386358 | TRAENA | Completed | Benznidazole, Placebo | Phase 3 | 910 participants | Other | Argentina |
| NCT00123916 | BENEFIT | Completed | Benznidazole, Placebo | Phase 3 | 2854 participants | Other | Argentina, Bolivia, Brazil, Colombia, El Salvador |
| NCT00323973 | Completed | Bisoprolol | Phase 3 | 500 participants | Other | Colombia | |
| NCT01755403 | CINEBENZ | Completed | Benznidazole | Phase 4 | 52 participants | Other | Spain |
| NCT01549236 | Pop PK Chagas | Completed | Benznidazole 12,5mg or 100mg | Phase 4 | 80 participants | Other | Argentina |
| NCT01557140 | Completed | RASi plus carvedilol | Phase 4 | 42 participants | Other | Brazil | |
| NCT03981523 | TESEO | Recruiting | Benznidazole, Nifurtimox | Phase 2 | 450 participants | Other, NIH | Bolivia |
| NCT03704181 | COACH | Recruiting | Colchicine 0.5 MG twice day for one year, Placebo Oral Tablet | Phase 2 | 60 participants | Other | Brazil |
| NCT04024163 | Recruiting | Benznidazole | Phase 3 | 164 participants | Industry, Other | Argentina, Bolivia, Colombia | |
| NCT00875173 | STCC | Recruiting | Selenium, Placebo (for Selenium) | Phase 3 | 130 participants | Other | Brazil |
| NCT03672487 | BETTY | Recruiting | Benznidazole, Placebo Oral Tablet | Phase 3 | 600 participants | Other | United States, Argentina |
| NCT03193749 | ATTACH | Recruiting | Amiodarone Hydrochloride, Placebo Oral Tablet | Phase 3 | 200 participants | Other | Colombia |
| NCT01650792 | CLINICS | Recruiting | Aspirin | Phase 4 | 500 participants | Other, NIH | Brazil |
| NCT04023227 | PARACHUTE-HF | Recruiting | Sacubitril/valsartan, Enalapril | Phase 4 | 900 participants | Industry | Argentina, Brazil |
| NCT01489228 | Unknown status | E1224, Benznidazole, Placebo | Phase 2 | 230 participants | Other/Industry | Bolivia | |
| NCT03191162 | MULTIBENZ | Unknown status | Benznidazole | Phase 2 | 240 participants | Other | Argentina, Brazil, Colombia, Spain |
| NCT03378661 | BENDITA | Unknown status | Benznidazole, E1224, E1224 Placebo, Benznidazole Placebo | Phase 2 | 210 participants | Other | Bolivia |
| NCT02498782 | Unknown status | Fexinidazole, Placebo | Phase 2 | 140 participants | Other | Bolivia | |
| NCT02369978 | CHICAMOCHA-3 | Unknown status | Nifurtimox, Benznidazole, Placebo | Phase 2, Phase 3 | 500 participants | Other | Colombia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansoldo, F.R.P.; Carta, F.; Angeli, A.; Cardoso, V.d.S.; Supuran, C.T.; Vermelho, A.B. Chagas Disease: Perspectives on the Past and Present and Challenges in Drug Discovery. Molecules 2020, 25, 5483. https://doi.org/10.3390/molecules25225483
Mansoldo FRP, Carta F, Angeli A, Cardoso VdS, Supuran CT, Vermelho AB. Chagas Disease: Perspectives on the Past and Present and Challenges in Drug Discovery. Molecules. 2020; 25(22):5483. https://doi.org/10.3390/molecules25225483
Chicago/Turabian StyleMansoldo, Felipe Raposo Passos, Fabrizio Carta, Andrea Angeli, Veronica da Silva Cardoso, Claudiu T. Supuran, and Alane Beatriz Vermelho. 2020. "Chagas Disease: Perspectives on the Past and Present and Challenges in Drug Discovery" Molecules 25, no. 22: 5483. https://doi.org/10.3390/molecules25225483
APA StyleMansoldo, F. R. P., Carta, F., Angeli, A., Cardoso, V. d. S., Supuran, C. T., & Vermelho, A. B. (2020). Chagas Disease: Perspectives on the Past and Present and Challenges in Drug Discovery. Molecules, 25(22), 5483. https://doi.org/10.3390/molecules25225483