
Recent Advances in the Reactions of Cyclic Carbynes

Abstract
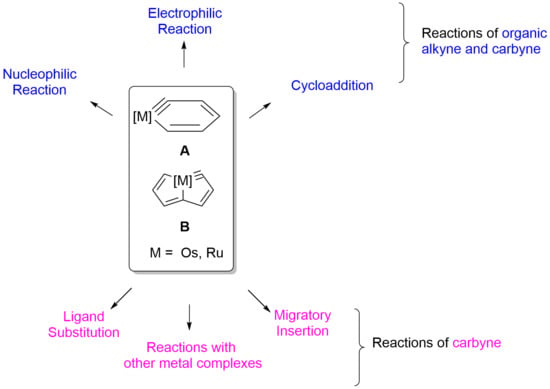
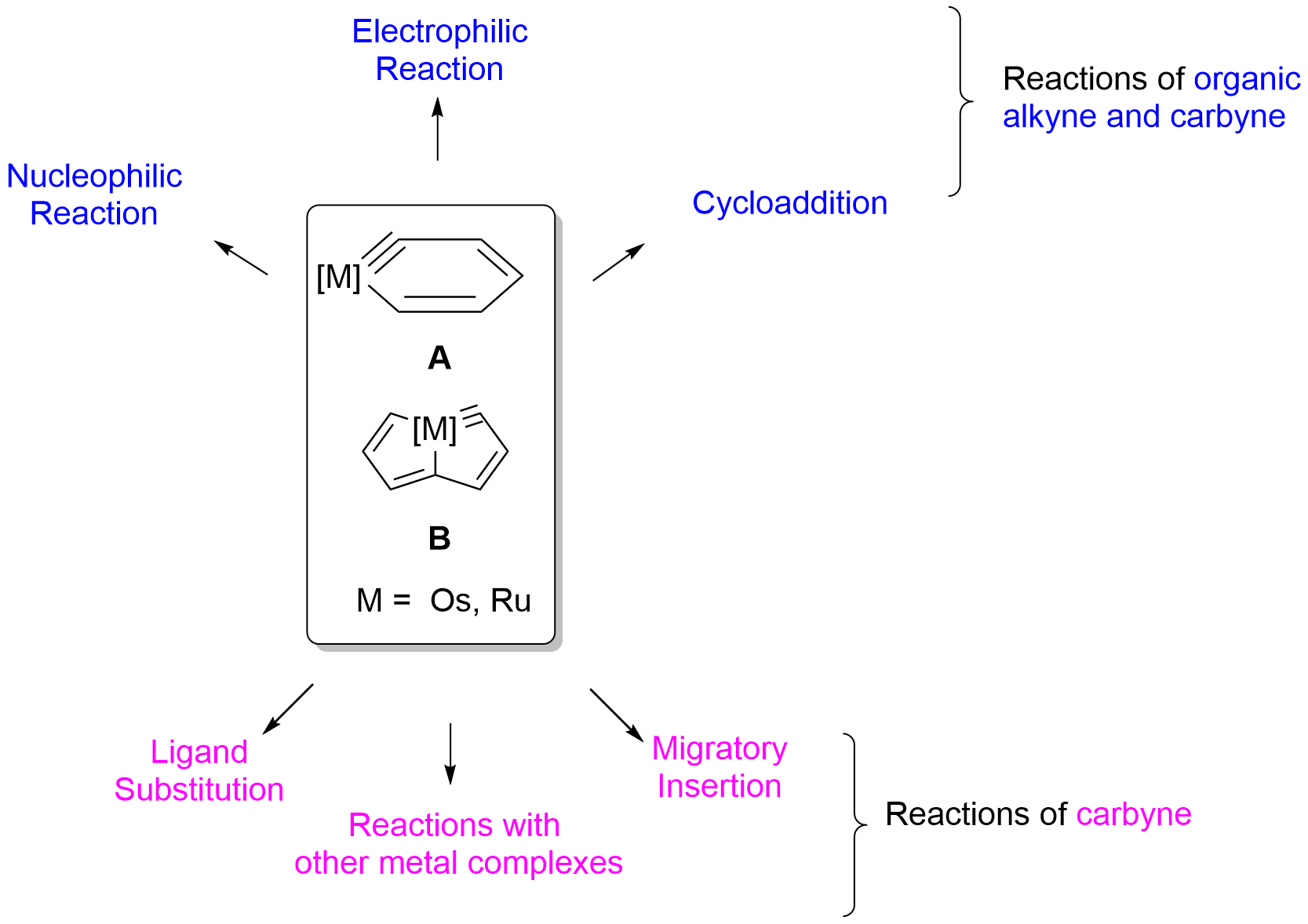{kind=link}
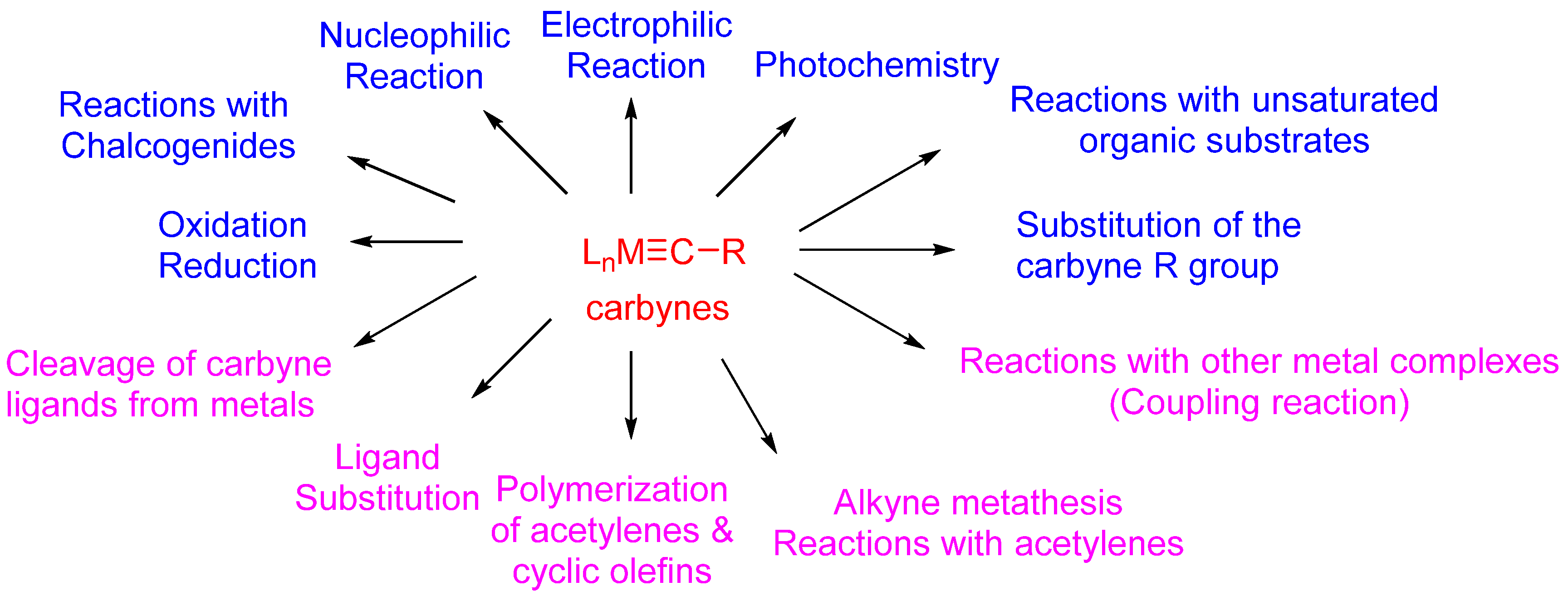{kind=link}
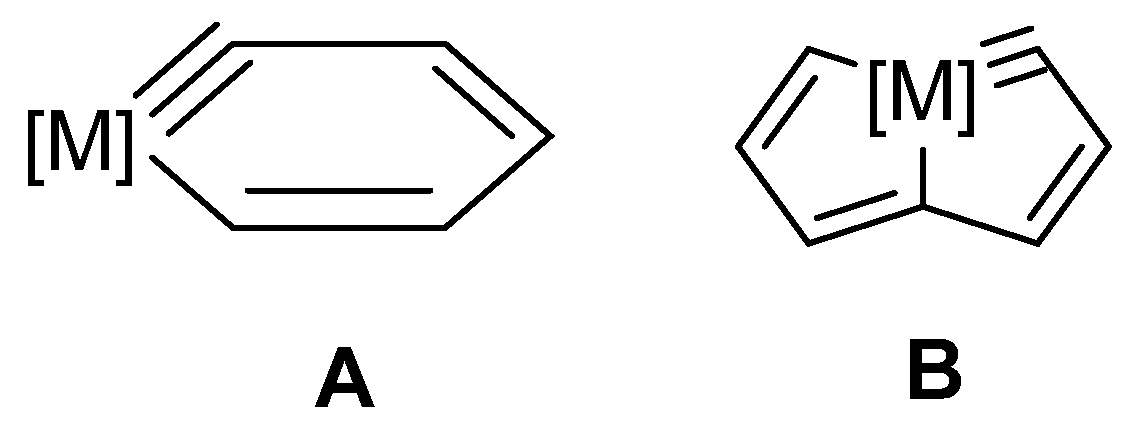{kind=link}
{kind=link}
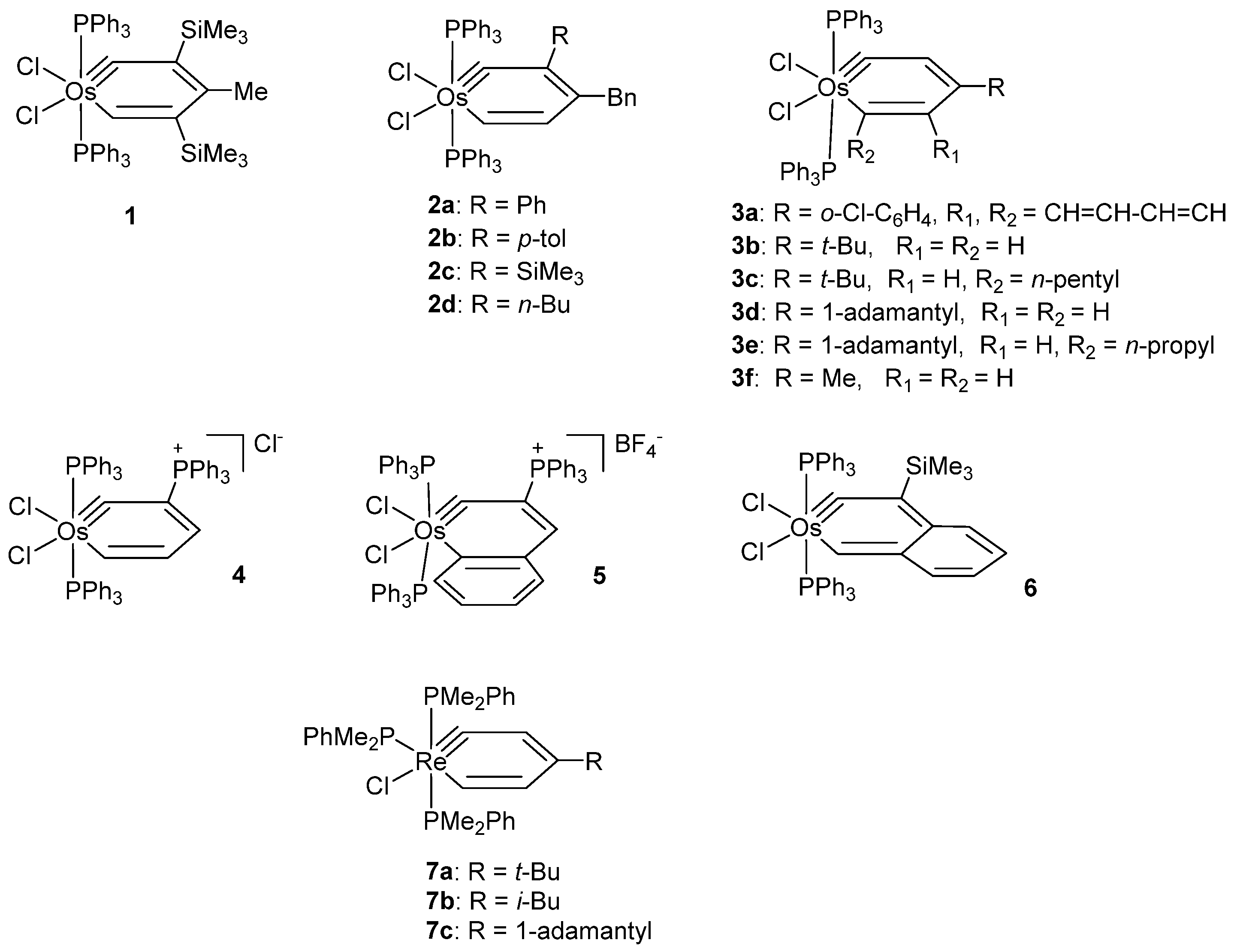{kind=link}
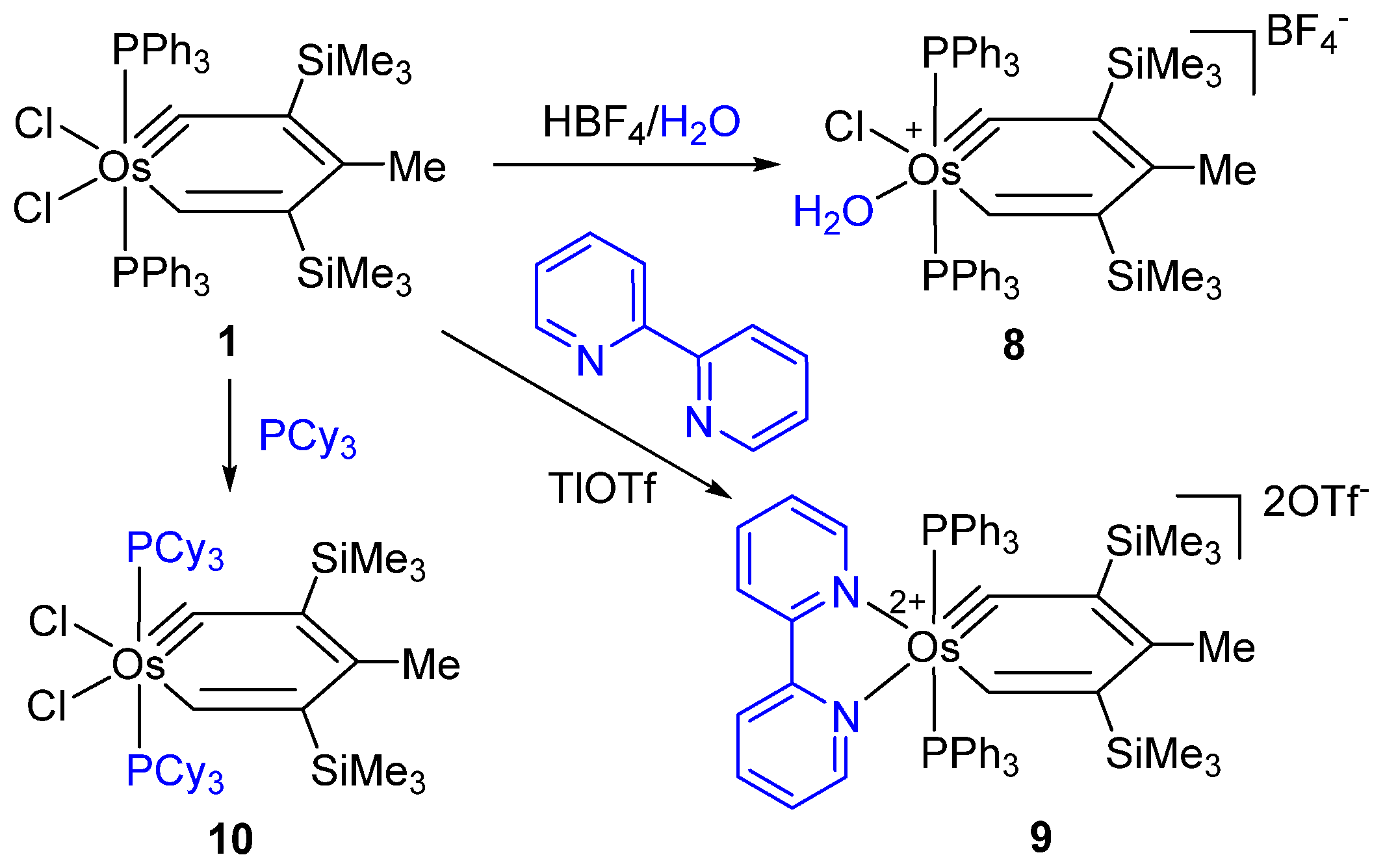{kind=link}
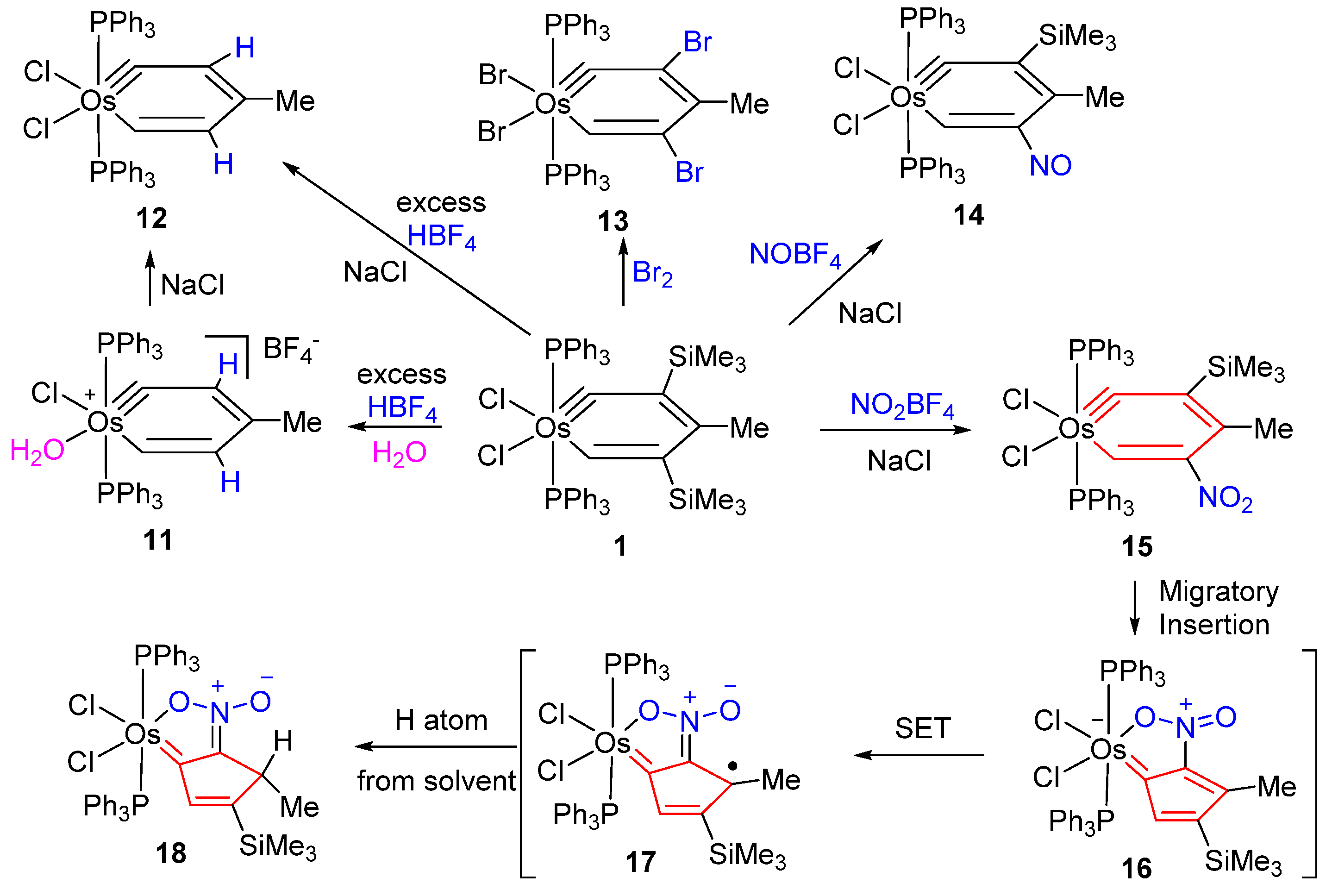{kind=link}
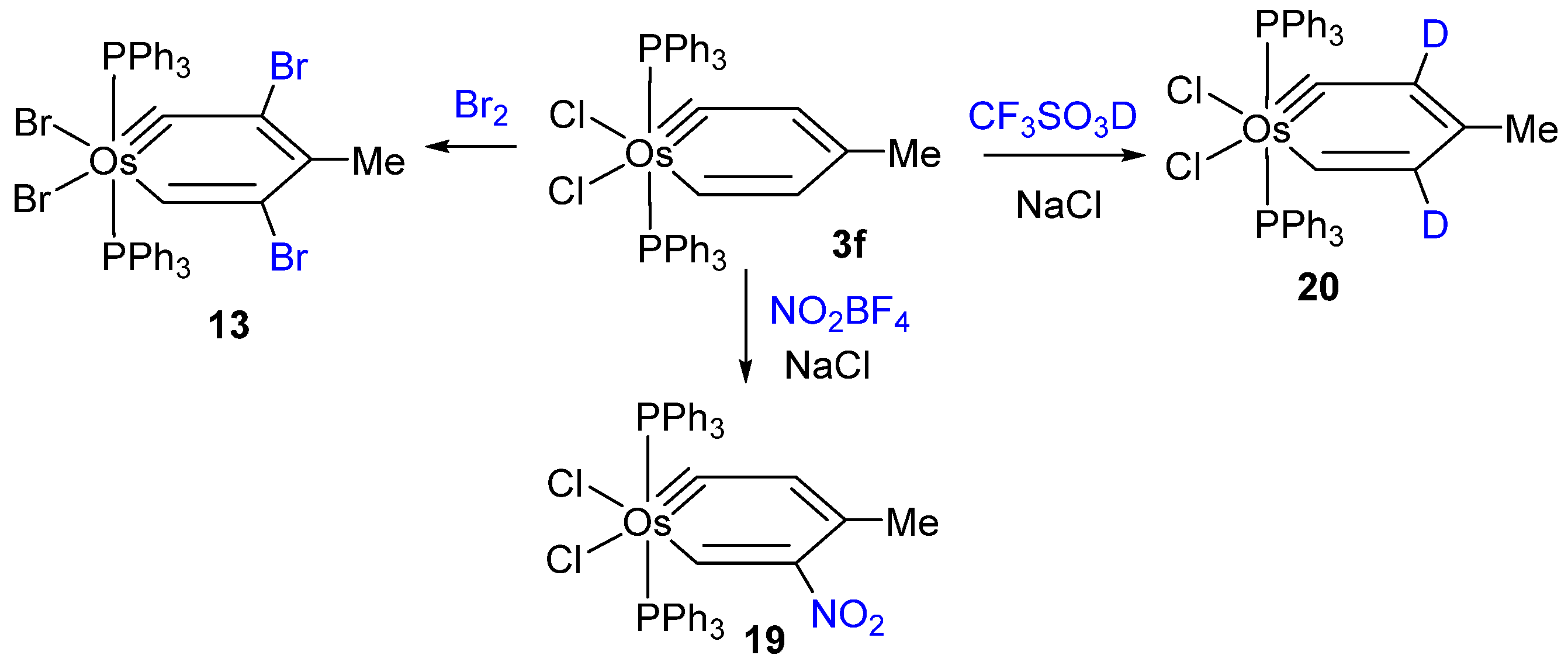{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Reactivity of Six-Membered Metallabenzynes
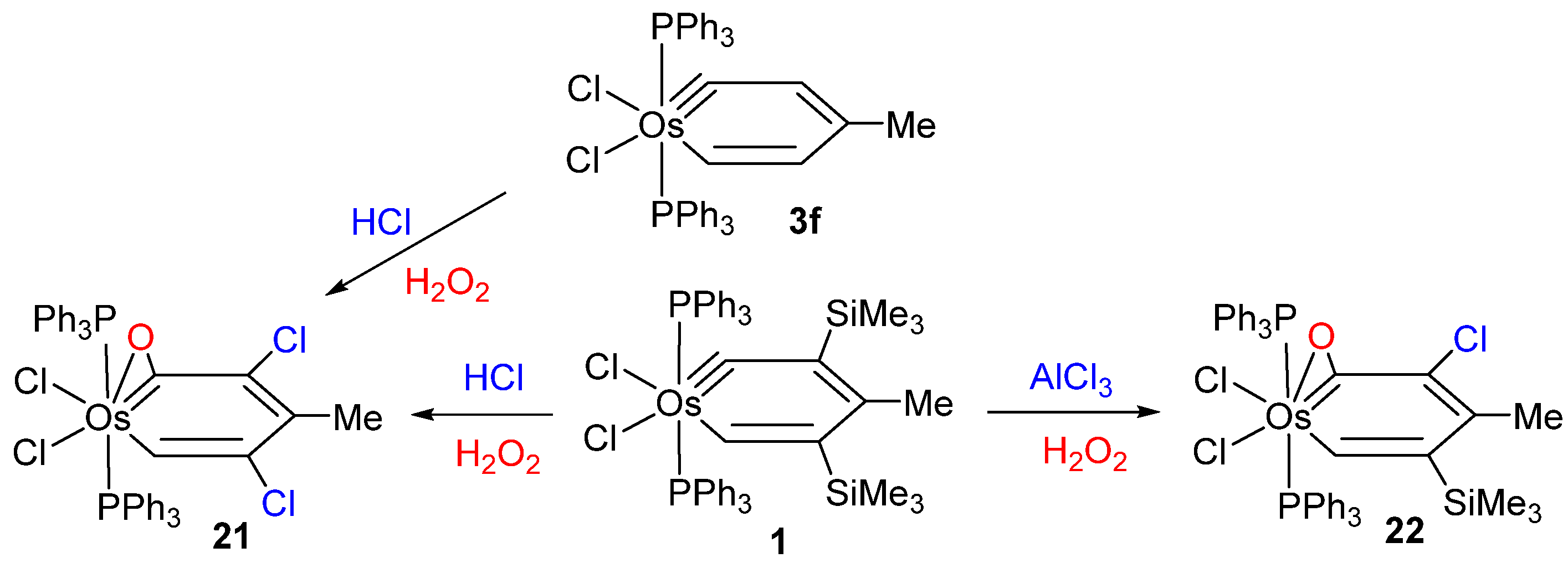
2.1. Electrophilic Substitution Reaction
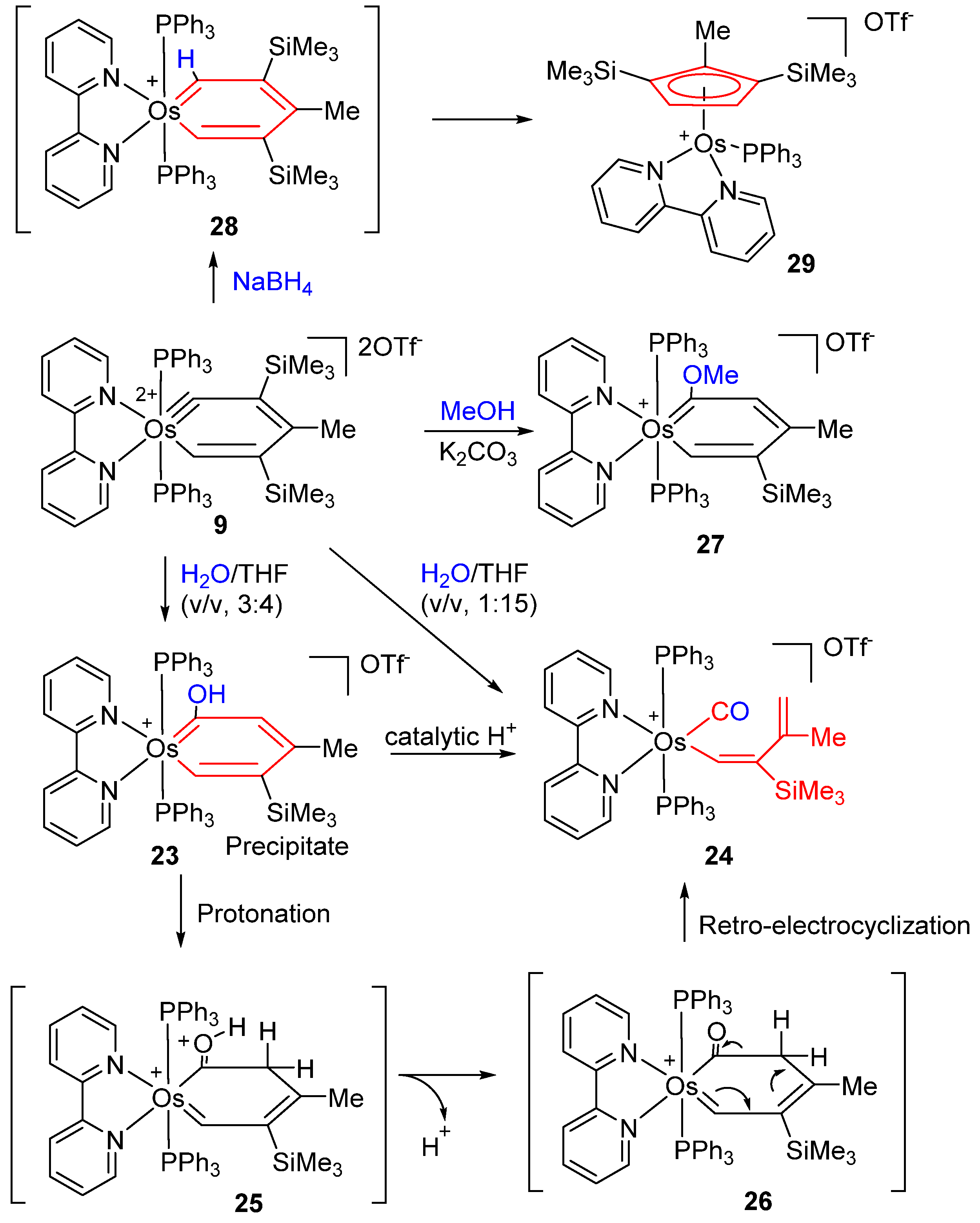
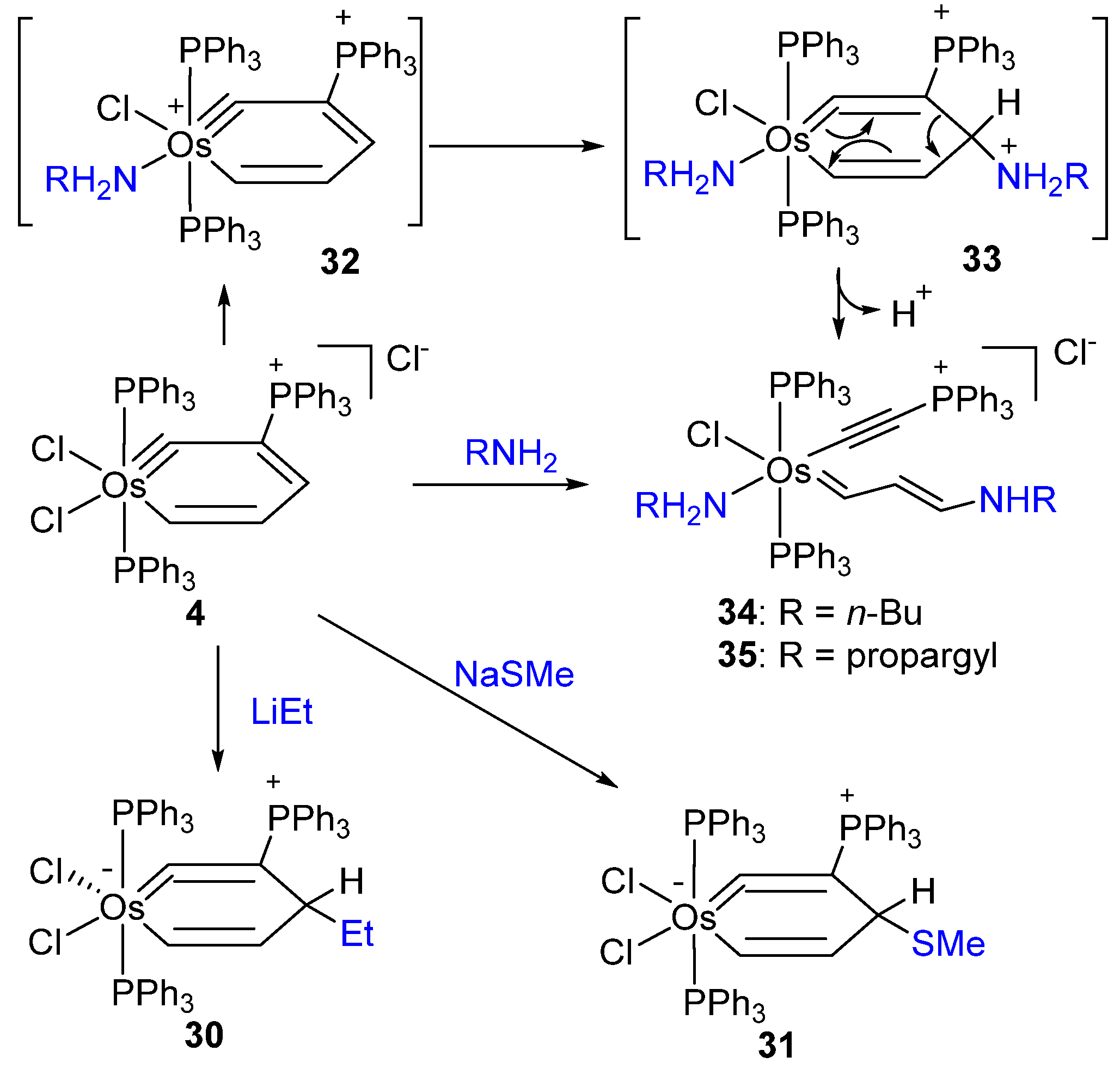
2.2. Nucleophilic Reaction
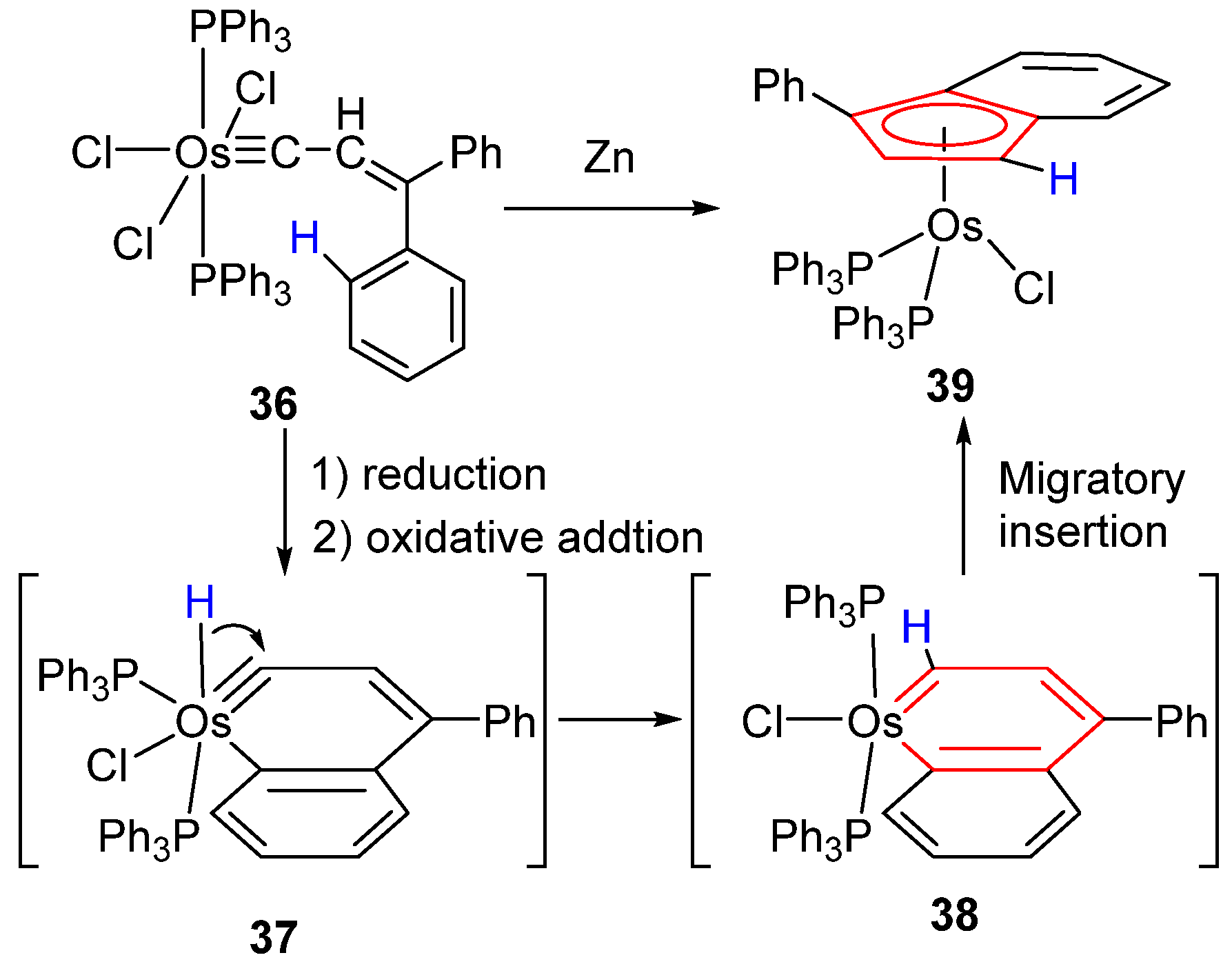
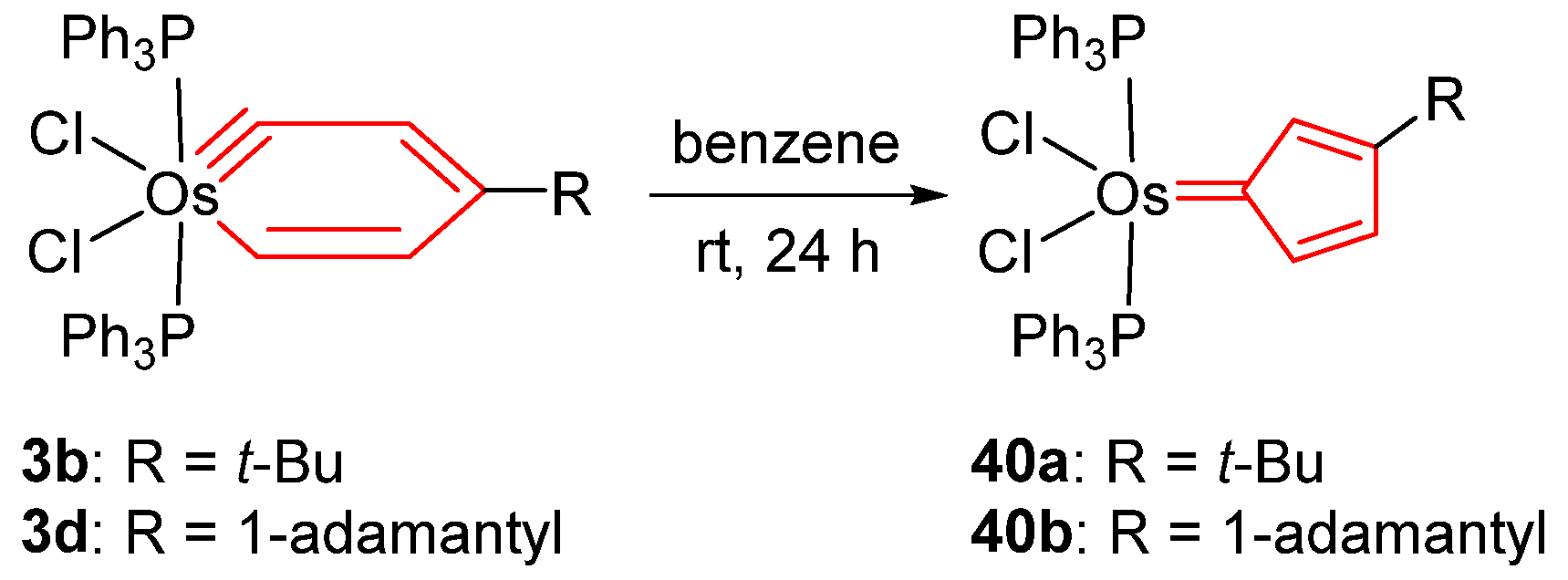
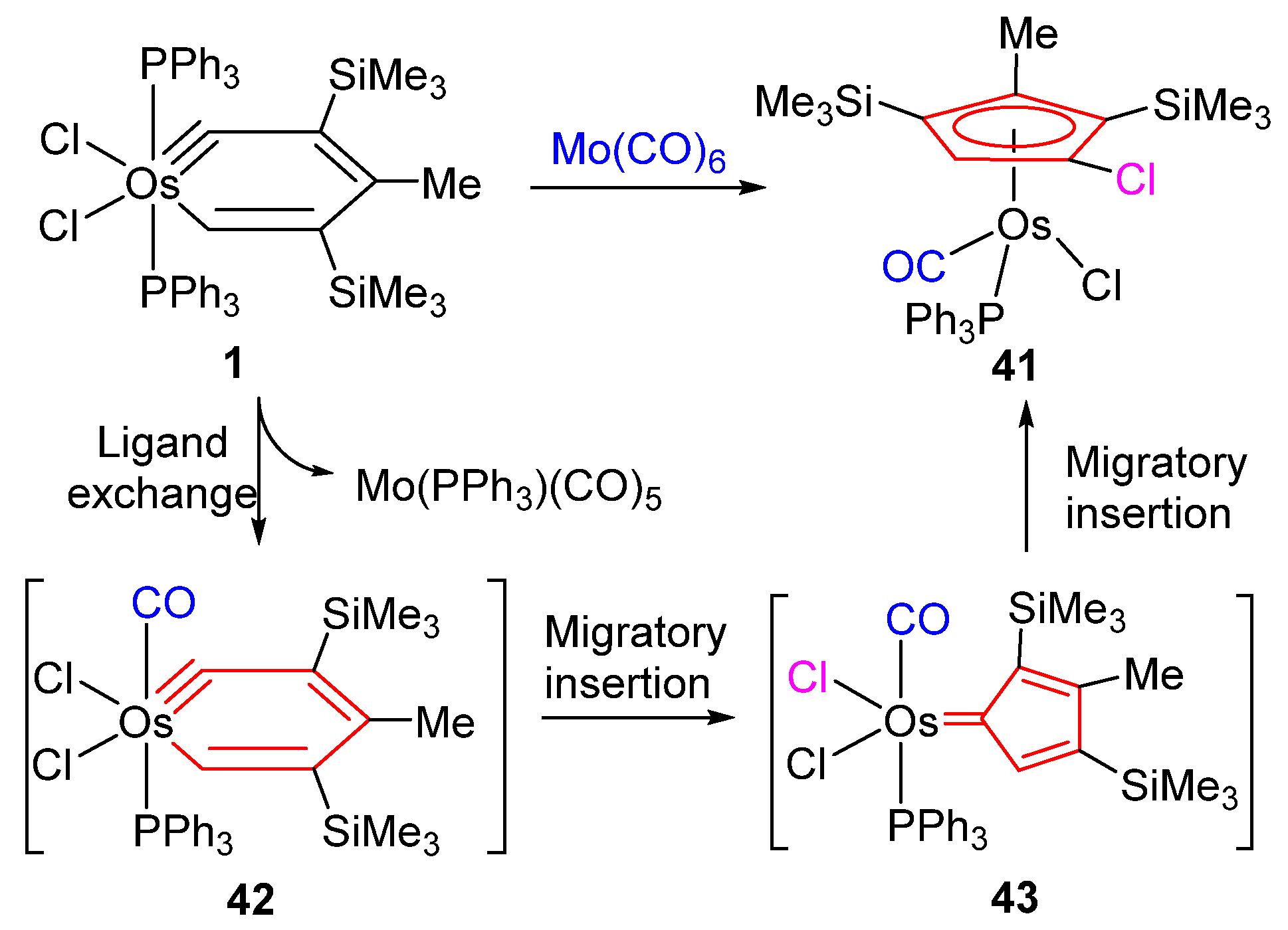
2.3. Migratory Insertion Reactions
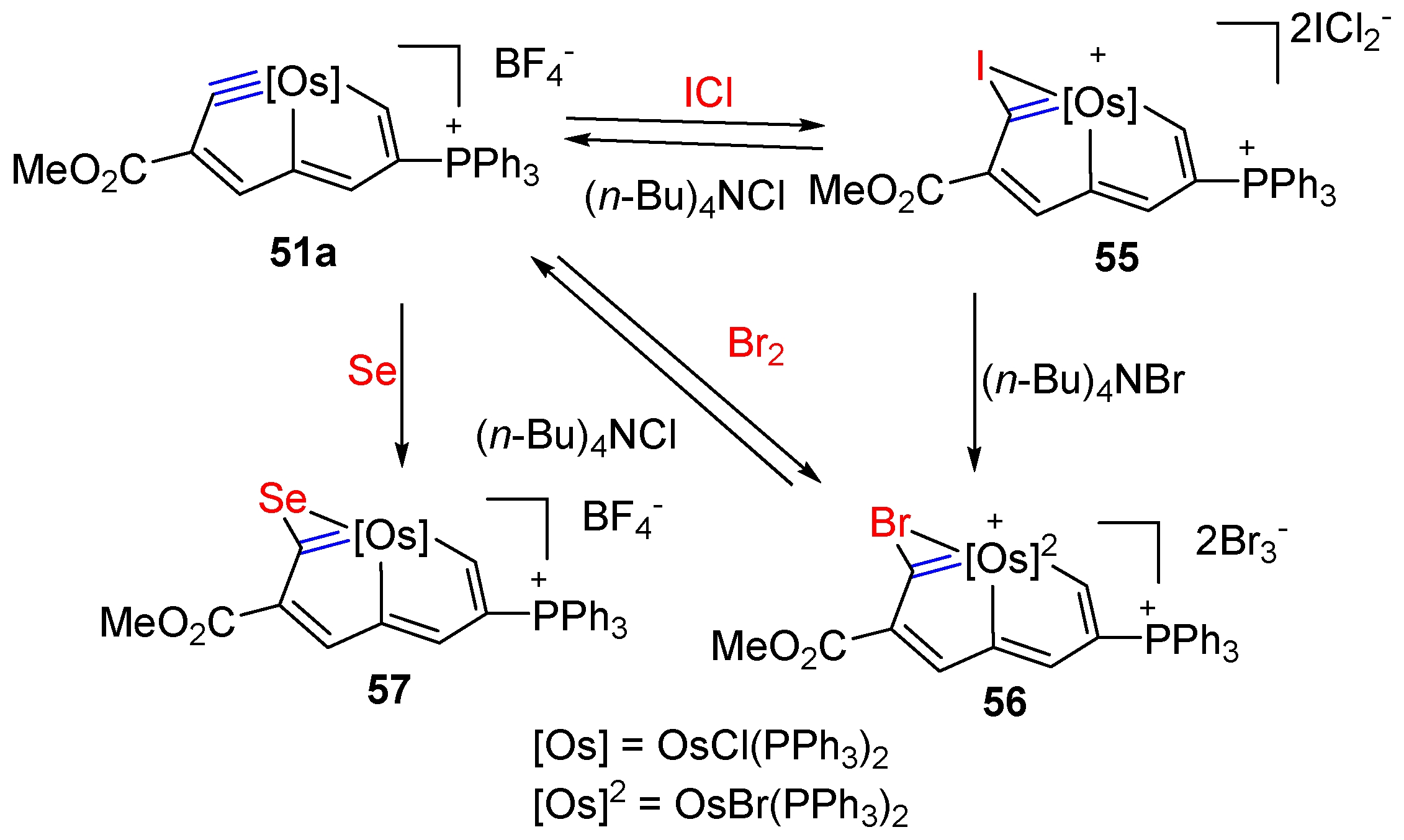
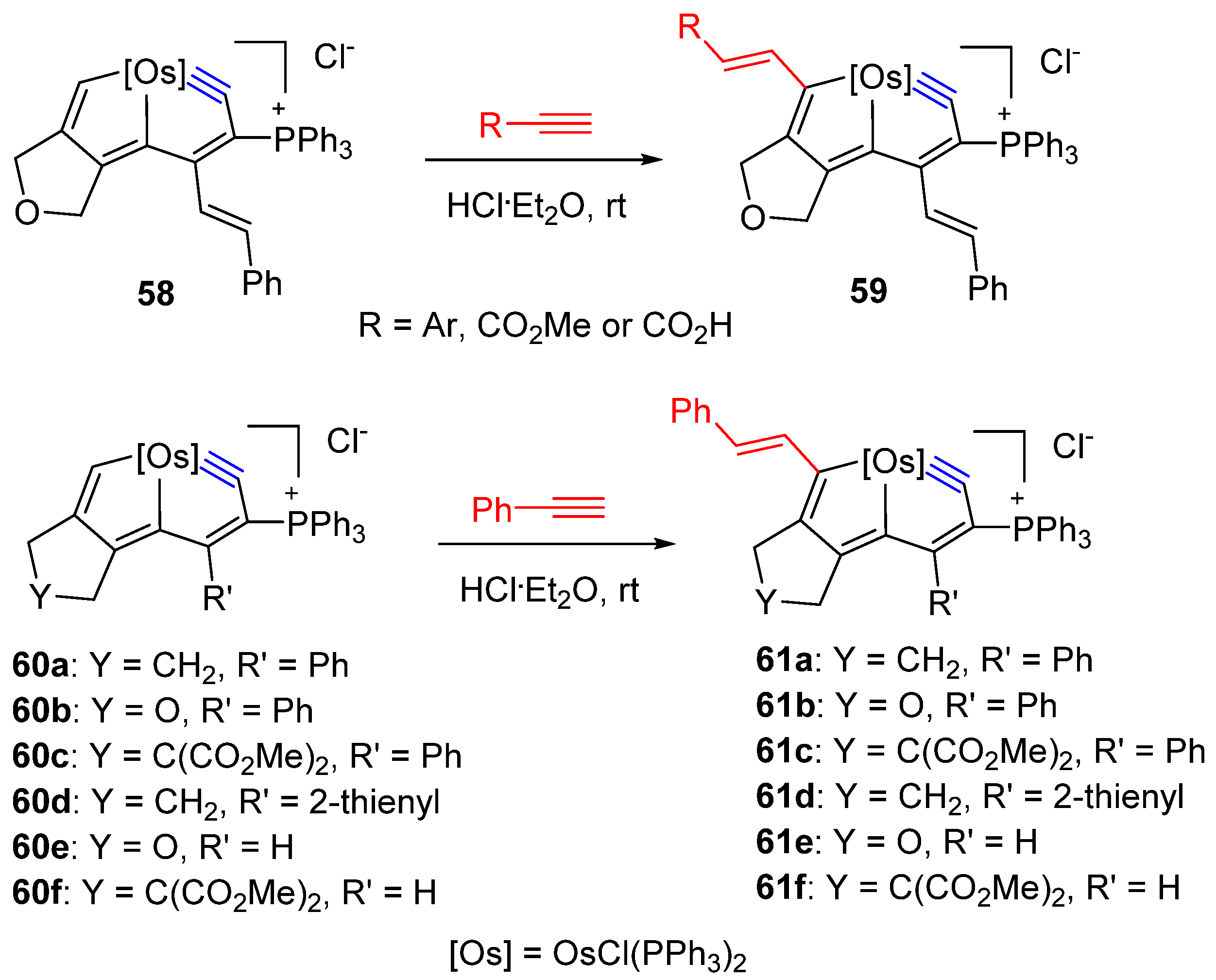
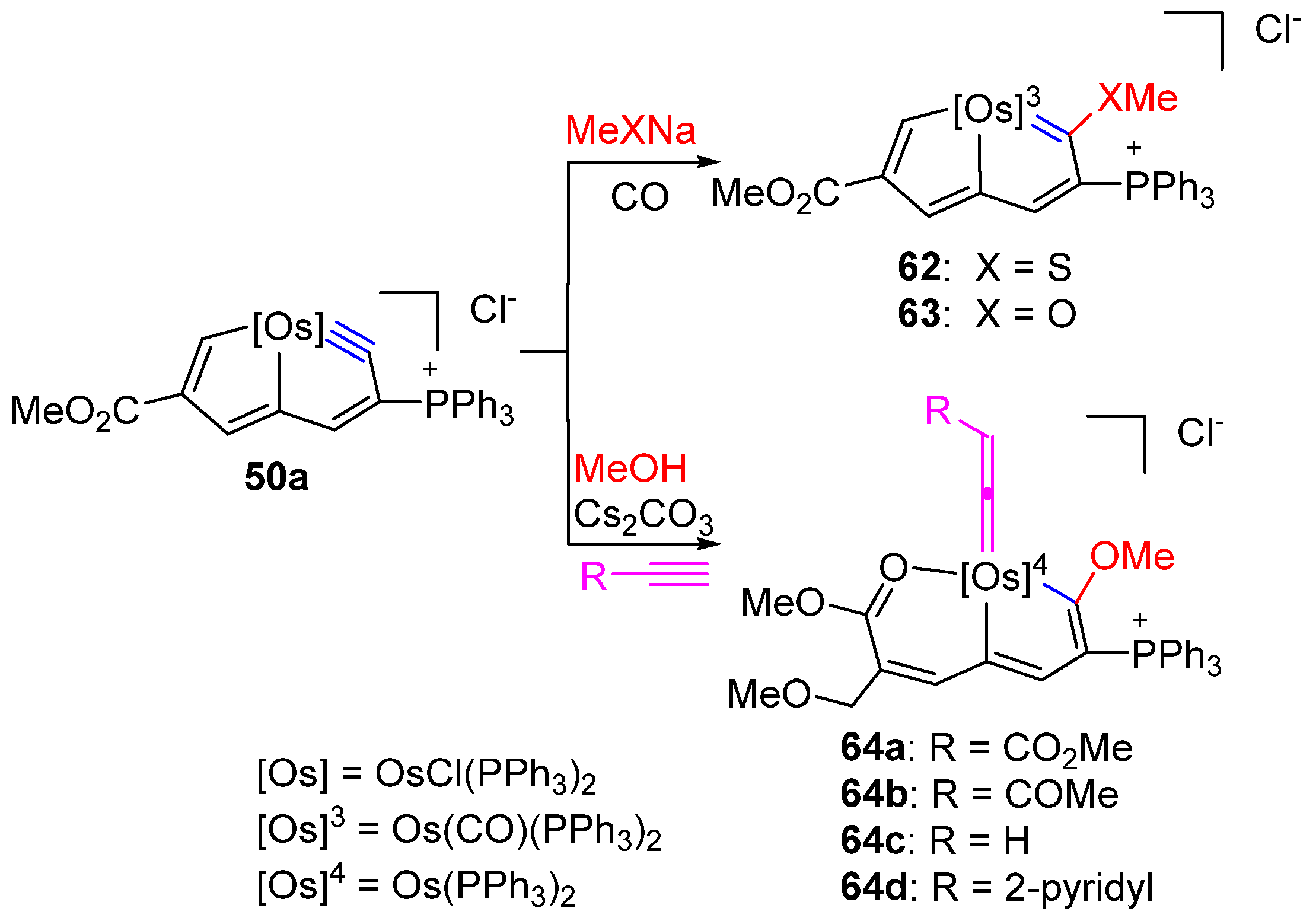
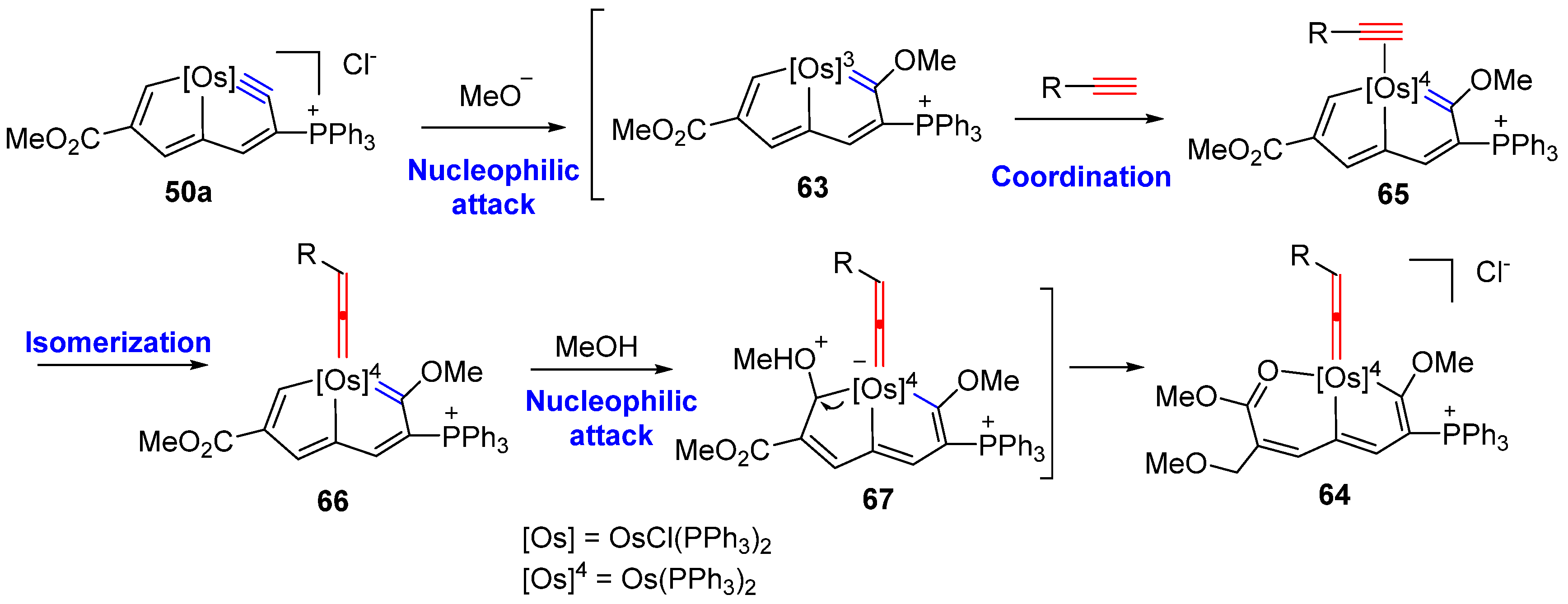
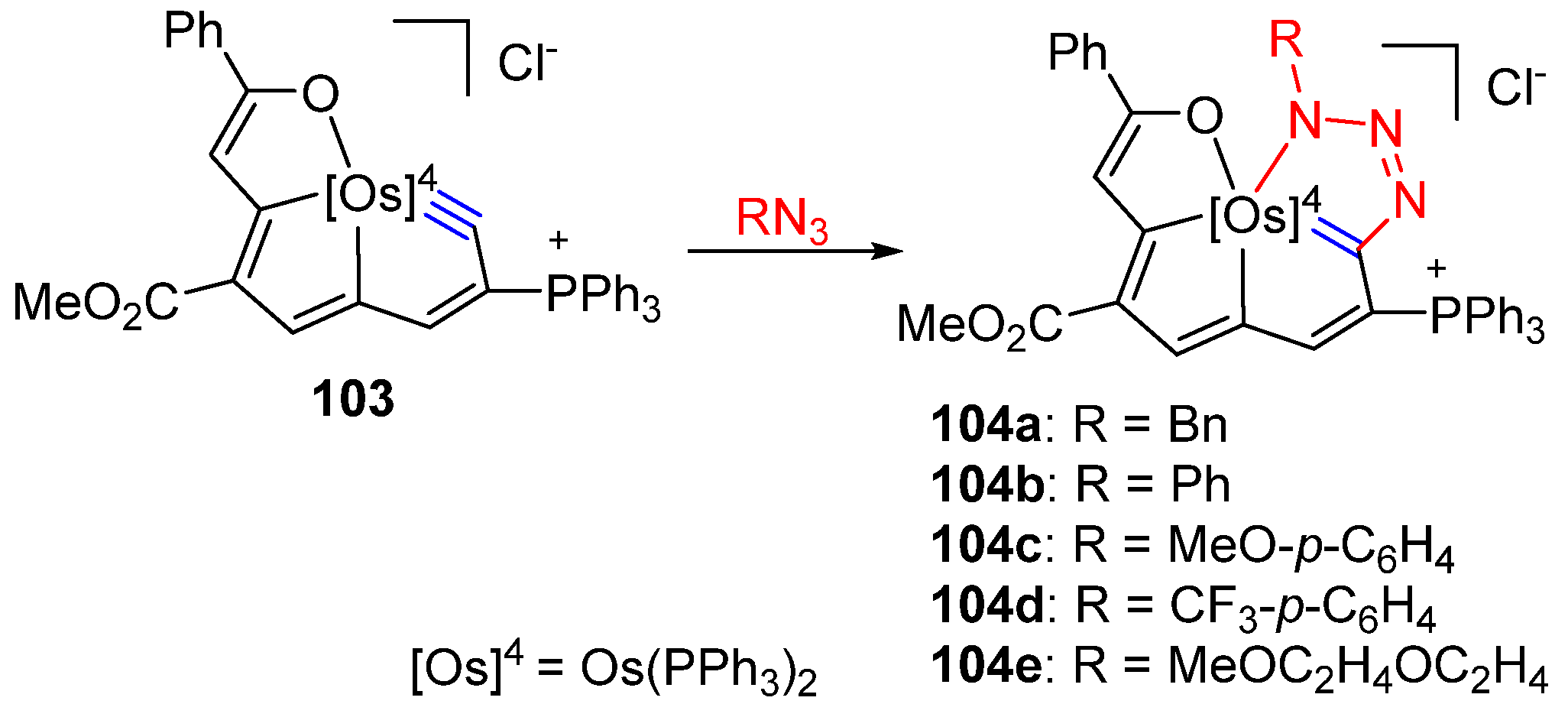
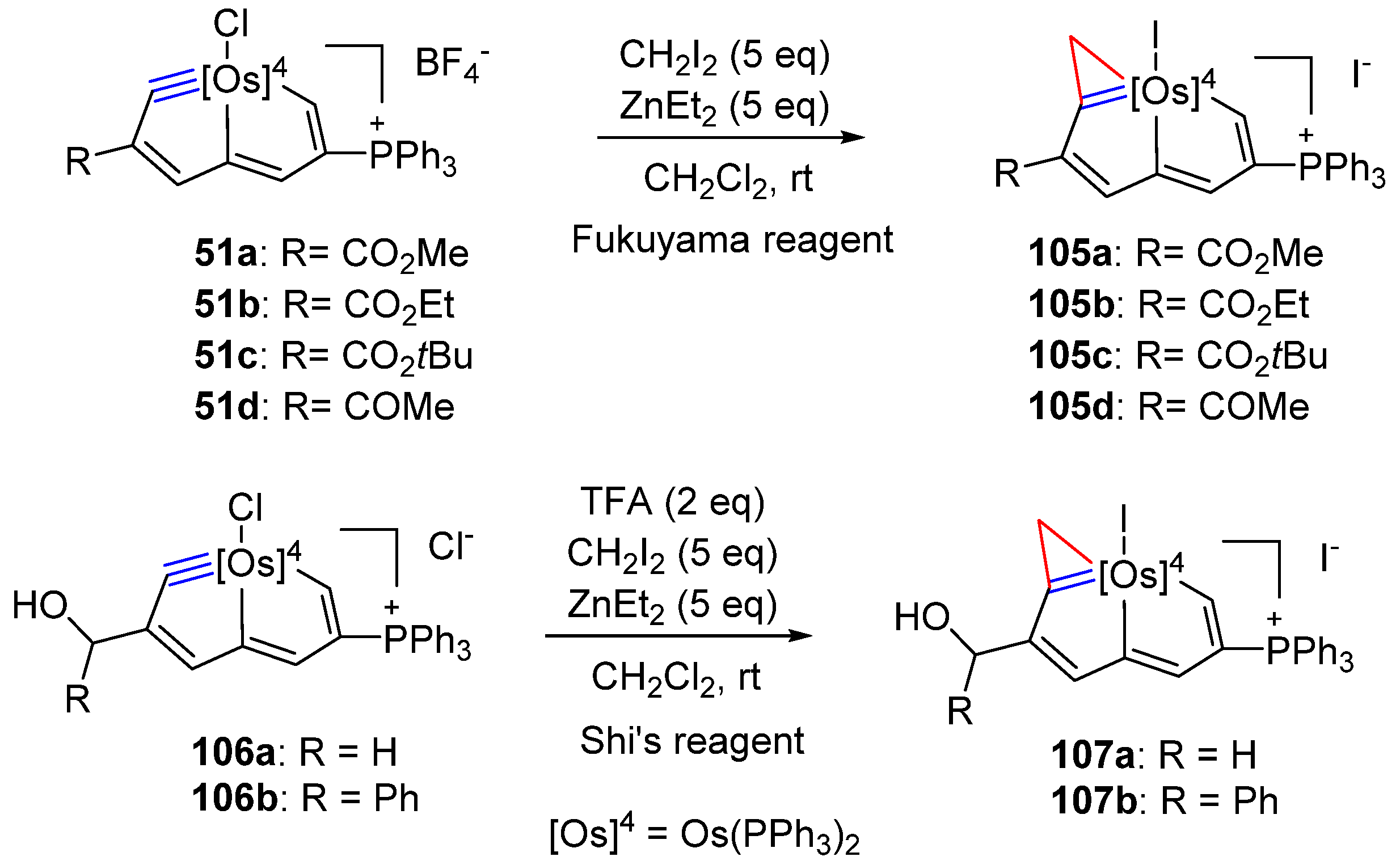
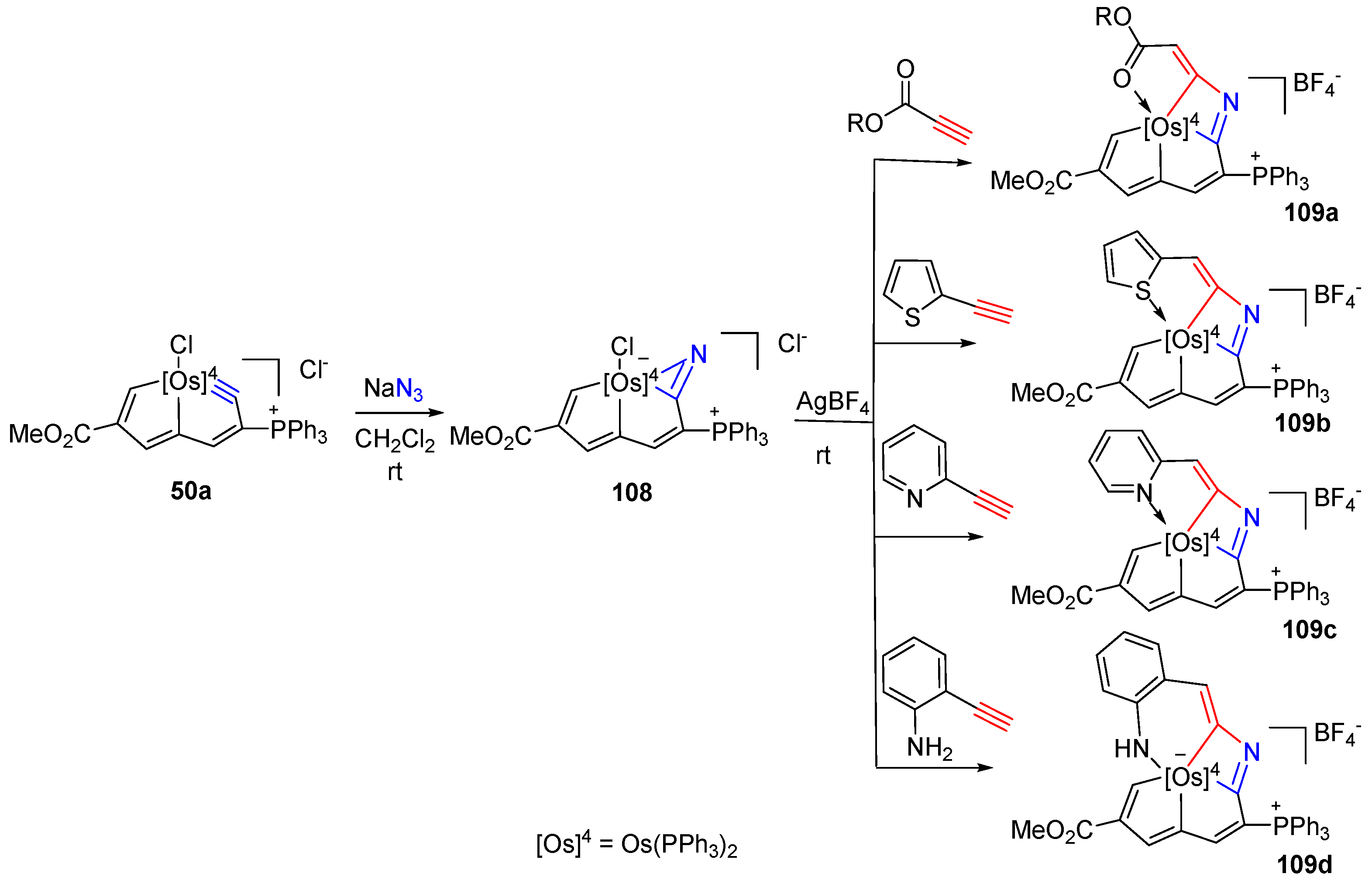
3. Reactivity of Five-Membered Metallapentalynes
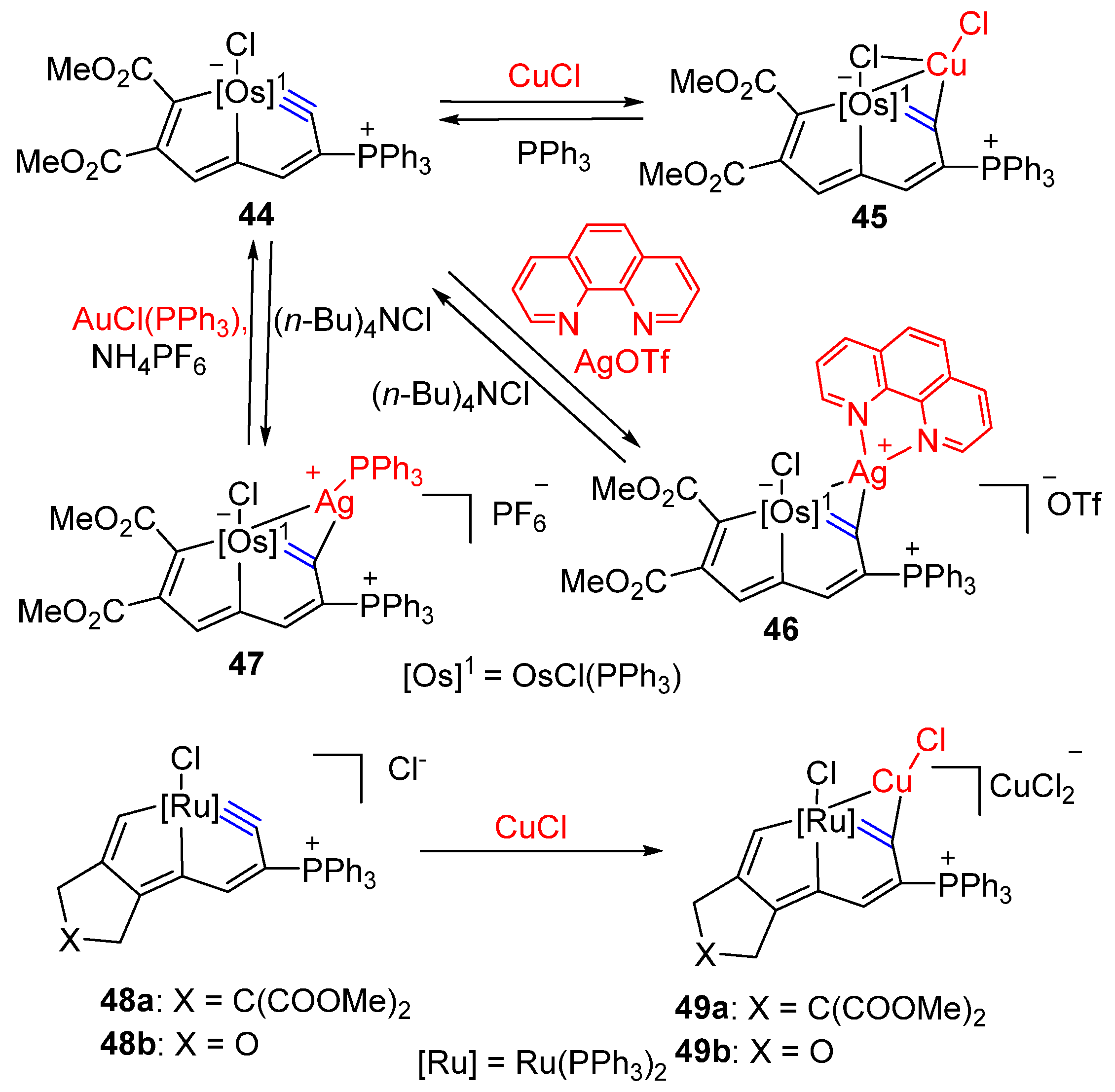
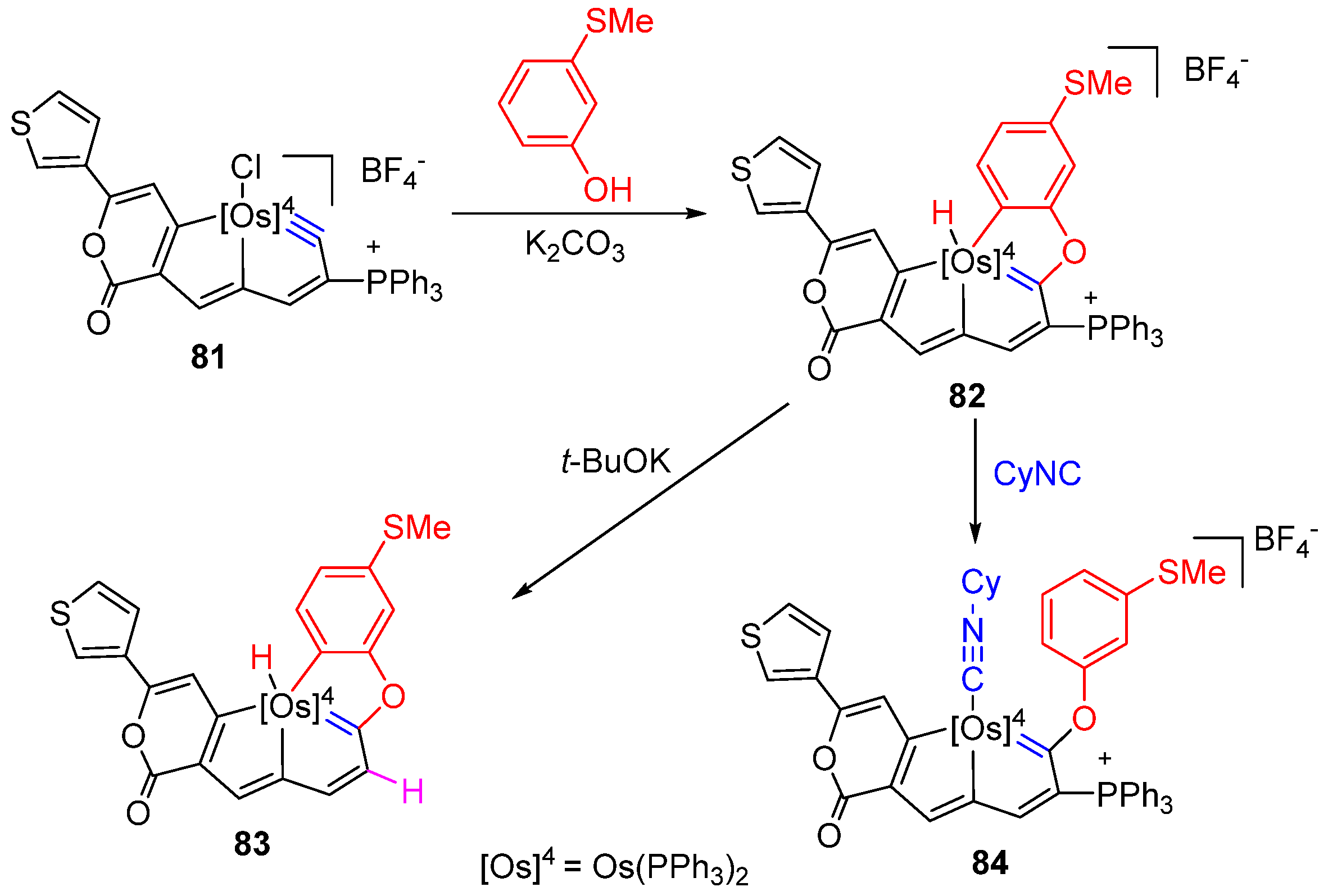
3.1. Formation of Osmapentalyne-Coinage Metal Complexes
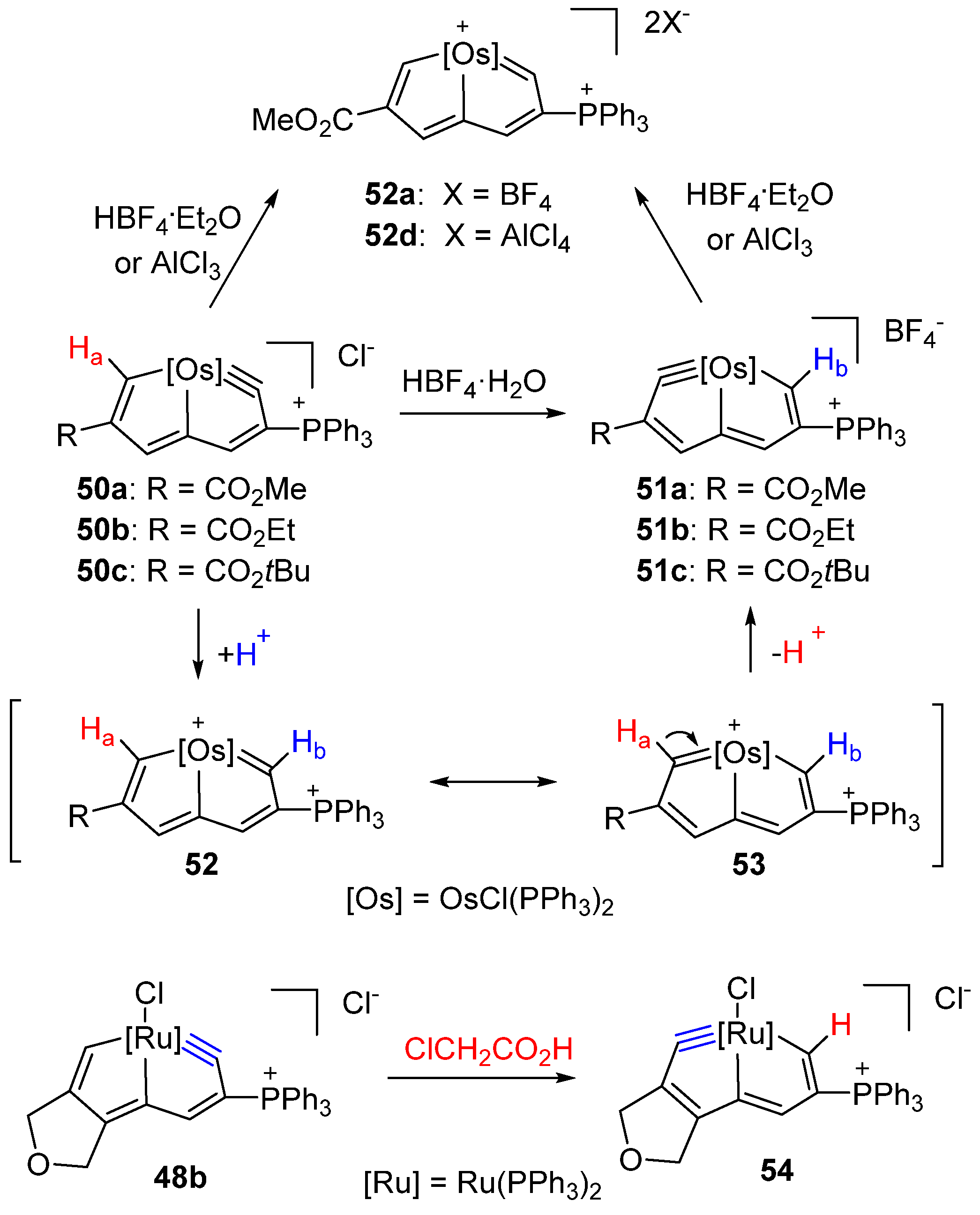
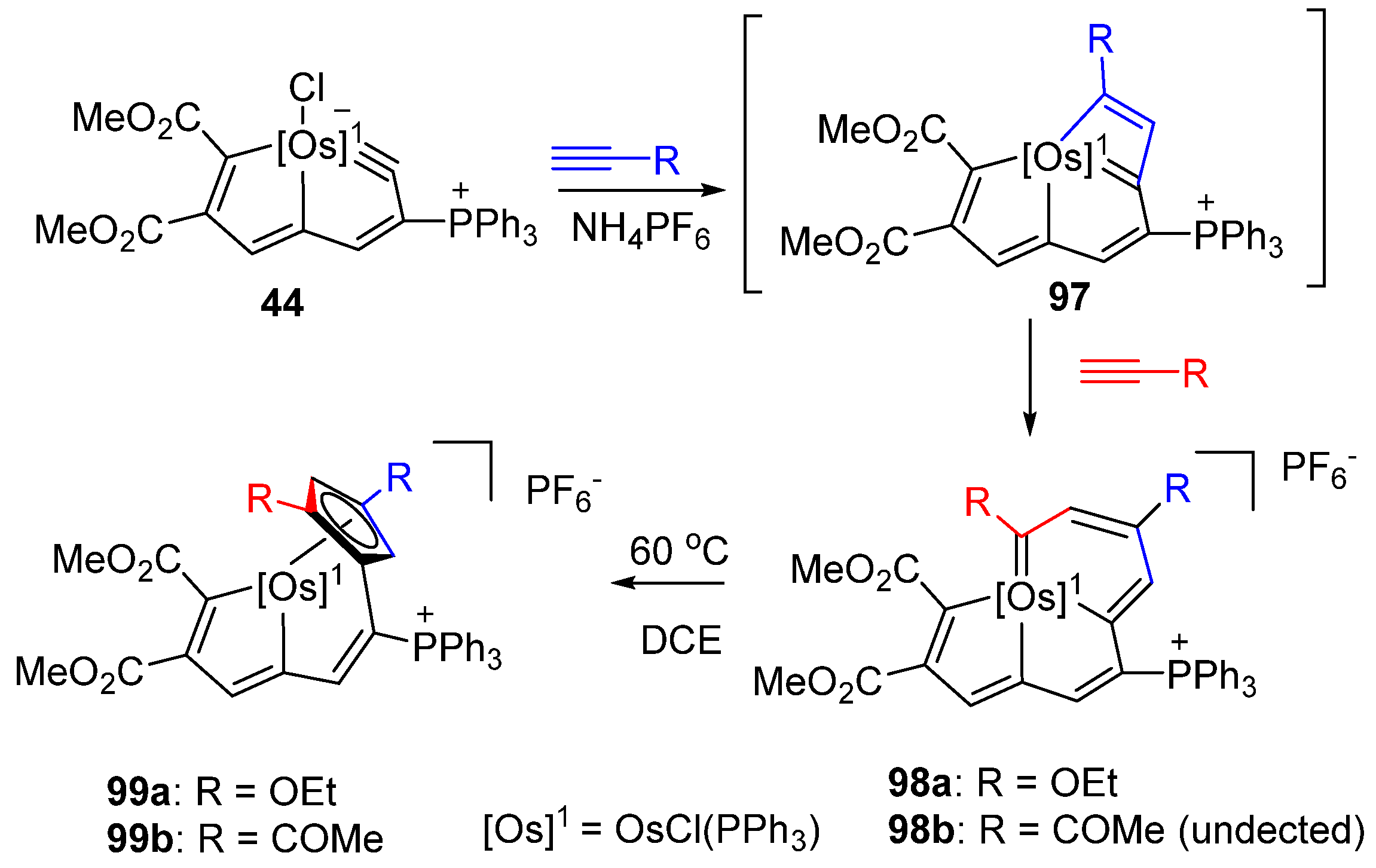
3.2. Electrophilic Reaction
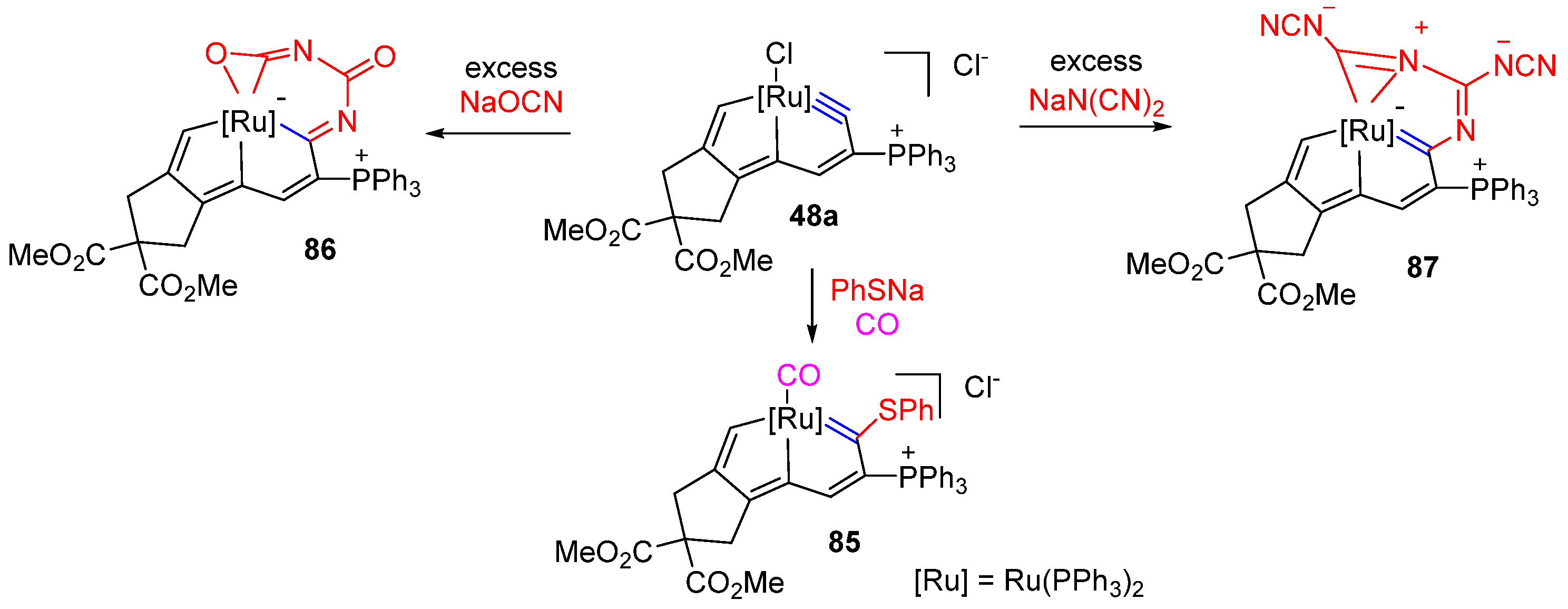
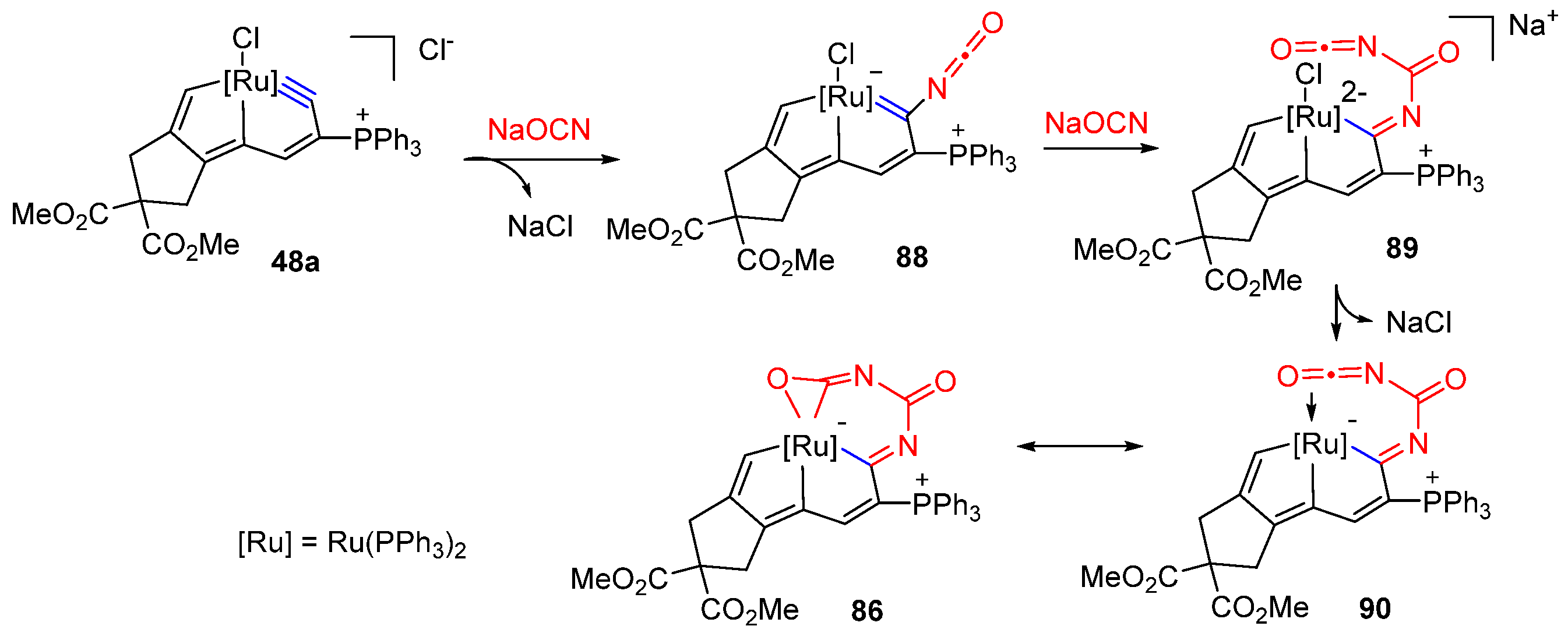
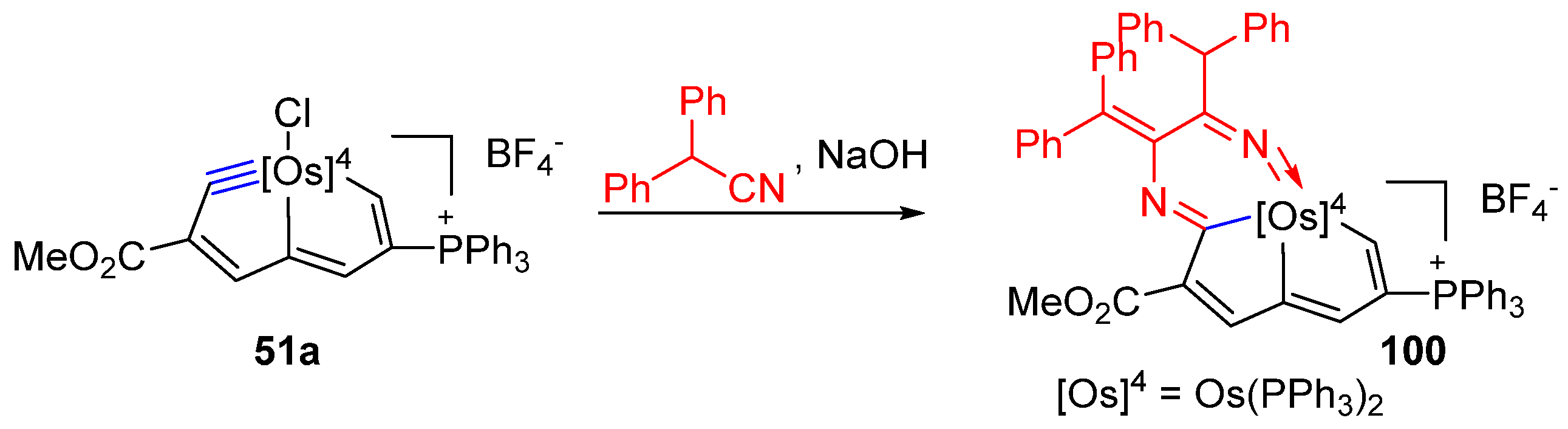
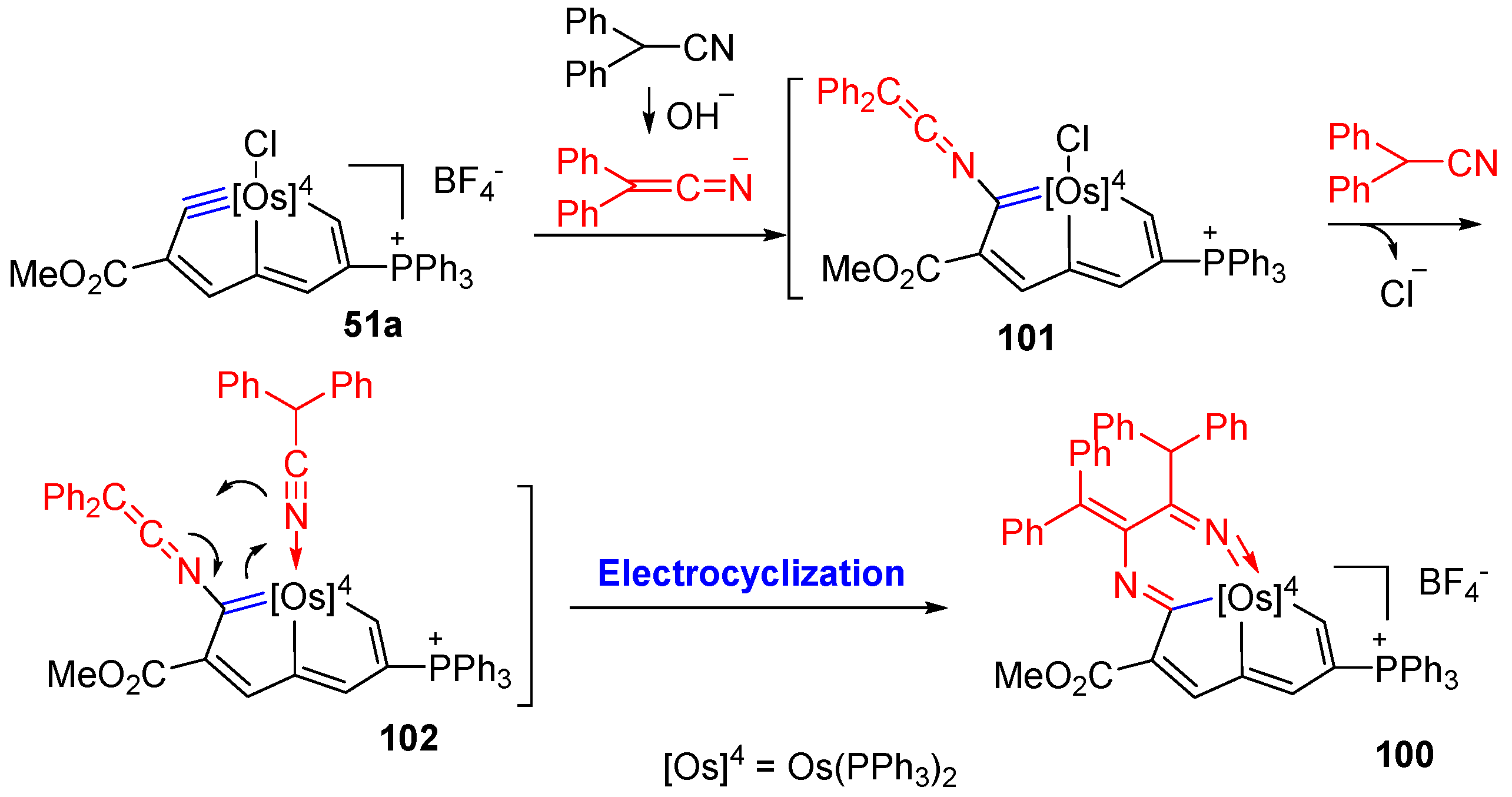
3.3. Nucleophilic Reaction
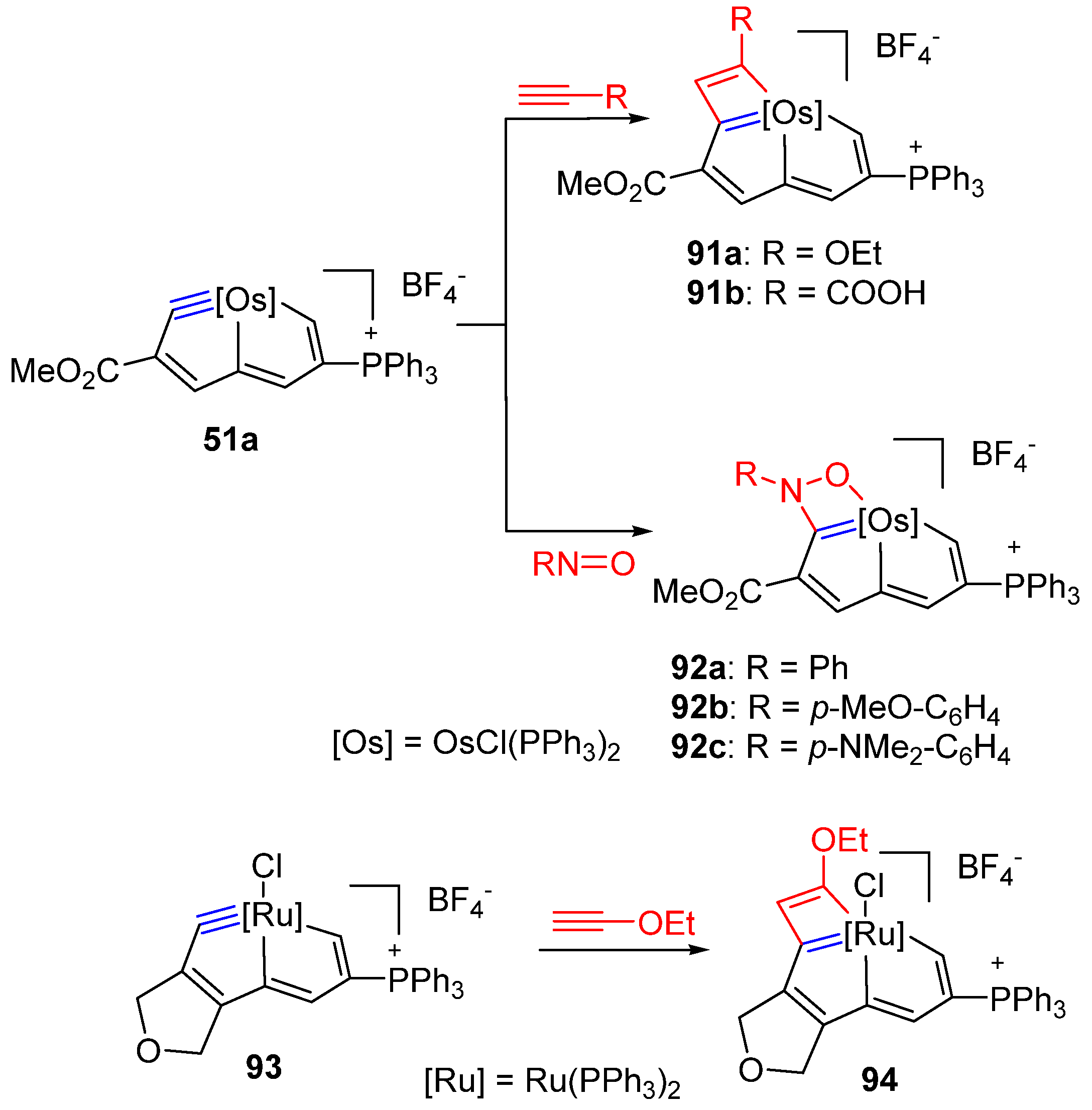
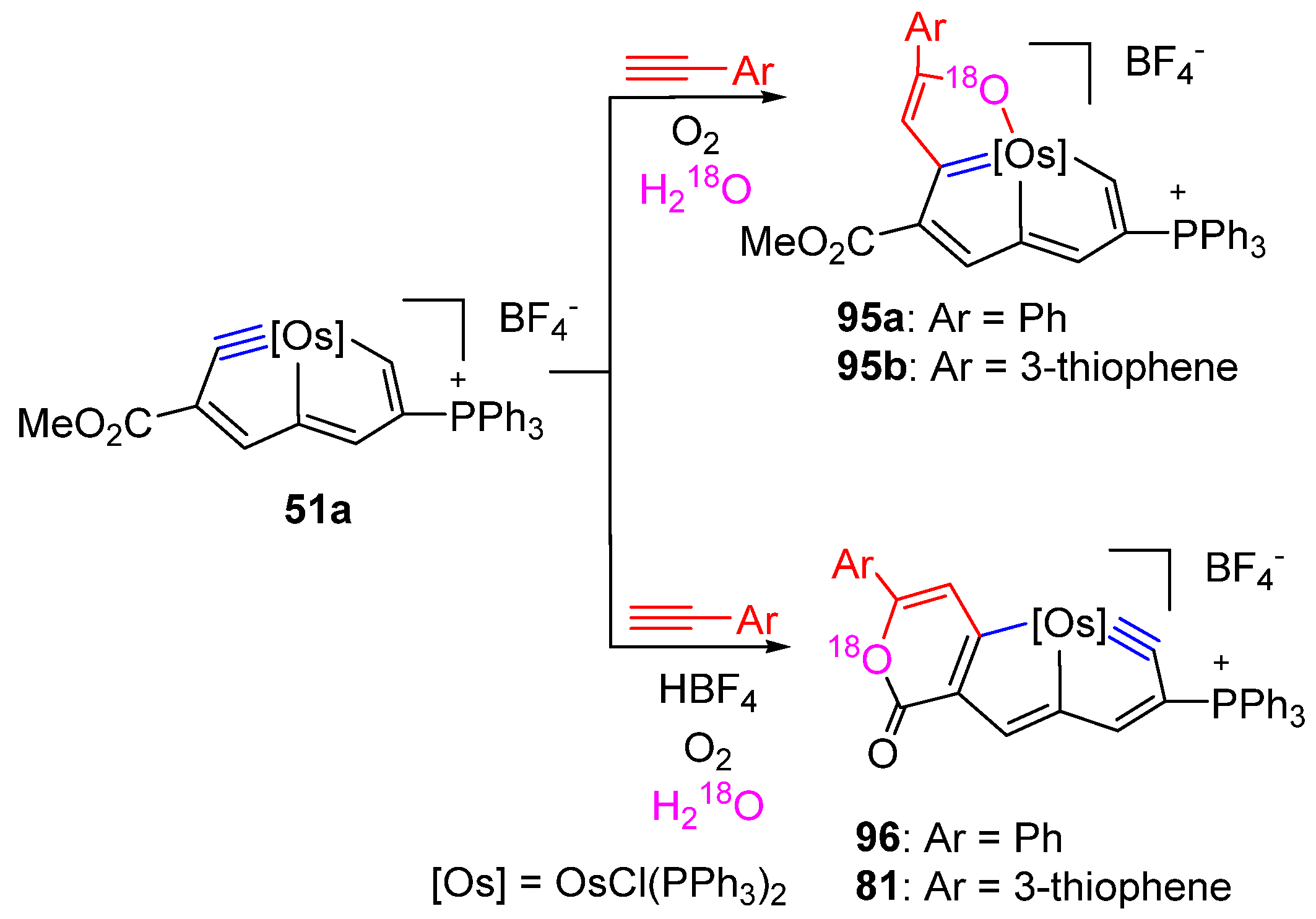
3.4. Cycloaddition Reactions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harvey, D.F.; Sigano, D.M. Carbene-Alkyne-Alkene Cyclization Reactions. Chem. Rev. 1996, 96, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.; Alabugin, I.V. Cyclizations of Alkynes: Revisiting Baldwin’s Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. [Google Scholar] [CrossRef] [PubMed]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.R.; Beke-Somfai, T.; Stalsmeden, A.S.; Kann, N. Ruthenium-Catalyzed Azide Alkyne Cycloaddition Reaction: Scope, Mechanism, and Applications. Chem. Rev. 2016, 116, 14726–14768. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom-Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3159. [Google Scholar] [CrossRef]
- Kim, H.P.; Angelici, R.J. Transition Metal Complexes with Terminal Carbyne Ligands. Adv. Organomet. Chem. 1987, 27, 51–111. [Google Scholar]
- Mayr, A.; Hoffmeister, H. Recent Advances in the Chemistry of Metal- Carbon Triple Bonds. Adv. Organomet. Chem. 1991, 32, 227–323. [Google Scholar]
- Engel, P.F.; Pfeffer, M. Carbon-Carbon and Carbon-Heteroatom Coupling Reactions of Metallacarbynes. Chem. Rev. 1995, 95, 2281–2309. [Google Scholar] [CrossRef]
- Bolaño, T.; Esteruelas, M.A.; Oñate, E. Osmiumecarbon multiple bonds: Reduction and CeC coupling reactions. J. Organomet. Chem. 2011, 696, 3911–3923. [Google Scholar] [CrossRef]
- Herndon, J.W. The chemistry of the carbon-transition metal double and triple bond: Annual survey covering the year 2018. Coord. Chem. Rev. 2019, 401, 213051. [Google Scholar] [CrossRef]
- Shi, C.; Jia, G. Chemistry of rhenium carbyne complexes. Coord. Chem. Rev. 2013, 257, 666–701. [Google Scholar] [CrossRef]
- Gampe, C.M.; Carreira, E.M. Arynes and Cyclohexyne in Natural Product Synthesis. Angew. Chem. Int. Ed. 2012, 51, 3766–3778. [Google Scholar] [CrossRef] [PubMed]
- Komarov, I.V. Organic molecules with abnormal geometric parameters. Russ. Chem. Rev. 2001, 70, 991–1016. [Google Scholar] [CrossRef]
- Roper, W.R. First Metallabenzenes and now a Stable Metallabenzyne. Angew. Chem. Int. Ed. 2001, 40, 2440–2441. [Google Scholar] [CrossRef]
- Jia, G. Progress in the Chemistry of Metallabenzynes. Acc. Chem. Res. 2004, 37, 479–486. [Google Scholar] [CrossRef]
- Landorf, C.W.; Haley, M.M. Recent Advances in Metallabenzene Chemistry. Angew. Chem. Int. Ed. 2006, 45, 3914–3936. [Google Scholar] [CrossRef]
- Jia, G. Recent progress in the chemistry of osmium carbyne and metallabenzyne complexes. Coord. Chem. Rev. 2007, 251, 2167–2187. [Google Scholar] [CrossRef]
- Chen, J.; Jia, G. Recent development in the chemistry of transition metal-containing metallabenzenes and metallabenzynes. Coord. Chem. Rev. 2013, 257, 2491–2521. [Google Scholar] [CrossRef]
- Jia, G. Our Journey to the Chemistry of Metallabenzynes. Organometallics 2013, 32, 6852–6866. [Google Scholar] [CrossRef]
- Chen, J.; He, G.; Jia, G. Synthesis and Chemical Properties of Metallabenzynes. Chin. J. Org. Chem. 2013, 33, 792. [Google Scholar] [CrossRef][Green Version]
- Zhang, H.; Zhou, X. Reactions of Metal-Carbon Bonds within Six-Membered Metallaaromatic Rings. Chem. Eur. J. 2018, 24, 8962–8973. [Google Scholar]
- Zhu, C.; Xia, H. Carbolong Chemistry: A Story of Carbon Chain Ligands and Transition Metals. Acc. Chem. Res. 2018, 51, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Frogley, B.J.; Wright, L.J. Recent Advances in Metallaaromatic Chemistry. Chem. Eur. J. 2018, 24, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Beweries, T.; Rosenthal, U. Breaking the rules. Nat. Chem. 2013, 5, 649–650. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, H.; Han, F.; Lin, R.; Lin, Z.; Xia, H. Synthesis and Characterization of a Metallapyridyne Complex. Angew. Chem. Int. Ed. 2012, 51, 9838–9841. [Google Scholar] [CrossRef]
- Bolaño, T.; Castarlenas, R.; Esteruelas, M.A.; Modrego, F.J.; Oñate, E. Hydride-Alkenylcarbyne to Alkenylcarbene Transformation in Bisphosphine-Osmium Complexes. J. Am. Chem. Soc. 2005, 127, 11184–11195. [Google Scholar] [CrossRef]
- Johs, P.M.; Roper, W.R.; Woodgate, S.D.; Wright, L.J. Thermal Rearrangement of Osmabenzenes to Osmium Cyclopentadienyl Complexes. Organometallics 2010, 29, 5358–5365. [Google Scholar]
- Schrock, R.R. Alkyne metathesis by molybdenum and tungsten alkylidyne complexes. Chem. Commun. 2013, 49, 5529–5531. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A. Alkyne Metathesis on the Rise. Angew. Chem. Int. Ed. 2013, 52, 2794–2819. [Google Scholar] [CrossRef]
- Wen, T.B.; Zhou, Z.Y.; Jia, G. Synthesis and Characterization of a Metallabenzyne. Angew. Chem. Int. Ed. 2001, 40, 1951–1954. [Google Scholar] [CrossRef]
- Wen, T.B.; Hung, W.Y.; Sung, H.H.Y.; Williams, I.D.; Jia, G. Syntheses of Metallabenzynes from an Allenylcarbene Complex. J. Am. Chem. Soc. 2005, 127, 2856–2857. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.B.; Lee, K.-H.; Chen, J.; Hung, W.Y.; Bai, W.; Li, H.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Preparation of Osmium η3-Allenylcarbene Complexes and Their Uses for the Syntheses of Osmabenzyne Complexes. Organometallics 2016, 35, 1514–1525. [Google Scholar] [CrossRef]
- Zhang, M.-X.; Xu, Z.; Lu, T.; Yin, J.; Liu, S.H. A Visible-Light-Induced Strategy to Construct Osmanaphthalynes, Osmaanthracyne, and Osmaphenanthryne. Chem. Eur. J. 2018, 24, 14891–14895. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Leung, T.-F.; Shi, C.; Lee, K.H.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Facile Synthesis of Polycyclic Metallaarynes. Chem. Sci. 2018, 9, 5994–5998. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xie, H.; Wang, H.; Wu, L.; Zhao, Q.; Chen, J.; Wen, T.B.; Cao, Z.; Xia, H. Selective Synthesis of Osmanaphthalene and Osmanaphthalyne by Intramolecular C-H Activation. Angew. Chem. Int. Ed. 2009, 48, 5461–5464. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, J.; Huang, Z.-A.; Cao, X.-Y.; Xia, H. Conversions of Osmabenzyne and Isoosmabenzene. Chem. Eur. J. 2012, 18, 11597–11603. [Google Scholar] [CrossRef]
- He, G.; Zhu, J.; Hung, W.Y.; Wen, T.B.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. A Metallanaphthalyne Complex from Zinc Reduction of a Vinylcarbyne Complex. Angew. Chem. Int. Ed. 2007, 46, 9065–9068. [Google Scholar] [CrossRef]
- Chen, J.; Shi, C.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Conversion of Metallabenzynes into Carbene Complexes. Angew. Chem. Int. Ed. 2011, 50, 7295–7299. [Google Scholar] [CrossRef]
- Chen, J.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Synthesis and Characterization of a Rhenabenzyne Complex. Angew. Chem. Int. Ed. 2011, 50, 10675–10678. [Google Scholar] [CrossRef]
- Chen, J.; Shi, C.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Synthesis and Characterization of a Rhenabenzyne Complexes. Chem. Eur. J. 2012, 18, 14128–14139. [Google Scholar] [CrossRef]
- Wen, T.B.; Ng, S.M.; Hung, W.Y.; Zhou, Z.Y.; Lo, M.F.; Shek, L.-Y.; Williams, I.D.; Lin, Z.; Jia, G. Protonation and Bromination of an Osmabenzyne: Reactions Leading to the Formation of New Metallabenzynes. J. Am. Chem. Soc. 2003, 125, 884–885. [Google Scholar] [CrossRef]
- Hung, W.Y.; Zhu, J.; Wen, T.B.; Yu, K.P.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Osmabenzenes from the Reactions of a Dicationic Osmabenzyne Complex. J. Am. Chem. Soc. 2006, 128, 13742–13752. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, K.-H.; Wen, T.; Gao, F.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Rearrangement of Metallabenzynes to Chlorocyclopentadienyl Complexes. Organometallics 2015, 34, 890–896. [Google Scholar] [CrossRef]
- Hung, W.Y.; Liu, B.; Shou, W.; Wen, T.B.; Shi, C.; Sung, H.H.-Y.; Williams, I.D.; Lin, Z.; Jia, G. Electrophilic Substitution Reactions of Metallabenzynes. J. Am. Chem. Soc. 2011, 133, 18350–18360. [Google Scholar] [CrossRef] [PubMed]
- Anusha, C.; De, S.; Parameswaran, P. Ring Contraction of Six-Membered Metallabenzynes to Five-Membered Metal–Carbene Complexes: A Comparison with Organic Analogues. Dalton Trans. 2013, 42, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; An, K.; Wang, X.; Zhu, J. Interconversion of Metallanaphthalynes and Indenylidene Complexes: A DFT Prediction. Organometallics 2013, 32, 6271–6276. [Google Scholar] [CrossRef]
- Zhu, C.; Li, S.; Luo, M.; Zhou, X.; Niu, Y.; Lin, M.; Zhu, J.; Cao, Z.; Lu, X.; Wen, T.; et al. Stabilization of Anti-Aromatic and Strained Five-Membered Rings with a Transition Metal. Nat. Chem. 2013, 5, 698–703. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, J.; Zhou, X.; Zhu, Q.; Yang, Y.; Wen, T.B.; Xia, H. Isolation of an Eleven-Atom Polydentate Carbon-Chain Chelate Obtained by Cycloaddition of a Cyclic Osmium Carbyne with an Alkyne. Angew. Chem. Int. Ed. 2018, 57, 3154–3157. [Google Scholar] [CrossRef]
- Zhuo, Q.; Lin, J.; Hua, Y.; Zhou, X.; Shao, Y.; Chen, S.; Chen, Z.; Zhu, J.; Zhang, H.; Xia, H. Multiyne Chains Chelating Osmium via Three Metalcarbon σ Bonds. Nat. Commun. 2017, 8, 1912. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, Y.; Wu, J.; Luo, M.; Fan, J.; Zhu, J.; Xia, H. Five-Membered Cyclic Metal Carbyne: Synthesis of Osmapentalynes by the Reactions of Osmapentalene with Allene, Alkyne, and Alkene. Angew. Chem. Int. Ed. 2015, 54, 7189–7192. [Google Scholar] [CrossRef]
- Hua, Y.; Lan, Q.; Fei, J.; Tang, C.; Lin, J.; Zha, H.; Chen, S.; Lu, Y.; Chen, J.; He, X.; et al. Metallapentalenofuran: Shifting Metallafuran Rings Promoted by Substituent Effects. Chem. Eur. J. 2018, 24, 14531–14538. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Q.; Zhang, H.; Hua, Y.; Kang, H.; Zhou, X.; Lin, X.; Chen, Z.; Lin, J.; Zhuo, K.; Xia, H. Constraint of a Ruthenium-Carbon Triple Bond to a Five-Membered Ring. Sci. Adv. 2018, 4, eaat0336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Y.; Shao, Y.; Hua, Y.; Zhang, H.; Lin, Y.-M.; Xia, H. Reactions of Cyclic Osmacarbyne with Coinage Metal Complexes. Organometallics 2018, 37, 1788–1794. [Google Scholar] [CrossRef]
- Zhu, C.; Luo, M.; Zhu, Q.; Zhu, J.; Schleyer, P.v.R.; Wu, J.I.-C.; Lu, X.; Xia, H. Planar Möbius Aromatic Pentalenes Incorporating 16 and 18 Valence Electron Osmiums. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Luo, M.; Zhu, C.; Chen, L.; Zhang, H.; Xia, H. Halogenation of Carbyne Complexes: Isolation of Unsaturated Metallaiodirenium Ion and Metallabromirenium Ion. Chem. Sci. 2016, 7, 1815–1818. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, J.; Hao, Y.; Zhu, C.; Zhuo, Q.; Xia, H.; Zhu, J. Rational Design and Synthesis of Unsaturated Se-Containing Osmacycles with σ-Aromaticity. Chem. Eur. J. 2018, 24, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, L.; Gao, X.; Hua, Y.; Peng, L.; Zhang, Y.; Yang, L.; Tan, Y.; He, F.; Xia, H. Addition of alkynes and osmium carbynes towards functionalized dπ–pπ conjugated systems. Nat Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Luo, M.; Deng, Z.; Ruan, Y.; Cai, Y.; Zhuo, K.; Zhang, H.; Xia, H. Reactions of Metallacyclopentadiene with Terminal Alkynes: Isolation and Characterization of Metallafulvenallene Complexes. Organometallics 2019, 38, 3053–3059. [Google Scholar] [CrossRef]
- Lin, J.; Xu, Q.; Lin, X.; Hua, Y.; Chen, D.; Ruan, Y.; Zhang, H.; Xia, H. The First OCCCO Pentadentate Chelates: Osmium Mediated Stepwise Oxidations of Terminal Alkynes by Pyridine N-Oxide. Chin. J. Chem. 2020, 38, 1273–1279. [Google Scholar] [CrossRef]
- Deng, Z.; Wu, P.; Cai, Y.; Sui, Y.; Chen, Z.; Zhang, H.; Wang, B.; Xia, H. Dioxygen Activation by Internally Aromatic Metallacycle: Crystallographic Structure and Mechanistic Investigations. iScience 2020, 23, 101379. [Google Scholar] [CrossRef]
- Luo, M.; Long, L.; Zhang, H.; Yang, Y.; Hua, Y.; Liu, G.; Lin, Z.; Xia, H. Reactions of Isocyanides with Metal Carbyne Complexes: Isolation and Characterization of Metallacyclopropenimine Intermediates. J. Am. Chem. Soc. 2017, 139, 1822–1825. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kang, H.; Zhuo, K.; Zhuo, Q.; Zhang, H.; Lin, Y.-M.; Xia, H. Alternation of Metal-Bridged Metallacycle Skeletons: From Ruthenapentalyne to Ruthenapentalene and Ruthenaindene Derivative. Chin. J. Chem. 2018, 36, 1156–1160. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, Q.; Fan, J.; Zhu, J.; He, X.; Cao, X.-Y.; Xia, H. A Metal-Bridged Tricyclic Aromatic System: Synthesis of Osmium Polycyclic Aromatic Complexes. Angew. Chem. Int. Ed. 2014, 53, 6232–6236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhu, C.; Deng, Z.; He, G.; Chen, J.; Zhu, J.; Xia, H. Synthesis and Characterization of Osmium Polycyclic Aromatic Complexes via Nucleophilic Reactions of Osmapentalyne. Chin. J. Chem. 2017, 35, 628–634. [Google Scholar] [CrossRef]
- Lu, Z.; Chen, J.; Xia, H. Synthesis of Olefinic Carbolong Complexes. Chin. J. Org. Chem. 2017, 37, 1181–1188. [Google Scholar] [CrossRef]
- Lu, Z.; Zhu, C.; Cai, Y.; Zhu, J.; Hua, Y.; Chen, Z.; Chen, J.; Xia, H. Metallapentalenofurans and Lactone-Fused Metallapentalynes. Chem. Eur. J. 2017, 23, 6426–6431. [Google Scholar] [CrossRef]
- Li, R.; Lu, Z.; Cai, Y.; Jiang, F.; Tang, C.; Chen, Z.; Zheng, J.; Pi, J.; Zhang, R.; Liu, J.; et al. Switching of Charge Transport Pathways via Delocalization Changes in Single-Molecule Metallacycles Junctions. J. Am. Chem. Soc. 2017, 139, 14344–14347. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, Y.; Luo, M.; Yang, C.; Wu, J.; Chen, L.; Liu, G.; Wen, T.; Zhu, J.; Xia, H. Stabilizing Two Classical Antiaromatic Frameworks: Demonstration of Photoacoustic Imaging and the Photothermal Effect in Metalla-aromatics. Angew. Chem. Int. Ed. 2015, 54, 6181–6185. [Google Scholar] [CrossRef]
- Deng, Z.; Zhu, C.; Hua, Y.; He, G.; Guo, Y.; Lu, R.; Cao, X.; Chen, J.; Xia, H. Synthesis and Characterization of Metallapentalenoxazetes by the [2+2] Cycloaddition of Metallapentalynes with Nitrosoarenes. Chem. Commun. 2019, 55, 6237–6240. [Google Scholar] [CrossRef]
- Lin, J.; Ding, L.; Zhuo, Q.; Zhang, H.; Xia, H. Formal [2+2+2] Cycloaddition Reaction of a Metal−Carbyne Complex with Nitriles: Synthesis of a Metallapyrazine Complex. Organometallics 2019, 38, 2264–2271. [Google Scholar] [CrossRef]
- Lu, Z.; Zhu, Q.; Cai, Y.; Chen, Z.; Zhuo, K.; Zhu, J.; Zhang, H.; Xia, H. Access to Tetracyclic Aromatics with Bridgehead Metals via Metalla-Click Reactions. Sci. Adv. 2020, 6, eaay2535. [Google Scholar] [CrossRef]
- Huang, F.; Zheng, X.; Lin, X.; Ding, L.; Zhuo, Q.; Wen, T.B.; Zhang, H.; Xia, H. Extension of the Simmons–Smith reaction to metal-carbynes: Efficient synthesis of metallacyclopropenes with σ-aromaticity. Chem. Sci. 2020, 11, 10159–10166. [Google Scholar] [CrossRef]
- Luo, M.; Hua, Y.; Zhuo, K.; Long, L.; Lin, X.; Deng, Z.; Lin, Z.; Zhang, H.; Chen, D.; Xia, H. Carbolong Chemistry: Planar CCCCX-Type (X = N, O, S) Pentadentate Chelates by Formal [3+1] Cycloadditions of Metalla-Azirines with Terminal Alkynes. CCS Chem. 2020, 2, 758–763. [Google Scholar] [CrossRef]
- Wu, F.; Huang, W.; Zhuo, K.; Hua, Y.; Lin, J.; He, G.; Chen, J.; Nie, L.; Xia, H. Carbolong Complexes as Photothermal Materials. Chin. J. Org. Chem. 2019, 39, 1743–1752. [Google Scholar] [CrossRef]
- Lu, Z.; Cai, Y.; Wei, Y.; Lin, Q.; Chen, J.; He, X.; Li, S.; Wu, W.; Xia, H. Photothermal Möbius Aromatic Metallapentalenofuran and Its NIR-Responsive Copolymer. Polym. Chem. 2018, 9, 2092–2100. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, C.; Wang, Y.; Lin, G.; Yang, Y.; Wang, X.; Zhu, J.; Chen, X.; Lu, X.; Liu, G.; et al. CCCCC pentadentate chelates with planar Möbius aromaticity and unique properties. Sci. Adv. 2016, 2, e1601031. [Google Scholar] [CrossRef]
- He, X.; He, X.; Li, S.; Zhuo, K.; Qin, W.; Dong, S.; Chen, J.; Ren, L.; Liu, G.; Xia, H. Amphipathic metal-containing macromolecules with photothermal properties. Polym. Chem. 2017, 8, 3674–3678. [Google Scholar] [CrossRef]
- Yang, C.; Lin, G.; Zhu, C.; Pang, X.; Zhang, Y.; Wang, X.; Li, X.; Wang, B.; Xia, H.; Liu, G.J. Metalla-aromatic loaded magnetic nanoparticles for MRI/photoacoustic imaging-guided cancer phototherapy. Mater. Chem. B 2018, 6, 2528–2535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, H.; Zhuo, K.; Hua, Y.; Chen, J.; He, X.; Weng, W.; Xia, H. “Carbolong” polymers with near infrared triggered, spatially resolved and rapid self-healing properties. Polym. Chem. 2019, 10, 386–394. [Google Scholar] [CrossRef]


































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Q.; Ding, J.; Du, Z.; Lai, Y.; Li, H.; Ouyang, M.-A.; Song, L.; Lin, R. Recent Advances in the Reactions of Cyclic Carbynes. Molecules 2020, 25, 5050. https://doi.org/10.3390/molecules25215050
Su Q, Ding J, Du Z, Lai Y, Li H, Ouyang M-A, Song L, Lin R. Recent Advances in the Reactions of Cyclic Carbynes. Molecules. 2020; 25(21):5050. https://doi.org/10.3390/molecules25215050
Chicago/Turabian StyleSu, Qian, Jipeng Ding, Zhihui Du, Yunrong Lai, Hongzuo Li, Ming-An Ouyang, Liyan Song, and Ran Lin. 2020. "Recent Advances in the Reactions of Cyclic Carbynes" Molecules 25, no. 21: 5050. https://doi.org/10.3390/molecules25215050
APA StyleSu, Q., Ding, J., Du, Z., Lai, Y., Li, H., Ouyang, M.-A., Song, L., & Lin, R. (2020). Recent Advances in the Reactions of Cyclic Carbynes. Molecules, 25(21), 5050. https://doi.org/10.3390/molecules25215050