
Substituted 2-Phenacylbenzoxazole Difluoroboranes: Synthesis, Structure and Properties


Abstract
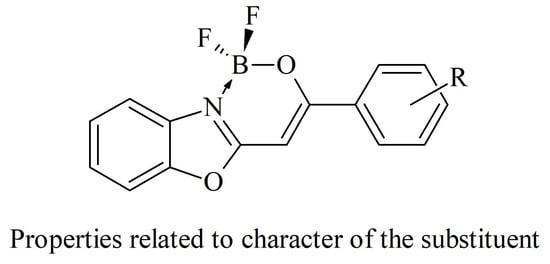
1. Introduction
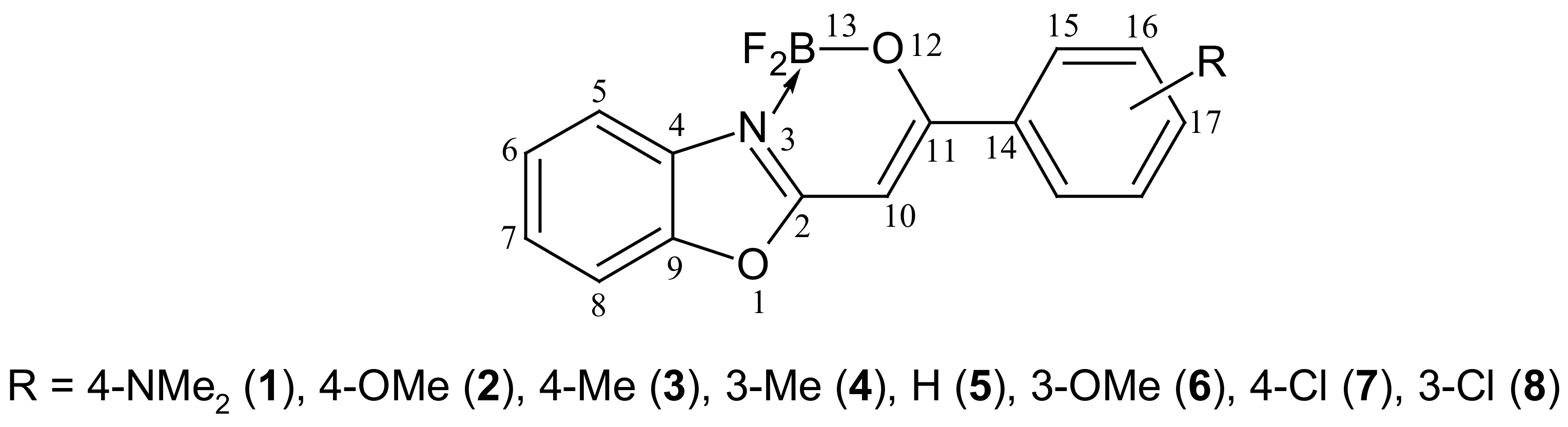
2. Results and Discussion
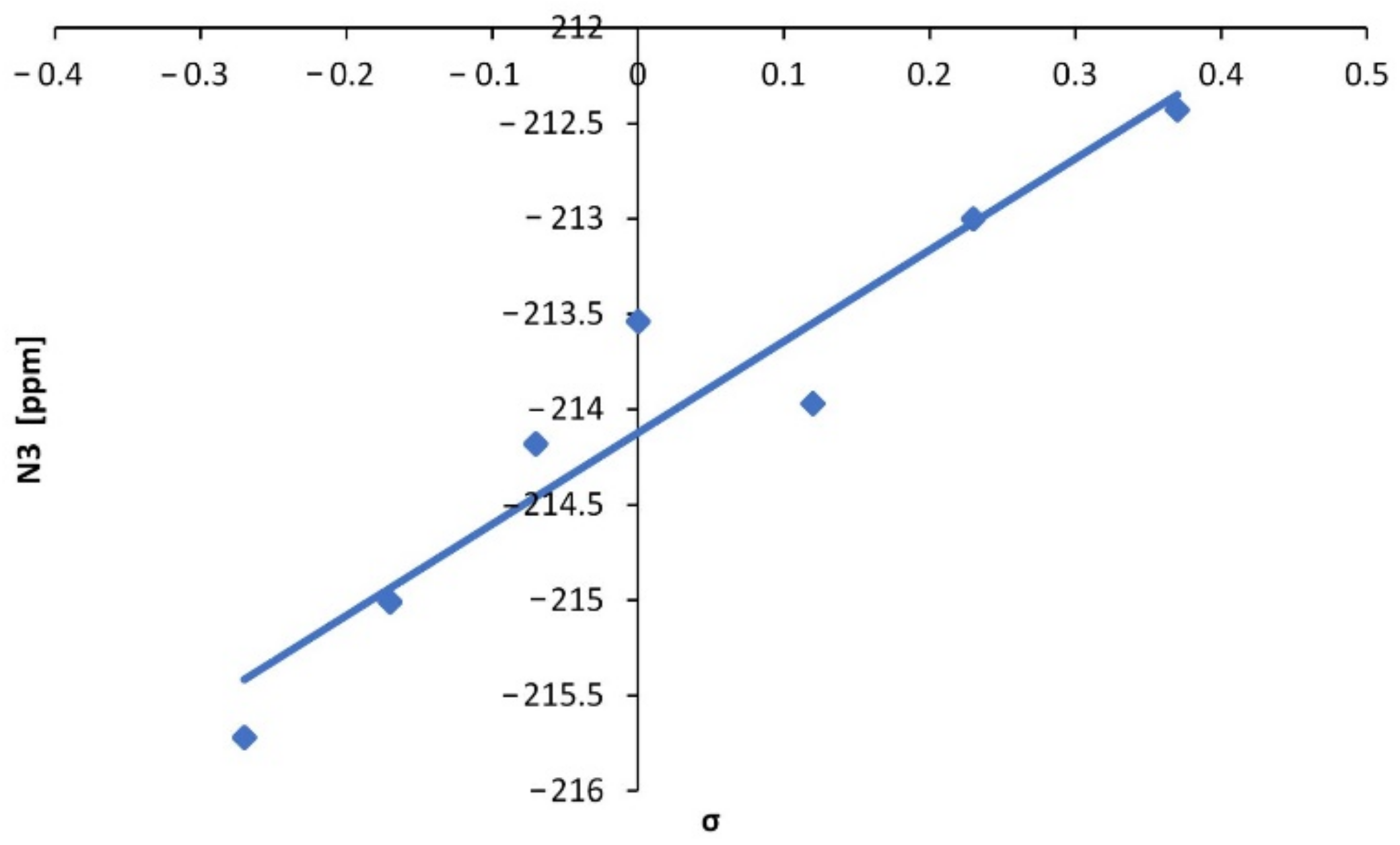
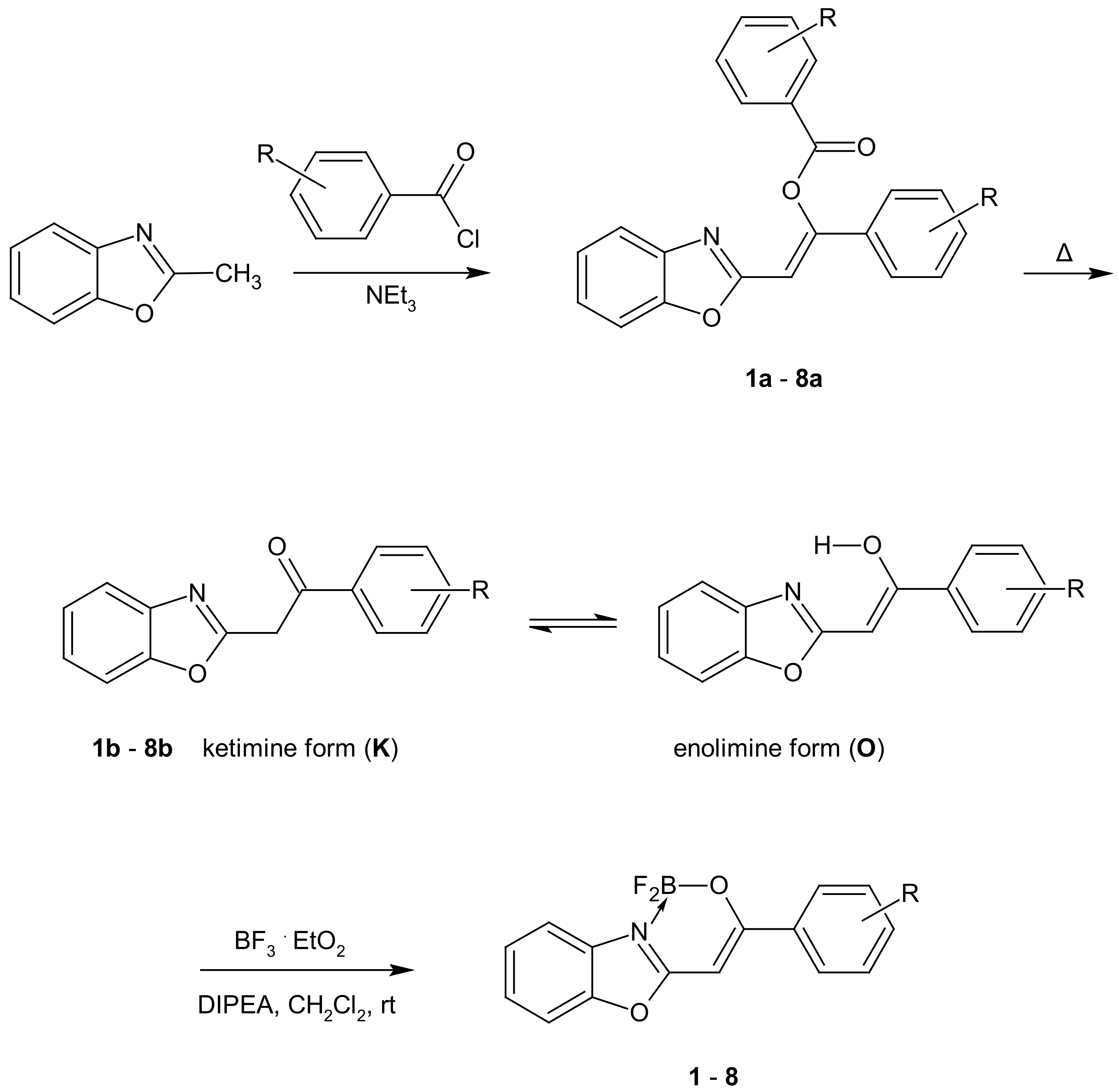
2.1. Synthesis and NMR Data
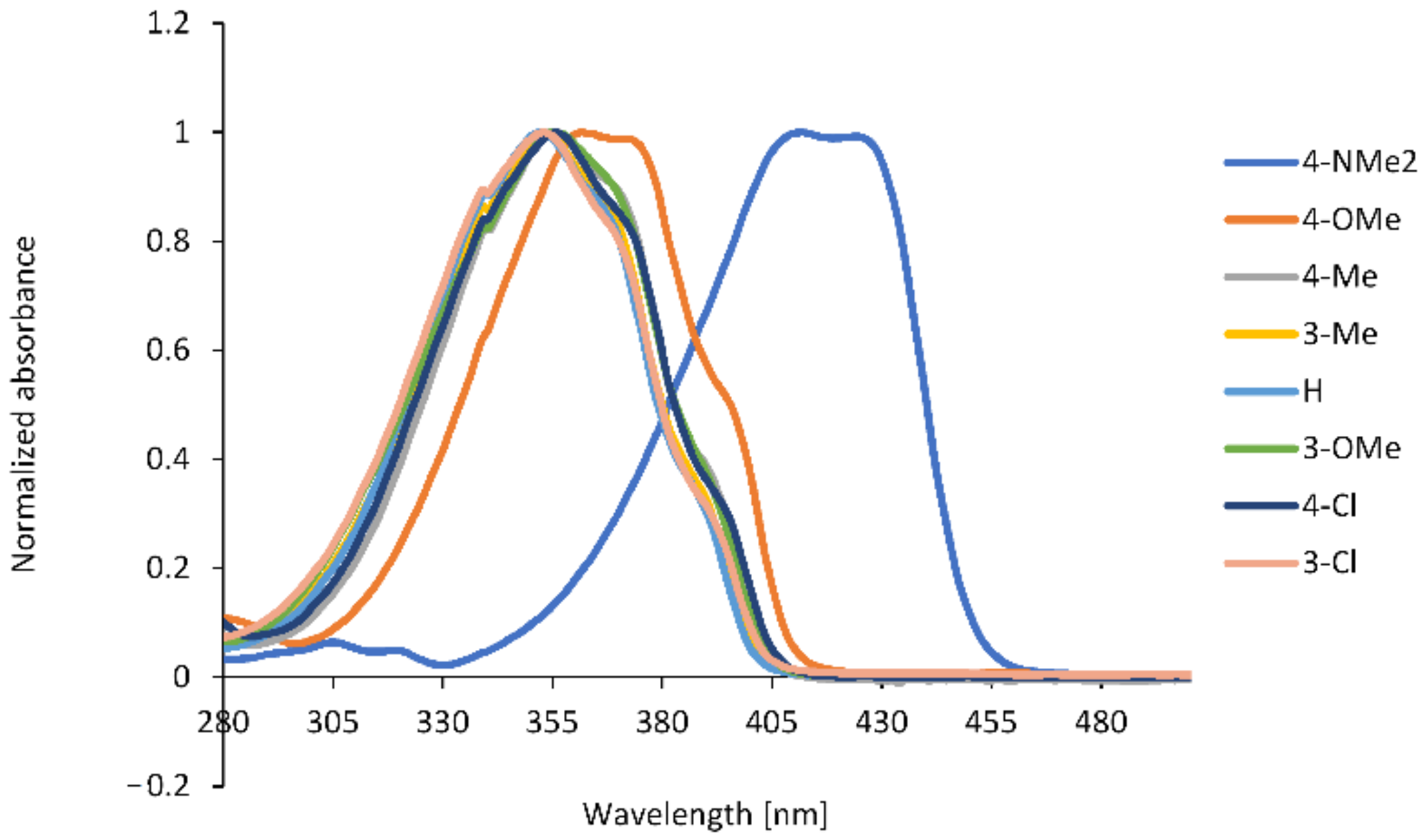
2.2. Spectroscopic Properties
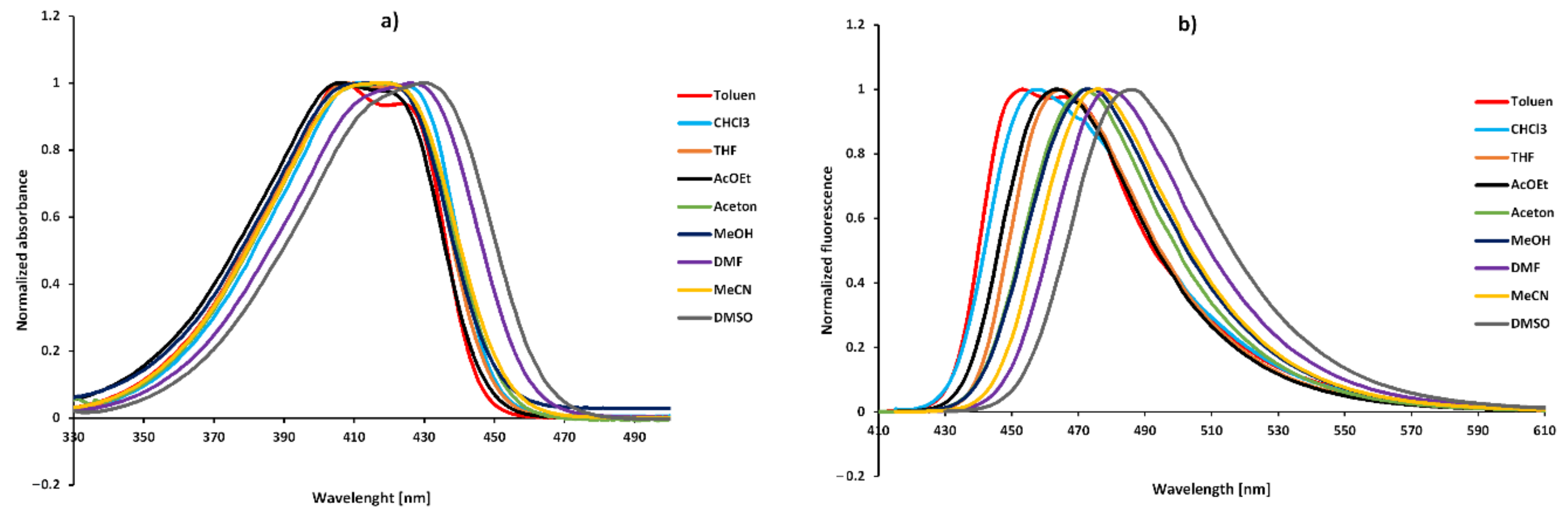
2.3. Solvatochromism
2.4. Computational Details
2.5. Vibrational Analysis
3. Experimental
3.1. Materials
3.2. Synthesis
Elemental Analysis Is as Follows
3.3. Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lu, H.; Mack, J.; Yang, Y.; Shen, Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. [Google Scholar] [CrossRef]
- Jean-Gérard, L.; Vasseur, W.; Scherninski, F.; Andrioletti, B. Recent advances in the synthesis of [a]-benzo-fused BODIPY fluorophores. Chem. Commun. 2018, 54, 12914–12929. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, K.; Yang, W.; Wang, Z.; Zhong, F. The triplet excited state of Bodipy: Formation, modulation and application. Chem. Soc. Rev. 2015, 44, 8904–8939. [Google Scholar] [CrossRef] [PubMed]
- Sansalone, L.; Tang, S.; Garcia-Amoros, J.; Zhang, Y.; Nonell, S.; Baker, J.D.; Captain, B.; Raymo, F.M. A photoactivatable far-red/near-infrared BODIPY to monitor cellular dynamics in vivo. ACS Sens. 2018, 3, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Boens, N.; Leen, V.; Dehaen, W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 2012, 41, 1130–1172. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Wu, J. Far-red and near infrared BODIPY dyes: Synthesis and applications for fluorescent pH probes and bio-imaging. Org. Biomol. Chem. 2014, 12, 3774–3791. [Google Scholar] [CrossRef] [PubMed]
- Telitel, S.; Blanchard, N.; Schweizer, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.-P.; Lalevee, J. BODIPY derivatives and boranil as new photoinitiating systems of cationic polymerization exhibiting a tunable absorption in the 400–600 nm spectral range. Polymer 2013, 54, 2071–2076. [Google Scholar] [CrossRef]
- Telitel, S.; Lalevee, J.; Blanchard, N.; Kavalli, T.; Tehfe, M.-A.; Schweizer, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.-P. Photopolymerization of cationic monomers and acrylate/divinylether blends under visible light using pyrromethene dyes. Macromolecules 2012, 45, 6864–6868. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Z.; Sun, J.; Xue, P.; Lu, R. Multi-color solid-state emission of β-iminoenolate boron complexes tuned by methoxyl groups: Aggregation-induced emission and mechanofluorochromism. RSC Adv. 2016, 6, 43755–43766. [Google Scholar] [CrossRef]
- Zhao, J.; Peng, J.; Chen, P.; Wang, H.; Xue, P.; Lu, R. Mechanofluorochromism of difluoroboron β-ketoiminate boron complexes functionalized with benzoxazole and benzothiazole. Dyes Pigm. 2018, 149, 276–283. [Google Scholar] [CrossRef]
- Grabarz, A.M.; Jędrzejewska, B.; Skotnicka, A.; Murugan, N.A.; Patalas, F.; Bartkowiak, W.; Jacquemin, D.; Ośmiałowski, B. The impact of the heteroatom in a five-membered ring on the photophysical properties of difluoroborates. Dyes Pigm. 2019, 170, 1074812. [Google Scholar] [CrossRef]
- Zakrzewska, A.; Zaleśny, R.; Kolehmainen, E.; Ośmiałowski, B.; Jędrzejewska, B.; Ågren, H.; Pietrzak, M. Substituent effects on the photophysical properties of fluorescent 2-benzoylmethylenequinoline difluoroboranes: A combined experimental and quantum chemical study. Dyes Pigm. 2013, 99, 957–965. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Zakrzewska, A.; Jędrzejewska, B.; Grabarz, A.M.; Zaleśny, R.; Bartkowiak, W.; Kolehmainen, E. Influence of substituent and benzoannulation on photophysical properties of 1-benzoylmethyleneisoquinoline difluoroborates. J. Org. Chem. 2015, 80, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Dzvinchuk, I.B.; Lozinskii, M.O.; Vypirailenko, A.V. C-Mono- and dibenzoylation of 2-methylbenzimidazole with use of benzoyl chloride. Zh. Org. Khim. 1994, 30, 909–914. [Google Scholar]
- Skotnicka, A.; Kolehmainen, E.; Czeleń, P.; Valkonen, A.; Gawinecki, R. Synthesis and structural characterization of substituted 2-phenacylbenzoxazoles. Int. J. Mol. Sci. 2013, 14, 4444–4460. [Google Scholar] [CrossRef]
- Kubota, Y.; Tanaka, S.; Funabiki, K.; Matsui, M. Synthesis and fluorescence properties of thiazole-boron complexes bearing a β-ketoiminate ligand. Org. Lett. 2012, 14, 4682–4685. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Xu, S.; Evans, R.E.; Liu, T.; Zhang, G.; Demas, J.N.; Trindle, C.O.; Frser, C.L. Aromatic difluoroboron β-diketonate complexes: Effects of π-conjugation and media on optical properties. Inorg. Chem. 2013, 52, 3597–3610. [Google Scholar] [CrossRef]
- Patil, R.S.; Patil, A.S.; Patil, V.S.; Jirimali, H.D.; Mahulikar, P.P. Synthesis, photophysical, solvatochromic and DFT studies of (Z)-2-(2-Phenyl-4H-benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ylidene)acetonitrile derivatives. J. Lumin. 2019, 210, 303–310. [Google Scholar] [CrossRef]
- Chitnis, D.; Thejokalyani, N.; Dhoble, S.J. Exploration of spectroscopic properties of solvated tris(thenoyltrifluoroacetonate)(2,2′-bipyridine)europium(III)red hybrid organic complex for solution processed OLEDs and displays. J. Lumin. 2017, 185, 61–71. [Google Scholar] [CrossRef]
- Vennila, P.; Govindaraju, M.; Venkatesh, G.; Kamal, C. Molecular structure, vibrational spectral assignments (FT-IR and FT-RAMAN), NMR, NBO, HOMO-LUMO and NLO properties of O-methoxybenzaldehyde based on DFT calculations. J. Mol. Struct. 2016, 1111, 151–156. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X. Spectroscopic Identification of Organic Compound, 6th ed.; John Willey & Sons: New York, NY, USA, 1998. [Google Scholar]
- Yoshida, H.; Takeda, K.; Okamura, J.; Ehara, A.; Matsurra, H. A new approach to vibrational analysis of large molecules by density functional theory: Wavenumber-linear scaling method. J. Phys. Chem. A 2002, 106, 3580–3586. [Google Scholar] [CrossRef]
- Meech, S.R.; Phillips, D. Photophysics of some common fluorescence standards. J. Photochem. 1983, 23, 193–217. [Google Scholar] [CrossRef]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef]
- Jones, G., II; Jackson, W.R.; Choi, C.Y.; Bergmark, W.R. Solvent effects on emission yield and lifetime for coumarin laser dyes. Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985, 89, 294–300. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ong, B.K.; Woon, K.L.; Ariffin, A. Evaluation of various density functionals for predicting the electrophosphorescent host HOMO, LUMO and triplet energies. Synth. Met. 2014, 195, 54–60. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Frisch, D.J.; Trucks, M.J.; Schlegel, G.W.; Scuseria, H.B.; Robb, G.E.; Cheeseman, M.A.; Scalmani, J.R.; Barone, G.; Petersson, V.; Nakatsuji, G.A.; et al. Gaussian 09 (Revision A. 02); Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Substituent | C10 | C11 | H10 | B13 | F13 | N13 |
|---|---|---|---|---|---|---|---|
| 1 | 4-NMe2 | 77.74 | 172.13 | 6.81 | 1.79 | −134.64 | - |
| 2 | 4-OMe | 78.85 | 172.23 | 6.37 | 1.80 | −135.59 | −215.72 |
| 3 | 4-Me | 79.74 | 172.63 | 6.44 | 1.84 | −135.34 | −215.01 |
| 4 | 3-Me | 80.33 | 172.70 | 6.47 | 1.85 | −135.14 | −214.18 |
| 5 | H | 82.11 | 170.79 | 6.49 | 1.85 | −135.07 | −213.54 |
| 6 | 3-OMe | 80.56 | 172.28 | 6.47 | 1.84 | −134.98 | −213.97 |
| 7 | 4-Cl | 80.63 | 171.01 | 6.45 | 1.81 | −134.98 | −213.00 |
| 8 | 3-Cl | 81.17 | 170.64 | 6.48 | 1.81 | −134.78 | −212.43 |
| No. | Substituent | λabs (nm) | λfl (nm) | ε (M−1·cm−1) | Stokes Shift (cm−1) | ϕfl (× 10−2) |
|---|---|---|---|---|---|---|
| 1 | 4-NMe2 | 410 | 458 | 42,500 | 2449 | 98.18 |
| 2 | 4-OMe | 362 | 433 | 32,900 | 4530 | 1.82 |
| 3 | 4-Me | 354 | 428 | 31,219 | 4884 | 0.66 |
| 4 | 3-Me | 352 | 423 | 22,961 | 4769 | 0.48 |
| 5 | H | 350 | 424 | 27,000 | 4905 | 0.54 |
| 6 | 3-OMe | 354 | 429 | 26,727 | 4938 | 0.57 |
| 7 | 4-Cl | 355 | 428 | 29,615 | 4805 | 0.58 |
| 8 | 3-Cl | 353 | 430 | 29,648 | 5073 | 0.50 |
| No. | Toluene | CHCl3 | THF | AcOEt | Acetone | MeOH | DMF | MeCN | DMSO | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | λabs (nm) | 407 | 410 | 408 | 407 | 420 | 419 | 426 | 415 | 431 |
| λflu (nm) | 453 | 458 | 465 | 462 | 427 | 473 | 479 | 475 | 486 | |
| Stokes (cm−1) | 2495 | 2449 | 3004 | 2925 | 2623 | 2725 | 2597 | 3043 | 2626 | |
| 2 | λabs (nm) | 362 | 362 | 361 | 359 | 360 | 358 | 363 | 359 | 375 |
| λflu (nm) | 450 | 433 | 432 | 435 | 432 | 431 | 435 | 463 | 438 | |
| Stokes (cm−1) | 5402 | 4530 | 4553 | 4867 | 4630 | 4731 | 4560 | 6257 | 3836 | |
| 3 | λabs (nm) | 355 | 354 | 354 | 352 | 352 | 351 | 355 | 351 | 356 |
| λflu (nm) | 434 | 428 | 429 | 432 | 426 | 471 | 431 | 455 | 431 | |
| Stokes (cm−1) | 5127 | 4884 | 4938 | 5261 | 4935 | 7259 | 4967 | 6512 | 4888 | |
| 4 | λabs (nm) | 354 | 352 | 353 | 351 | 351 | 349 | 353 | 350 | 355 |
| λflu (nm) | 435 | 423 | 427 | 433 | 430 | 424 | 429 | 448 | 430 | |
| Stokes (cm−1) | 5261 | 4769 | 4909 | 5395 | 5234 | 5068 | 5019 | 6250 | 4913 | |
| 5 | λabs (nm) | 354 | 350 | 351 | 349 | 349 | 349 | 352 | 349 | 354 |
| λflu (nm) | 433 | 424 | 425 | 447 | 433 | 425 | 427 | 447 | 456 | |
| Stokes (cm−1) | 5154 | 4905 | 5071 | 6282 | 5559 | 5124 | 4990 | 6282 | 4993 | |
| 6 | λabs (nm) | 356 | 354 | 354 | 352 | 352 | 351 | 355 | 351 | 357 |
| λflu (nm) | 436 | 429 | 428 | 431 | 432 | 426 | 432 | 434 | 455 | |
| Stokes (cm−1) | 5154 | 4938 | 4884 | 5207 | 5261 | 5016 | 5021 | 5448 | 4916 | |
| 7 | λabs (nm) | 357 | 355 | 355 | 352 | 353 | 352 | 355 | 352 | 358 |
| λflu (nm) | 458 | 428 | 456 | 448 | 456 | 463 | 459 | 455 | 460 | |
| Stokes (cm−1) | 6431 | 4805 | 6239 | 6088 | 5601 | 6545 | 6382 | 6447 | 6462 | |
| 8 | λabs (nm) | 354 | 353 | 352 | 351 | 351 | 350 | 353 | 350 | 354 |
| λflu (nm) | 456 | 430 | 454 | 454 | 454 | 454 | 456 | 452 | 459 | |
| Stokes (cm−1) | 6319 | 5037 | 6383 | 6464 | 6464 | 6545 | 6399 | 6447 | 6462 |
| No. | Substituent | HOMO (eV) | LUMO (eV) | Energy gap (eV) | η (eV) |
|---|---|---|---|---|---|
| 1 | 4-NMe2 | −5.664 | −2.090 | 3.574 | 1.787 |
| 2 | 4-OMe | −6.168 | −2.323 | 3.846 | 1.923 |
| 3 | 4-Me | −6.328 | −2.423 | 3.906 | 1.953 |
| 4 | 3-Me | −6.389 | −2.454 | 3.935 | 1.967 |
| 5 | H | −9.438 | −2.504 | 3.934 | 1.967 |
| 6 | 3-OMe | −6.390 | −2.425 | 3.964 | 1.982 |
| 7 | 4-Cl | −6.523 | −2.663 | 3.860 | 1.930 |
| 8 | 3-Cl | −6.595 | −2.684 | 3.910 | 1.955 |
Sample Availability: Samples of the compounds 1–8 are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skotnicka, A.; Czeleń, P. Substituted 2-Phenacylbenzoxazole Difluoroboranes: Synthesis, Structure and Properties. Molecules 2020, 25, 5420. https://doi.org/10.3390/molecules25225420
Skotnicka A, Czeleń P. Substituted 2-Phenacylbenzoxazole Difluoroboranes: Synthesis, Structure and Properties. Molecules. 2020; 25(22):5420. https://doi.org/10.3390/molecules25225420
Chicago/Turabian StyleSkotnicka, Agnieszka, and Przemysław Czeleń. 2020. "Substituted 2-Phenacylbenzoxazole Difluoroboranes: Synthesis, Structure and Properties" Molecules 25, no. 22: 5420. https://doi.org/10.3390/molecules25225420
APA StyleSkotnicka, A., & Czeleń, P. (2020). Substituted 2-Phenacylbenzoxazole Difluoroboranes: Synthesis, Structure and Properties. Molecules, 25(22), 5420. https://doi.org/10.3390/molecules25225420