
Water-Soluble O-, S- and Se-Functionalized Cyclic Acetyl-triaza-phosphines. Synthesis, Characterization and Application in Catalytic Azide-alkyne Cycloaddition

 ,
,  ,
,  and
and 
Abstract
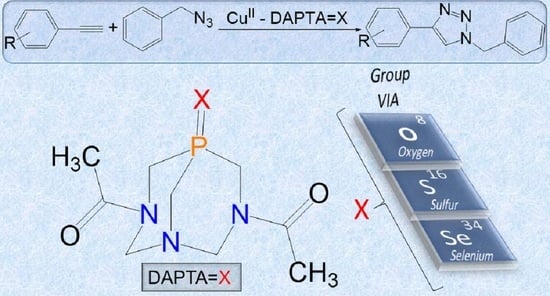
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of 1–3
2.2. Single Crystal X-ray Diffraction Analysis
2.3. Hirshfeld Structural Comparison of DAPTA and Derivatives 1–3
2.4. Catalytic Performances of 1–3 in CuAAC Reaction
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis of Compounds 1–3
3.2.1. Synthesis 3,7-Diacetyl-1,3,7-Triaza-5-phosphabicyclo[3.3.1]nonane-5-oxide (DAPTA=O, 1)
3.2.2. Synthesis 3,7-Diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane-5-sulfide (DAPTA=S, 2) and 3,7-Diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane-5-selenide (DAPTA=Se, 3)
3.3. X-ray Structure Determination of Compounds
3.4. General Procedure for the Synthesis of 1,2,3-Triazoles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A. Green solvents for sustainable organic synthesis: State of the art. Green Chem. 2005, 7, 267–278. [Google Scholar] [CrossRef]
- Capello, C.; Fischer, U.; Hungerbühler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Shaughnessy, K.H. Hydrophilic ligands and their application in aqueous-phase metal-catalyzed reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar] [CrossRef] [PubMed]
- Ounkham, W.L.; Frost, B.J. Introduction to Aqueous Organometallic Chemistry and Catalysis. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2017; pp. 1–26. ISBN 9781119951438. [Google Scholar]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Coordination chemistry of 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane (PTA) and derivatives. Part III. Variations on a theme: Novel architectures, materials and applications. Coord. Chem. Rev. 2018, 355, 328–361. [Google Scholar] [CrossRef]
- Scalambra, F.; Lorenzo-Luis, P.; Ríos, I.D.L.; Romerosa, A. New Findings in Metal Complexes with Antiproliferative Activity Containing 1, 3, 5-Triaza-7-phosphaadamantane (PTA) and Derivative Ligands. Eur. J. Inorg. Chem. 2019, 1529–1538. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Ortiz, C.G.; Kamplain, J.W. A New Water-Soluble Phosphine Derived from 1,3,5-Triaza-7-phosphaadamantane (PTA), 3,7-Diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane. Structural, Bonding, and Solubility Properties. Organometallics 2004, 23, 1747–1754. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. 3,7-Diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane (DAPTA) and derivatives: Coordination chemistry and applications. Coord. Chem. Rev. 2020, 213614. [Google Scholar] [CrossRef]
- Siele, V.I. Some reactions of 1,3,5-Triaza-7-phosphaadamantane and its 7-Oxide. J. Heterocycl. Chem. 1977, 14, 337–339. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Guedes da Silva, M.F.C.; Śliwa, E.I.; Smoleński, P.; Kuznetsov, M.L.; Pombeiro, A.J.L. Copper(II) and Sodium(I) Complexes based on 3,7-Diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane-5-oxide: Synthesis, Characterization, and Catalytic Activity. Chem. Asian J. 2018, 13, 2868–2880. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Guedes da Silva, M.F.C.; Sokolnicki, J.; Smoleński, P.; Pombeiro, A.J.L. Hydrosoluble Cu(I)-DAPTA complexes: Synthesis, characterization, luminescence thermochromism and catalytic activity for microwave-assisted three-component azide-alkyne cycloaddition click reaction. Dalton Trans. 2018, 47, 7290–7299. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.G.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Copper complexes bearing C-scorpionate ligands: Synthesis, characterization and catalytic activity for azide-alkyne cycloaddition in aqueous medium. Inorg. Chim. Acta 2018, 483, 371–378. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Guedes da Silva, M.F.C.; Mahmudov, K.T.; Pombeiro, A.J.L. Arylhydrazone ligands as Cu-protectors and -catalysis promoters in the azide–alkyne cycloaddition reaction. Dalton Trans. 2019, 48, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Alyea, E.C.; Shahnazarian, N. A 31P nmr study of the water soluble derivatives of l, 3, 5-triaza-7-phosphaadamantane (PTA). Phosphorus. Sulfur. Silicon Relat. Elem. 1990, 48, 37–40. [Google Scholar] [CrossRef]
- Allen, L.C. Electronegativity is the Average One-Electron Energy of the Valence-Shell Electrons in Ground-State Free Atoms. J. Am. Chem. Soc. 1989, 111, 9003–9014. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Díez, J.; Gimeno, J.; Suárez, F.J.; Vincent, C. (Iminophosphorane)copper(I) complexes as highly efficient catalysts for 1,3-dipolar cycloaddition of azides with terminal and 1-iodoalkynes in water: One-pot multi-component reaction from alkynes and in situ generated azides. Eur. J. Inorg. Chem. 2012, 5854–5863. [Google Scholar] [CrossRef]
- Kamijo, S.; Jin, T.; Huo, Z.; Yamamoto, Y. A One-Pot Procedure for the Regiocontrolled Synthesis of Allyltriazoles via the Pd-Cu Bimetallic Catalyzed Three-Component Coupling Reaction of Nonactivated Terminal Alkynes, Allyl Carbonate, and Trimethylsilyl Azide. J. Org. Chem. 2004, 69, 2386–2393. [Google Scholar] [CrossRef]
- Li, F.; Hor, T.S.A. Facile Synthesis of Nitrogen Tetradentate Ligands and Their Applications in Cu I -Catalyzed N-Arylation and Azide-Alkyne Cycloaddition. Chem. A Eur. J. 2009, 15, 10585–10592. [Google Scholar] [CrossRef]
- Gonda, Z.; Novák, Z. Highly active copper-catalysts for azide-alkynecycloaddition. Dalton Trans. 2010, 39, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, M.; Fokin, V.V. Enhanced reactivity of dinuclear copper(I) acetylides in dipolar cycloadditions. Organometallics 2007, 26, 4389–4391. [Google Scholar] [CrossRef]
- Straub, B.F. µ-Acetylide and µ-alkenylidene ligands in “click” triazole syntheses. Chem. Commun. 2007, 3868–3870. [Google Scholar] [CrossRef]
- Rodionov, V.O.; Fokin, V.V.; Finn, M.G. Mechanism of the Ligand-Free CuI-Catalyzed Azide-Alkyne Cycloaddition Reaction. Angew. Chem. Int. Ed. 2005, 44, 2210–2215. [Google Scholar] [CrossRef]
- Nolte, C.; Mayer, P.; Straub, B.F. Isolation of a copper(I) triazolide: A “click” intermediate. Angew. Chem. Int. Ed. 2007, 46, 2101–2103. [Google Scholar] [CrossRef]
- Worrell, B.T.; Malik, J.A.; Fokin, V. V Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef]
- Brotherton, W.S.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Dalal, N.S.; Zhu, L. Apparent Copper(II)-Accelerated Azide−Alkyne Cycloaddition. Org. Lett. 2009, 11, 4954–4957. [Google Scholar] [CrossRef]
- Kamata, K.; Nakagawa, Y.; Yamaguchi, K.; Mizuno, N. 1,3-Dipolar Cycloaddition of Organic Azides to Alkynes by a Dicopper-Substituted Silicotungstate. J. Am. Chem. Soc. 2008, 130, 15304–15310. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Oishi, T.; Katayama, T.; Mizuno, N. A Supported Copper Hydroxide on Titanium Oxide as an Efficient Reusable Heterogeneous Catalyst for 1,3-Dipolar Cycloaddition of Organic Azides to Terminal Alkynes. Chem. A Eur. J. 2009, 15, 10464–10472. [Google Scholar] [CrossRef]
- Kuang, G.-C.; Guha, P.M.; Brotherton, W.S.; Simmons, J.T.; Stankee, L.A.; Nguyen, B.T.; Clark, R.J.; Zhu, L. Experimental Investigation on the Mechanism of Chelation-Assisted, Copper(II) Acetate-Accelerated Azide–Alkyne Cycloaddition. J. Am. Chem. Soc. 2011, 133, 13984–14001. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, W.S. Development of Copper (II)-Mediated Azide-Alkyne Cycloaddition Reactions Using Chelating Azides; Florida State University: Tallahassee, FL, USA, 2012. [Google Scholar]
- Eisch, J.J.; Gopal, H.; Rhee, S.-G. Organometallic compounds of Group III. Regiochemistry and stereochemistry in the hydralumination of heterosubstituted acetylenes. Interplay of inductive and resonance effects in electron-rich alkynes. J. Org. Chem. 1975, 40, 2064–2069. [Google Scholar] [CrossRef]
- Bruker AXS Inc. Bruker, APEX2; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SADABS. Program for Empirical Absorption Correction; University of Gottingen: Göttingen, Germany, 2000. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR 97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. IUCr A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Rajgopal, K.; Kantam, M.L. Copper(II)-Promoted Regioselective Synthesis of 1,4-Disubstituted 1,2,3-Triazoles in Water. Synlett 2006, 957–959. [Google Scholar] [CrossRef]
- Lőrincz, K.; Kele, P.; Novák, Z. The Sequential Sonogashira-Click Reaction: A Versatile Route to 4-Aryl-1,2,3-triazoles. Synthesis 2009, 3527–3532. [Google Scholar] [CrossRef]
- Ozkal, E.; Özçubukçu, S.; Jimeno, C.; Pericàs, M.A. Covalently immobilized tris(triazolyl)methanol–Cu(i) complexes: Highly active and recyclable catalysts for CuAAC reactions. Catal. Sci. Technol. 2012, 2, 195–200. [Google Scholar] [CrossRef]
- Wang, W.; Wu, J.; Xia, C.; Li, F. Reusable ammonium salt-tagged NHC–Cu(i) complexes: Preparation and catalytic application in the three component click reaction. Green Chem. 2011, 13, 3440–3445. [Google Scholar] [CrossRef]
- Park, I.S.; Kwon, M.S.; Kim, Y.; Lee, J.S.; Park, J. Heterogeneous copper catalyst for the cycloaddition of azides and alkynes without additives under ambient conditions. Org. Lett. 2008, 10, 497–500. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAPTA [8] | 1 [8] | 2 | 3 | ||||
|---|---|---|---|---|---|---|---|
| P-O | 1.491(3) | P1-S1 | 1.9524(7) | P1-Se1 | 2.096(1) | ||
| P-C | 1.71(1) | P-C | 1.797(3) | P1-C1 | 1.824(3) | P1-C1 | 1.821(7) |
| 1.74(1) | 1.817(3) | P1-C2 | 1.812(2) | P1-C2 | 1.817(4) | ||
| 1.74(2) | 1.828(3) | P1-C3 | 1.819(2) | P1-C3 | 1.824(5) | ||
| N4-C5 | 1.471(3) | N1-C4 | 1.486(6) | ||||
| N3-C5 | 1.448(3) | N2-C4 | 1.447(8) | ||||
| N3-C4 | 1.452(3) | N2-C5 | 1.429(6) | ||||
| N5-C4 | 1.469(3) | N3-C5 | 1.482(5) | ||||
| N4-C8 | 1.356(4) | N1-C6 | 1.350(6) | ||||
| N5-C6 | 1.359(3) | N3-C8 | 1.362(6) | ||||
| C6-O6 | 1.227(3) | C6-O1 | 1.221(6) | ||||
| C8-O7 | 1.212(4) | C8-O2 | 1.211(6) | ||||
| S1-P1-C1 | 114.61(9) | Se1-P1-C1 | 114.1(2) | ||||
| S1-P1-C2 | 119.89(8) | Se1-P1-C2 | 120.8(2) | ||||
| S1-P1-C3 | 112.98(9) | Se1-P1-C3 | 112.8(2) | ||||
| C-P-C | 93.8(7) | C-P-C | 100.4(1) | C1-P1-C2 | 100.3(1) | C1-P1-C2 | 100.0(2) |
| 98.4(7) | 100.7(1) | C2-P1-C3 | 99.9(1) | C2-P1-C3 | 100.0(2) | ||
| 104.0(7) | 106.8(1) | C1-P1-C3 | 107.3(1) | C1-P1-C3 | 107.3(2) | ||
| C4-N3-C5 | 115.8(2) | C4-N1-C6 | 124.6(4) | ||||
| C5-N4-C8 | 126.1(2) | C4-N2-C5 | 119.9(4) | ||||
| C4-N5-C6 | 120.0(2) | C5-N3-C8 | 116.1(4) | ||||
| P-C-N | 115.1(9) | P-C-N | 106.2(2) | P1-C2-N3 | 106.9(2) | P1-C2-N2 | 105.7(3) |
| 118(1) | 111.8(2) | P1-C1-N5 | 112.1(2) | P1-C3-N3 | 112.9(4) | ||
| 120.0(9) | 114.5(2) | P1-C3-N4 | 113.5(2) | P1-C1-N1 | 114.2(4) | ||

| Entry. | Cu Salt | Compound | Change from the “Standard Conditions” | Isolated Yield |
|---|---|---|---|---|
| 1 | CuI | - | None | 0 |
| 2 | CuBr | - | None | 0 |
| 3 | CuCl | - | None | 0 |
| 4 | CuI | 1 | None | 62 |
| 5 | CuBr | 1 | None | 41 |
| 6 | CuCl | 1 | None | 15 |
| 7 | CuI | 2 | None | 56 |
| 8 | CuBr | 2 | None | 36 |
| 9 | CuCl | 2 | None | 7 |
| 10 | CuI | 3 | None | 44 |
| 11 | CuBr | 3 | None | 27 |
| 12 | CuCl | 3 | None | 11 |
| 13 | CuSO4·5H2O | 1 | None | 16 |
| 14 | CuSO4·5H2O | 2 | None | 10 |
| 15 | CuSO4·5H2O | 3 | None | 12 |
| 16 | Cu(NO3)2·3H2O | 1 | None | 39 |
| 17 | Cu(NO3)2·3H2O | 2 | None | 37 |
| 18 | Cu(NO3)2·3H2O | 3 | None | 31 |
| 19 | CuBr2 | 1 | None | 56 |
| 20 | CuBr2 | 2 | None | 48 |
| 21 | CuBr2 | 3 | None | 50 |
| 22 | Cu(CH3COO)2·H2O | 1 | None | 73 |
| 23 | Cu(CH3COO)2·H2O | 2 | None | 58 |
| 24 | Cu(CH3COO)2·H2O | 3 | None | 55 |
| 25 | Cu(CH3COO)2·H2O | 1 | 0.5 mol% of Cu and 1 mol% of 1, 48 h | 31 |
| 26 | Cu(CH3COO)2·H2O | 1 | H2O + MeOH (1:1) solvent mixture | 69 |
| 27 | Cu(CH3COO)2·H2O | 1 | H2O + EtOH (1:1) solvent mixture | 75 |
| 28 | Cu(CH3COO)2·H2O | 1 | H2O + tBuOH (1:1) solvent mixture | 78 |
| 29 | Cu(CH3COO)2·H2O | 1 | H2O + DMF (1:1) solvent mixture | 88 |
| 30 | Cu(CH3COO)2·H2O | 1 | H2O + MeCN (1:1) solvent mixture | 97 |
| 31 | Cu(CH3COO)2·H2O | 1 | H2O + MeCN (1:1) solvent mixture, 80 °C, 8 h | >99 |

| Entry | Cat. Load (mol%) | Time (h) | Temp. (°C) | Isolated Yield (%) | TON b |
|---|---|---|---|---|---|
| 1 | 0.5 | 24 | 25 | 54 | 108 |
| 2 | 1 | 24 | 25 | 81 | 81 |
| 3 | 2 | 24 | 25 | 93 | 47 |
| 4 | 5 | 24 | 25 | >99 | 20 |
| 5 | 1 | 3 | 25 | 34 | 34 |
| 6 | 1 | 8 | 25 | 49 | 49 |
| 7 | 1 | 12 | 25 | 57 | 57 |
| 8 | 1 | 48 | 25 | 88 | 88 |
| 9 | 1 | 6 | 80 | >99 | 100 |

| Entry | R | Product | Catalyst | Time (h) | Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 | H | 5a | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | >99 |
| 2 | 4 (1 mol%) | 6 | >99 | ||
| 3 | 3-Me | 5b | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 93 |
| 4 | 4 (1 mol%) | 6 | 95 | ||
| 5 | 3-OMe | 5c | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | >99 |
| 6 | 4 (1 mol%) | 6 | >99 | ||
| 7 | 4-Me | 5d | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 91 |
| 8 | 4 (1 mol%) | 6 | 94 | ||
| 9 | 4-Et | 5e | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 88 |
| 10 | 4 (1 mol%) | 6 | 92 | ||
| 11 | 4-F | 5f | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 85 |
| 12 | 4 (1 mol%) | 6 | 87 | ||
| 13 | 4-tBu | 5g | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 95 |
| 14 | 4 (1 mol%) | 6 | 97 | ||
| 15 | 4-NH2 | 5h | Cu(CH3COO)2·H2O (1 mol%) + 1 (2 mol%) | 8 | 81 |
| 16 | 4 (1 mol%) | 6 | 86 |
Sample Availability: Samples of the compounds 1–4 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, A.G.; Smoleński, P.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Water-Soluble O-, S- and Se-Functionalized Cyclic Acetyl-triaza-phosphines. Synthesis, Characterization and Application in Catalytic Azide-alkyne Cycloaddition. Molecules 2020, 25, 5479. https://doi.org/10.3390/molecules25225479
Mahmoud AG, Smoleński P, Guedes da Silva MFC, Pombeiro AJL. Water-Soluble O-, S- and Se-Functionalized Cyclic Acetyl-triaza-phosphines. Synthesis, Characterization and Application in Catalytic Azide-alkyne Cycloaddition. Molecules. 2020; 25(22):5479. https://doi.org/10.3390/molecules25225479
Chicago/Turabian StyleMahmoud, Abdallah G., Piotr Smoleński, M. Fátima C. Guedes da Silva, and Armando J. L. Pombeiro. 2020. "Water-Soluble O-, S- and Se-Functionalized Cyclic Acetyl-triaza-phosphines. Synthesis, Characterization and Application in Catalytic Azide-alkyne Cycloaddition" Molecules 25, no. 22: 5479. https://doi.org/10.3390/molecules25225479
APA StyleMahmoud, A. G., Smoleński, P., Guedes da Silva, M. F. C., & Pombeiro, A. J. L. (2020). Water-Soluble O-, S- and Se-Functionalized Cyclic Acetyl-triaza-phosphines. Synthesis, Characterization and Application in Catalytic Azide-alkyne Cycloaddition. Molecules, 25(22), 5479. https://doi.org/10.3390/molecules25225479