
Total Synthesis of Cyclopiamide A Using Palladium-Catalyzed Domino Cyclization


Abstract
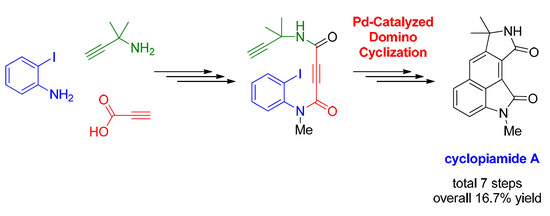
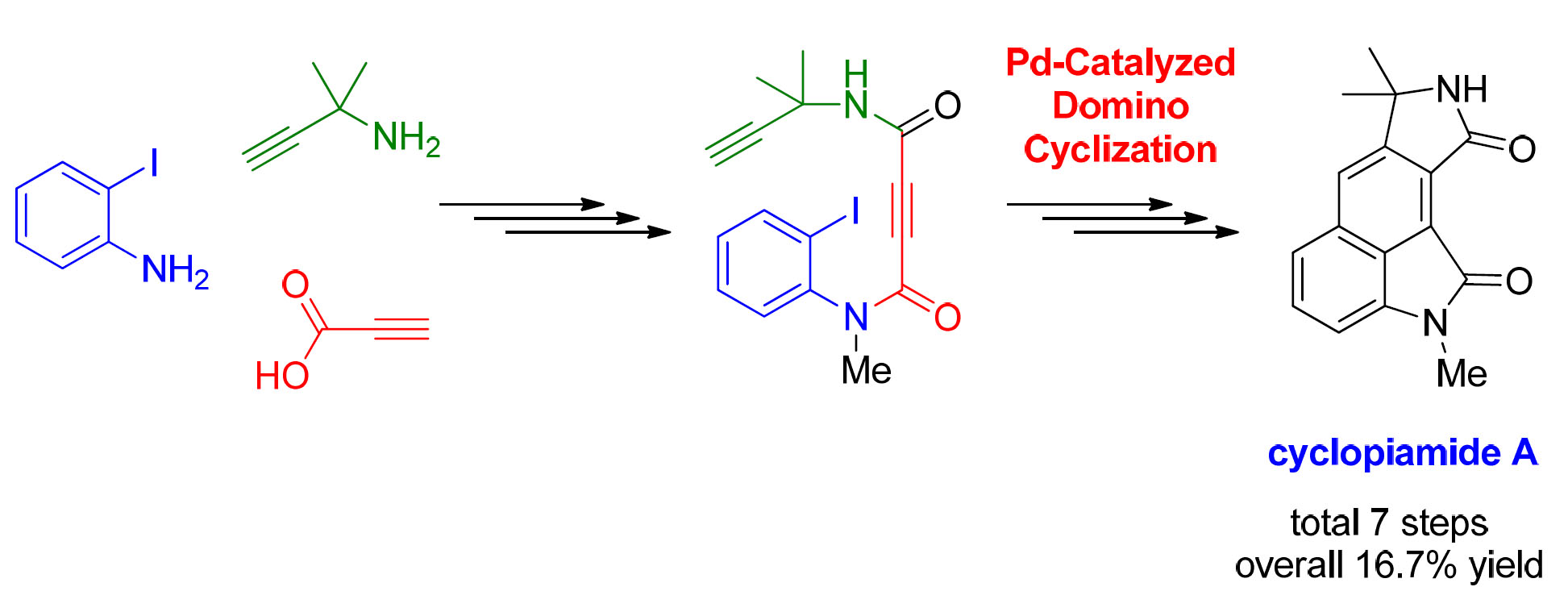
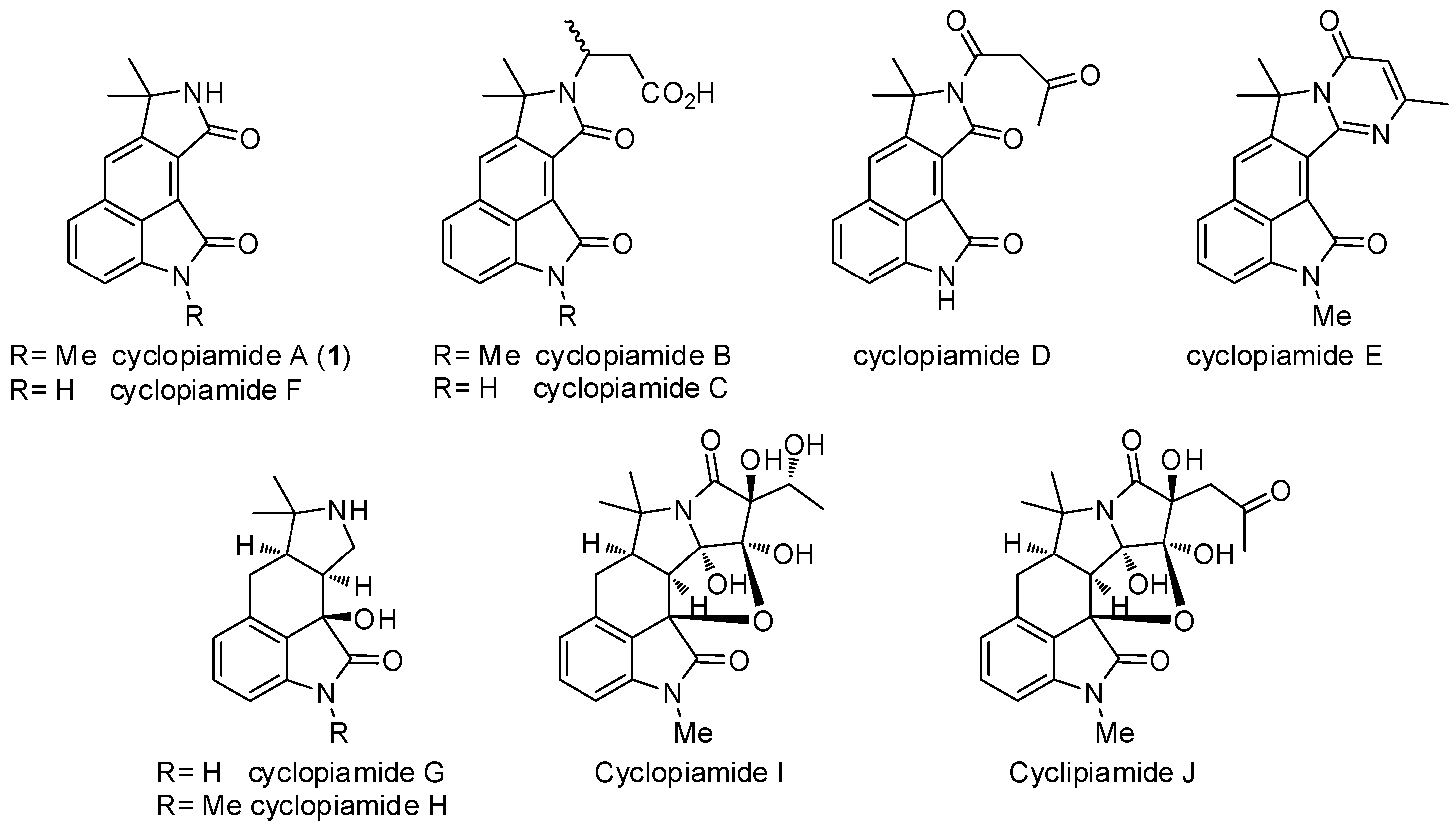
1. Introduction
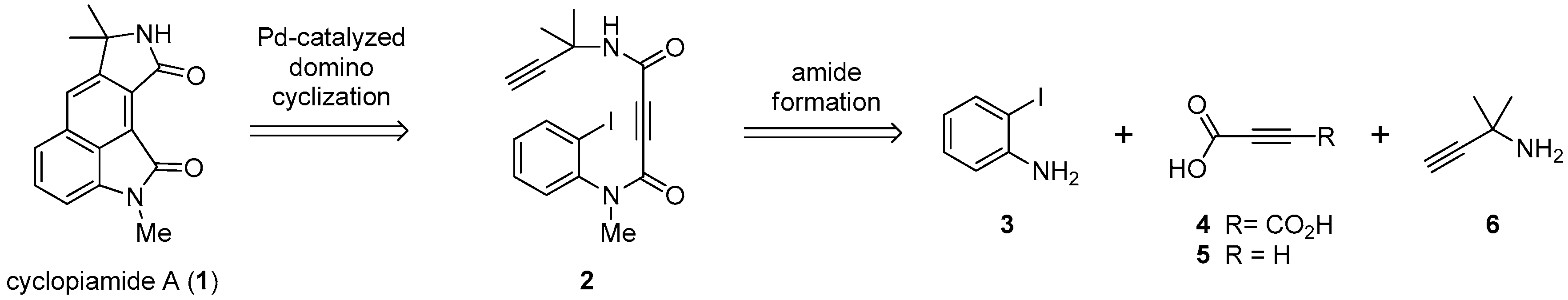
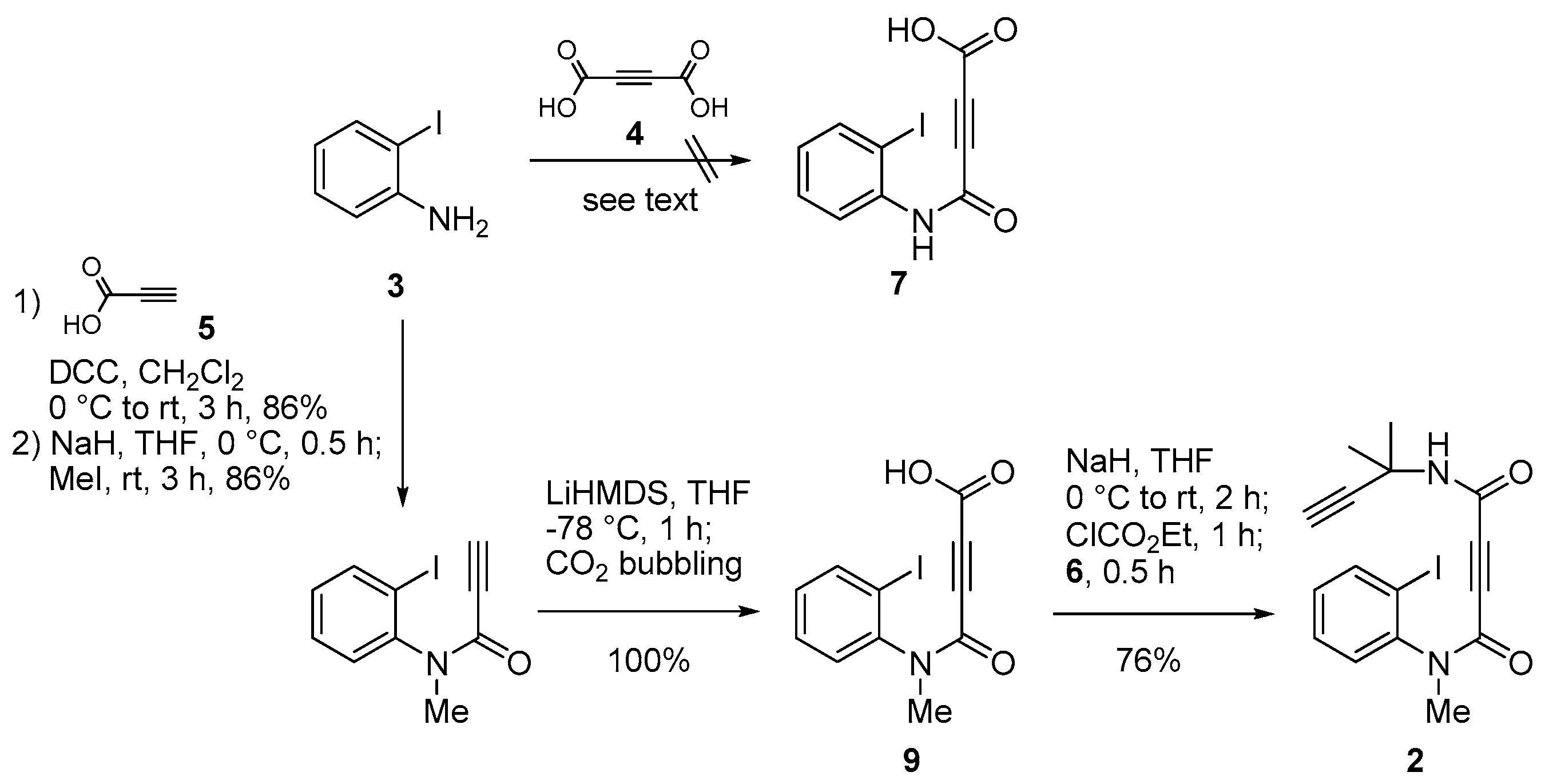
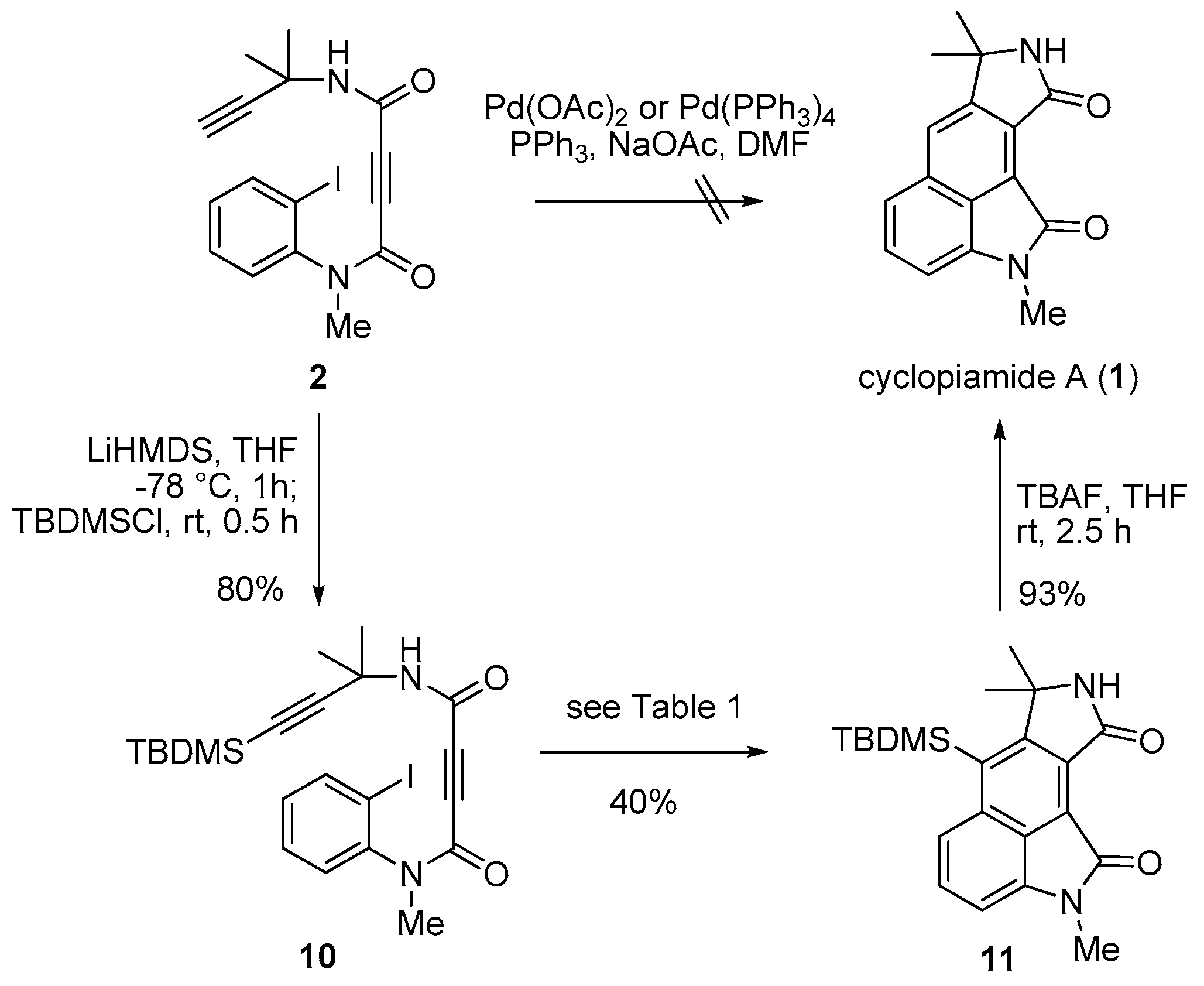
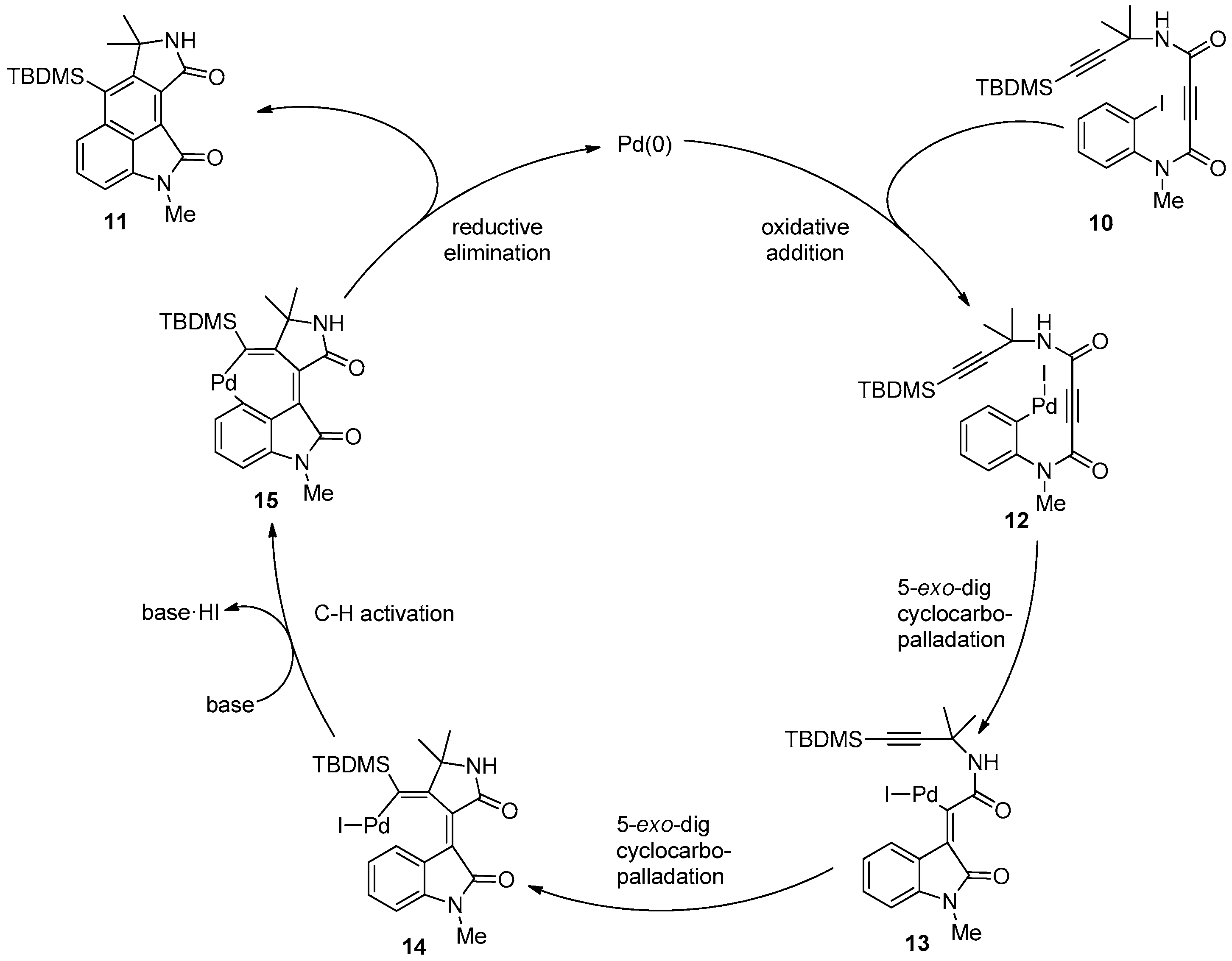
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis of 1,2 and 8–11
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Holzapel, C.W.; Bredenkamp, M.W.; Snyman, R.M.; Boeyenes, J.C.A.; Callen, C. Cyclopyamide, an Isoindolo[4,6-cd]indole from Penicillium cyclopium. Phytochemistry 1990, 29, 639–642. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Nong, X.; Wei, X.; Qu, S. Oxindole Alkaloids from the Fungus Penicillium commune DFFSC026 Isolated from Deep-See-Derived Sediments. Tetrahedron 2015, 71, 610–615. [Google Scholar] [CrossRef]
- Uka, V.; Moore, G.G.; Arroyo-Manzanares, N.; Nebija, D.; De Saeger, S.; Di Mavungu, J.D. Unravelling the Diversity of the Cyclopiazonic Acid Family of Mytotoxins in Aspergillus flavus by UHPLC Triple-TOF HRMS. Toxins 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Nakhla, M.C.; Weeks, K.N.; Villalobos, M.N.; Wood, J.L. Total Synthesis of Cyclopiamide A and Speradine, E. Tetrahedron 2018, 74, 5085–5088. [Google Scholar] [CrossRef]
- Dong, G.R.; Park, S.; Lee, D.; Shin, K.J.; Seo, J.H. Synthesis of 3-(Diarylmethylene)oxindoles via a Palladium-Catalyzed One-Pot Reaction: Sonogashira-Heck-Suzuki-Miyaura Combined Reaction. Synlett 2013, 24, 1993–1997. [Google Scholar] [CrossRef]
- Park, S.; Shin, K.J.; Seo, J.H. Palladium-Catalyzed Tandem Approach to 3-(Diarylmethylene)oxindoles Using Microwave Irradiation. Synlett 2015, 26, 2296–2300. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.; Shin, K.J.; Oh, E.; Seo, J.H. Sequential One-Pot versus Tandem Multicomponent Approach to 3-(Diarylmethylene)oxindoles. Molecules 2017, 26, 2296–2300. [Google Scholar]
- Yu, Y.; Shin, K.J.; Seo, J.H. Stereoselective Synthesis of 3-(1,3-Diarylallylidene)oxindoles via a Palladium-Catalyzed Tandem Reaction. J. Org. Chem. 2017, 82, 1864–1871. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.; Shin, K.J.; Seo, J.H. Palladium-Catalyzed One-Pot Approach to 3-(1,3-Diarylprop-2-yn-1-ylidene)oxindoles. Heterocycles 2018, 96, 1795–1807. [Google Scholar]
- Czifrák, K.; Hadady, Z.; Docsa, T.; Gergely, P.; Schmidt, J.; Wessjohann, L.; Somsák, L. Synthesis of N-(β-d-glucopyranosyl)monoamides of Dicarboxylic Acids as Potential Inhibitors of Glycogen Phosphorylase. Carbohydr. Res. 2006, 341, 947–956. [Google Scholar] [CrossRef]
- Heyl, D.; Fessner, W.-D. Facile Direct Synthesis of Acetylenedicarboxamides. Synthesis 2014, 46, 1463–1468. [Google Scholar]
- Li, G.; Zhou, G.; Zhang-Negrerie, D.; Du, Y.; Huang, J.; Zhao, K. Palladium(II) Acetate Dual C-H Functionalization and C-C bond Formation: A Domino Reaction for the Synthesis of Functionalized (E)-Bisindole-2-ones from Diarylbut-2-ynediamides. Adv. Synth. Catal. 2016, 358, 3534–3540. [Google Scholar] [CrossRef]
- Alvaro, M.; Garcia, H.; Miranda, M.A.; Primo, J. Preparation and Photolysis of Diaryl Esters of Acetylenedicarboxylic Acid. Tetrahedron 1992, 48, 3437–3444. [Google Scholar] [CrossRef]
- Overman, L.E.; Watson, D.A. Diastereoselection in the Formation of Spirocyclic Oxindoles by the Intramolecular Heck Reaction. J. Org. Chem. 2006, 71, 2587–2599. [Google Scholar] [CrossRef]
- Arthius, M.; Pontikis, R.; Florent, J.-C. Tandem Heck-Suzuki-Miyaura Reaction: Application to the Synthesis of Constrained Analogues of Combretastatin A-4. Tetrahedron Lett. 2007, 48, 6397–6400. [Google Scholar] [CrossRef]
- Gherbovet, O.; Sánchez-Murica, P.A.; Garcia Alvarez, M.C.; Bignon, J.; Thoret, S.; Gago, F.; Roussi, F. Synthesis and Evaluation of Hybrid Molecules Targeting the Vinca Domain of Tublin. Org. Bioorg. Chem. 2015, 13, 3144–3154. [Google Scholar] [CrossRef]
- Grigg, R.; Loganathan, V.; Sridharan, V. Palladium Catalyzed Cascade Alkyne-Arene Vinylation/Alkylation Approach to Polyfused Heterocycles. Tetrahedron Lett. 1996, 37, 3399–3402. [Google Scholar] [CrossRef]
- Tietze, L.F.; Lotz, F. Pd-Catalyzed Domino Arylation/CH activation for the Synthesis of Acenaphthylenes. Eur. J. Org. Chem. 2006, 2006, 4676–4684. [Google Scholar] [CrossRef]
- Abdur Rahman, S.M.; Sonoda, M.; Ono, M.; Miki, K.; Tobe, Y. Novel Synthesis of Bridged Phenylthienyenthenes and Dithienylethenes via Pd-Catalyzed Double-Cyclization Reactions of Diarylhexadienynes. Org. Lett. 2006, 8, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Leibeling, M.; Pawliczek, M.; Kratzert, D.; Stalke, D.; Werz, D.B. A Ridge Walk between Reaction Modes: An Unprecedented Pd-Catalyzed Domino Sequence of Diynyl-Substituted Bromoarenes. Org. Lett. 2012, 14, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Hungerland, T.; Eichhorst, C.; Düfert, A.; Maaβ, C.; Stalke, D. Effcient Synthesis of Helical Tetrasubstituted Alkenes as Potential Molecular Switches: A Two-Component Palladium-Catalyzed Triple Domino Process. Angew. Chem. Int. Ed. 2013, 52, 3668–3671. [Google Scholar] [CrossRef] [PubMed]
- Joussot, J.; Schoenfelder, A.; Suffert, J.; Blond, G. Synthesis of Original Polycycles Containing Five-, Six- and Seven-Membered Rings Through Cyclocarbopalladations/C-H Activation Cascade Reactions. C. R. Chim. 2017, 20, 665–681. [Google Scholar] [CrossRef]
- Cai, Q.; Yan, J.; Ding, K. A CuAAC/Ullmann C-C Coupling Tandem Reaction: Copper-Catalyzed Reactions of Organic Azides with N-(2-Iodoaryl)propiolamides or 2-Iodo-N-(prop-2-ynyl)benzamides. Org. Lett. 2012, 14, 3332–3335. [Google Scholar] [CrossRef]
- Lin, W.-J.; Shia, K.-S.; Song, J.-S.; Wu, M.-H.; Li, W.-T. Synthesis of (E)-Oxindolyldene Acetate Using Tandem Palladium-Catalyzed Heck and Alkoxycarbonylation Reactions. Org. Biomol. Chem. 2016, 14, 220–228. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Pd Catalyst | Phosphine Ligand | Base | Temp (°C) | Time (h) | Yield (%) 2 |
|---|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | PPh3 | NaOAc | 60 | 3 | 29 |
| 2 | Pd(OAc)2 | PPh3 | NaOAc | rt | 24 | 23 |
| 3 | Pd(OAc)2 | PPh3 | NaOAc | 100 | 1 | 27 |
| 4 | Pd(OAc)2 | – | NaOAc | 60 | 2 | 20 |
| 5 | Pd(OAc)2 | dppf | NaOAc | 60 | 2 | 29 |
| 6 | Pd(OAc)2 | P(o-tol)3 | NaOAc | 100 | 21 | 18 |
| 7 3 | Pd(OAc)2 | t-BuXPhos | NaOAc | 60 | 3 | 21 |
| 8 3 | Pd(OAc)2 | PPh3 | NaOAc | 60 | 3.5 | 31 |
| 9 3 | Pd(OAc)2 | PPh3 | K2CO3 | 60 | 3 | 24 |
| 10 3 | Pd(OAc)2 | PPh3 | K3PO4 | 60 | 4 | 31 |
| 11 3 | Pd(OAc)2 | PPh3 | Cs2CO3 | 60 | 3 | 16 |
| 12 3 | Pd(PPh3)4 | PPh3 | NaOAc | 60 | 4 | 30 |
| 13 | Pd(PPh3)4 | – | NaOAc | 60 | 4 | 35 |
| 14 4 | Pd(PPh3)4 | – | NaOAc | 60 | 4.5 | 40 |
Sample Availability: Samples of the compounds 1,2 and 8–11 are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Shin, K.J.; Seo, J.H. Total Synthesis of Cyclopiamide A Using Palladium-Catalyzed Domino Cyclization. Molecules 2020, 25, 4903. https://doi.org/10.3390/molecules25214903
Park S, Shin KJ, Seo JH. Total Synthesis of Cyclopiamide A Using Palladium-Catalyzed Domino Cyclization. Molecules. 2020; 25(21):4903. https://doi.org/10.3390/molecules25214903
Chicago/Turabian StylePark, Sunhwa, Kye Jung Shin, and Jae Hong Seo. 2020. "Total Synthesis of Cyclopiamide A Using Palladium-Catalyzed Domino Cyclization" Molecules 25, no. 21: 4903. https://doi.org/10.3390/molecules25214903
APA StylePark, S., Shin, K. J., & Seo, J. H. (2020). Total Synthesis of Cyclopiamide A Using Palladium-Catalyzed Domino Cyclization. Molecules, 25(21), 4903. https://doi.org/10.3390/molecules25214903