How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices


Abstract

1. Introduction
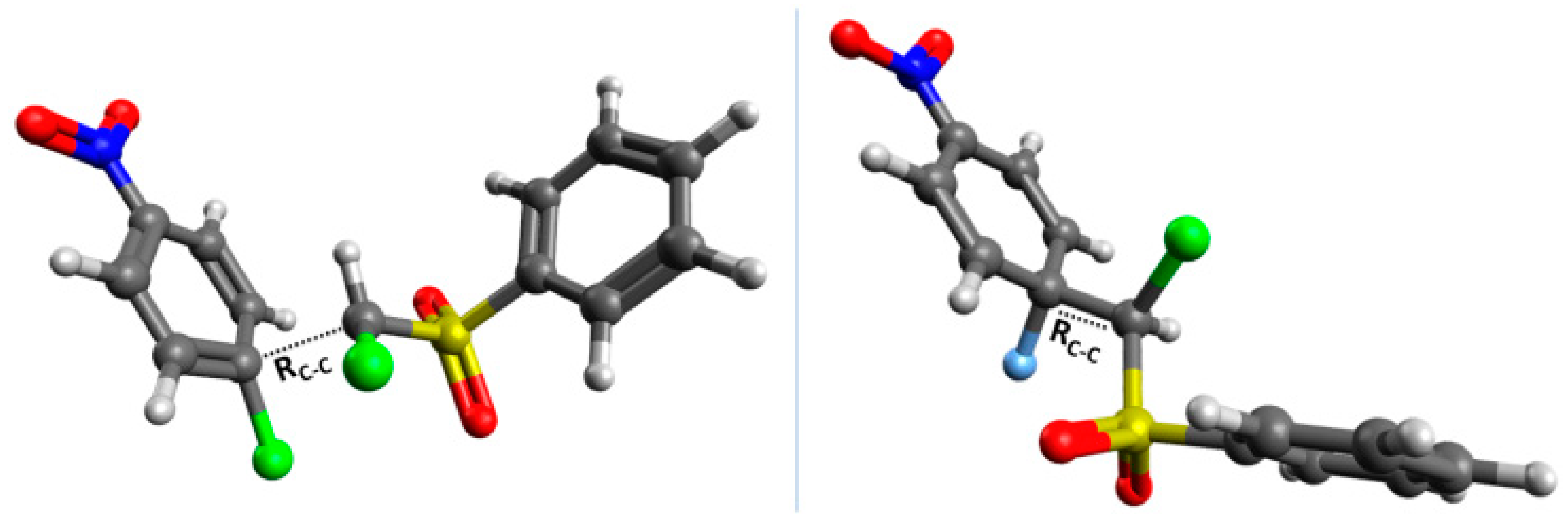
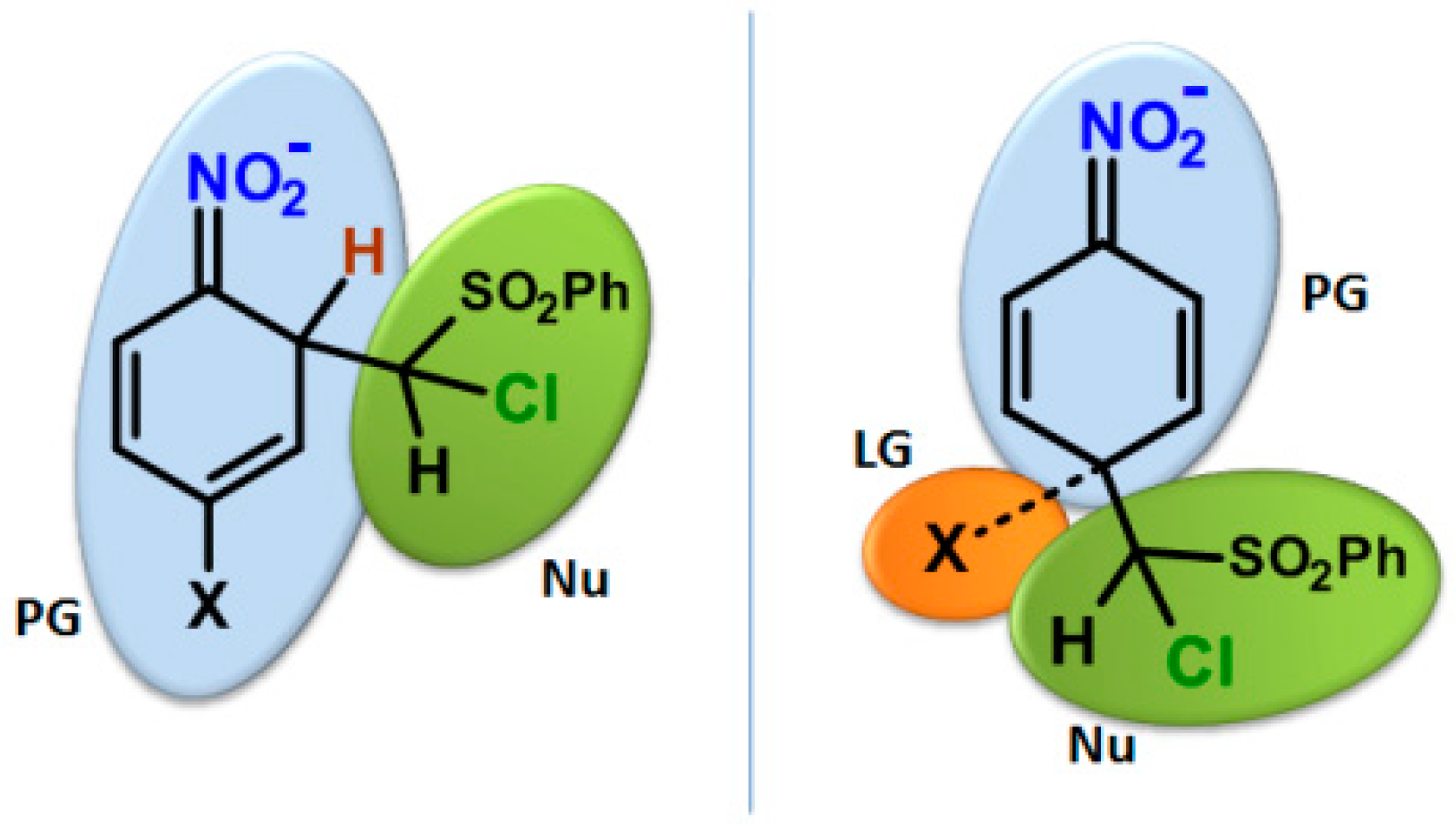
2. Results and Discussion
2.1. Aromaticity
2.2. Electrophilicity and Nucleophilicity
2.3. Kinetic Isotope Effect
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, J. Aromatic Nucleophilic Substitution; Elsevier: New York, NY, USA, 1968. [Google Scholar]
- Caron, S.; Ghosh, A. Practical Synthetic Organic Chemistry; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2011; pp. 237–253. [Google Scholar]
- Crampton, M.R. Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Bunnett, J.F. Mechanism and Reactivity in Aromatic Nucleophilic Substitution Reactions. Quart. Rev. Chem. Soc. 1958, 12, 1–16. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Nudelman, N.S.; MacCormack, P. Theoretical Calculations of Chemical Interactions. Part 5. 1,1-Disubstituted 2,4,6-Trinitrocyclohexadienylide (’Meisenheimer’) Complexes. J. Chem. Soc. Perkin Trans. 2 1987, 227–229. [Google Scholar] [CrossRef]
- Dotterer, S.K.; Harris, R.L. Mndo Study of Nucleophilic Aromatic Substitution. J. Org. Chem. 1988, 53, 777–779. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Gluz, E.B.; Glukhovtsev, M.N.; Minkin, V.I. Theoretical Study of Mechanisms of Aromatic Nucleophilic Substitution in the Gas Phase. J. Mol. Struct. Theochem. 1993, 284, 123–137. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Bach, R.D.; Laiter, S. Single-Step and Multistep Mechanisms of Aromatic Nucleophilic Substitution of Halobenzenes and Halonitrobenzenes with Halide Anions: Ab Initio Computational Study. J. Org. Chem. 1997, 62, 4036–4046. [Google Scholar] [CrossRef]
- Um, I.H.; Min, S.W.; Dust, J.M. Choice of Solvent (Mecn Vs H2o) Decides Rate-Limiting Step in Snar Aminolysis of 1-Fluoro-2,4-Dinitrobenzene with Secondary Amines: Importance of Brønsted-Type Analysis in Acetonitrile. J. Org. Chem. 2007, 72, 8797–8803. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, V.L. Solvent Effects and Mechanism for a Nucleophilic Aromatic Substitution from Qm/Mm Simulations. Org. Lett. 2004, 6, 2881–2884. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Aptula, A.O.; Patlewicz, G.Y. Chemistry-Based Risk Assessment for Skin Sensitization: Quantitative Mechanistic Modeling for the Snar Domain. Chem. Res. Toxicol. 2011, 24, 1003–1011. [Google Scholar] [CrossRef]
- Imoto, M.; Matsui, Y.; Takeda, M.; Tamaki, A.; Taniguchi, H.; Mizuno, K.; Ikeda, H. A Probable Hydrogen-Bonded Meisenheimer Complex: An Unusually High Snar Reactivity of Nitroaniline Derivatives with Hydroxide Ion in Aqueous Media. J. Org. Chem. 2011, 76, 6356–6361. [Google Scholar] [CrossRef]
- Jose, K.B.; Cyriac, J.; Moolayil, J.T.; Sebastian, V.S.; George, M. The Mechanism of Aromatic Nucleophilic Substitution Reaction between Ethanolamine and Fluoro-Nitrobenzenes: An Investigation by Kinetic Measurements and Dft Calculations. J. Phys. Org. Chem. 2011, 24, 714–719. [Google Scholar] [CrossRef]
- Ormazábal-Toledo, R.; Contreras, R.; Campodónico, P.R. Reactivity Indices Profile: A Companion Tool of the Potential Energy Surface for the Analysis of Reaction Mechanisms. Nucleophilic Aromatic Substitution Reactions as Test Case. J. Org. Chem. 2013, 78, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal-Toledo, R.; Contreras, R.; Tapia, R.A.; Campodonico, P.R. Specific Nucleophile-Electrophile Interactions in Nucleophilic Aromatic Substitutions. Org. Biomol. Chem. 2013, 11, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.; Andres, J.; Safont, V.S.; Campodonico, P.; Santos, J.G. A Theoretical Study on the Relationship between Nucleophilicity and Ionization Potentials in Solution Phase. J. Phys. Chem. A 2003, 107, 5588–5593. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G.; Uggerud, E. Rate-Determining Factors in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 2010, 75, 2971–2980. [Google Scholar] [CrossRef]
- Von Richter, V. Technical Translations. Chem. Ber. 1871, 4, 459. [Google Scholar]
- Davis, R.B.; Pizzini, L.C. Condensation of Aromatic Nitro Compounds with Acrylacetonitriles.1,2 Ii. Some P-Substituted Nitrobenzenes. J. Org. Chem. 1960, 25, 1884–1888. [Google Scholar] [CrossRef]
- Mąkosza, M.; Staliński, K. Oxidative Nucleophilic Substitution of Hydrogen in Nitroarenes. Chem. Eur. J. 1997, 3, 2025–2031. [Google Scholar] [CrossRef]
- Malykhin, E.V.; Kolesnichenko, G.A.; Shteingarts, V.D. Reaction of aromatic-compounds with nucleophilic-reagents in the liquid ammonium medium. 5. mechanism and hydroxylation orientation of para-substituted nitrobenzene with potassium hydroxide. Zh. Org. Khim. 1985, 21, 1150–1159. [Google Scholar]
- Goliński, J.; Makosza, M. “Vicarious” Nucleophilic Substitution of Hydrogen in Aromatic Nitro Compounds. Tetrahedron Lett. 1978, 19, 3495–3498. [Google Scholar] [CrossRef]
- Mąkosza, M.; Winiarski, J. Vicarious Nucleophilic Substitution of Hydrogen. Acc. Chem. Res. 1987, 20, 282–289. [Google Scholar] [CrossRef]
- Mąkosza, M. Nucleophilic Substitution of Hydrogen in Nitroarenes: A New Chapter of Aromatic Chemistry. Synthesis 2011, 2011, 2341–2356. [Google Scholar] [CrossRef]
- Mąkosza, M. Nucleophilic Substitution of Hydrogen in Electron-Deficient Arenes, a General Process of Great Practical Value. Chem. Soc. Rev. 2010, 39, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Chupakhin, O.N.; Charushin, V.N.; Van der Plas, H.C. Nucleophilic Aromatic Substitution of Hydrogen; Academic Press: New York, NY, USA, 1994. [Google Scholar]
- Mąkosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen in Heterocyclic Chemistry. Chem. Rev. 2004, 104, 2631–2666. [Google Scholar] [CrossRef]
- Antoniak, D.; Barbasiewicz, M. Corey–Chaykovsky Cyclopropanation of Nitronaphthalenes: Access to Benzonorcaradienes and Related Systems. Org. Lett. 2019, 21, 9320–9325. [Google Scholar] [CrossRef]
- Khutorianskyi, V.V.; Klepetářová, B.; Beier, P. Vicarious Nucleophilic Chloromethylation of Nitroaromatics. Org. Lett. 2019, 21, 5443–5446. [Google Scholar] [CrossRef]
- Brześkiewicz, J.; Loska, R.; Mąkosza, M. A-Chlorobenzylation of Nitroarenes Via Vicarious Nucleophilic Substitution with Benzylidene Dichloride: Umpolung of the Friedel–Crafts Reaction. J. Org. Chem. 2018, 83, 8499–8508. [Google Scholar] [CrossRef]
- Mąkosza, M. Reactions of Nucleophiles with Nitroarenes: Multifacial and Versatile Electrophiles. Chem. Eur. J. 2014, 20, 5536–5545. [Google Scholar] [CrossRef]
- Ormazábal-Toledo, R.; Richter, S.; Robles-Navarro, A.; Maulén, B.; Matute, R.A.; Gallardo-Fuentes, S. Meisenheimer Complexes as Hidden Intermediates in the Aza-Snar Mechanism. Org. Biomol. Chem. 2020, 18, 4238–4247. [Google Scholar] [CrossRef]
- Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification. J. Am. Chem. Soc. 2016, 138, 7276–7281. [Google Scholar] [CrossRef]
- Mąkosza, M. How Does Nucleophilic Aromatic Substitution in Nitroarenes Really Proceed: General Mechanism. Synthesis 2017, 49, 3247–3254. [Google Scholar] [CrossRef]
- Makosza, M. Nucleophilic substitution in nitroarenes: A general corrected mechanism. ChemTexts 2019, 5, 10. [Google Scholar] [CrossRef]
- Lemek, T.; Mąkosza, M.; Stephenson, D.S.; Mayr, H. Direct Observation of the Intermediate in Vicarious Nucleophilic Substitutions of Hydrogen. Angew. Chem. Int. Ed. 2003, 42, 2793–2795. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, J.; Krygowski, T.M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Gwaltney, S.R.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Charge-Transfer Mechanism for Electrophilic Aromatic Nitration and Nitrosation Via the Convergence of (Ab Initio) Molecular-Orbital and Marcus−Hush Theories with Experiments. J. Am. Chem. Soc. 2003, 125, 3273–3283. [Google Scholar] [CrossRef]
- Mąkosza, M.; Mortier, J. The Discovery and Development of Metal-Free Arylation Reactions with Unsymmetrical Diaryliodonium Salts; Mortier, J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; Chapter 11; pp. 269–298. [Google Scholar]
- Makosza, M.; Stalinski, K.; Klepka, C. Oxidative Nucleophilic Substitution of Hydrogen in Nitrobenzene with 2-Phenylpropionitrile Carbanion and Potassium Permanganate Oxidant. Chem. Commun. 1996, 837–838. [Google Scholar] [CrossRef]
- Wróbel, Z.; Mąkosza, M. New Simple Synthesis of N-Hydroxy 2-Vinylindoles. Synlett 1993, 1993, 597–598. [Google Scholar] [CrossRef]
- Mąkosza, M. Current Trends in Organic Synthesis, Proceedings of the Fourth International Conference on Organic Synthesis, Tokyo, Japan, 22–27 August 1982; Nozaki, H., Ed.; Pergamon: Oxford, UK, 1983; pp. 401–412. [Google Scholar] [CrossRef]
- Wróbel, Z.; Kwast, A. Simple Synthesis of N-Aryl-2-Nitrosoanilines in the Reaction of Nitroarenes with Aniline Anion Derivatives. Synthesis 2010, 2010, 3865–3872. [Google Scholar] [CrossRef]
- Swain, C.G.; Scott, C.B. Quantitative Correlation of Relative Rates. Comparison of Hydroxide Ion with Other Nucleophilic Reagents toward Alkyl Halides, Esters, Epoxides and Acyl Halides1. J. Am. Chem. Soc. 1953, 75, 141–147. [Google Scholar] [CrossRef]
- Edwards, J.O. Correlation of Relative Rates and Equilibria with a Double Basicity Scale. J. Am. Chem. Soc. 1954, 76, 1540–1547. [Google Scholar] [CrossRef]
- Ritchie, C.D. Nucleophilic Reactivities toward Cations. Acc. Chem. Res. 1972, 5, 348–354. [Google Scholar] [CrossRef]
- Ritchie, C.D. Cation–Anion Combination Reactions. 26. A Review. Can. J. Chem. 1986, 64, 2239–2250. [Google Scholar] [CrossRef]
- Lucius, R.; Mayr, H. Constant Selectivity Relationships of Addition Reactions of Carbanions. Angew. Chem. Int. Ed. 2000, 39, 1995–1997. [Google Scholar] [CrossRef]
- Mayr, H. Cc Bond Formation by Addition of Carbenium Ions to Alkenes: Kinetics and Mechanism. Angew. Chem. Int. Ed. Engl. 1990, 29, 1371–1384. [Google Scholar] [CrossRef]
- Mayr, H.; Bug, T.; Gotta, M.F.; Hering, N.; Irrgang, B.; Janker, B.; Kempf, B.; Loos, R.; Ofial, A.R.; Remennikov, G.; et al. Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles. J. Am. Chem. Soc. 2001, 123, 9500–9512. [Google Scholar] [CrossRef]
- Mayr, H.; Müller, K.H.; Rau, D. Comparison of the Electrophilicities of Cationic Metal Π Complexes and of Ordinary Carbenium Ions. Angew. Chem. Int. Ed. Engl. 1993, 32, 1630–1632. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R. Electrophilicities of Iminium Ions. Tetrahedron Lett. 1997, 38, 3503–3506. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R.; Sauer, J.; Schmied, B. [2++4] Cycloadditions of Iminium Ions − Concerted or Stepwise Mechanism of Aza Diels−Alder Reactions? Eur. J. Org. Chem. 2000, 2000, 2013–2020. [Google Scholar] [CrossRef]
- Roth, M.; Mayr, H. The Coexistence of the Reactivity–Selectivity Principle and of Linear Free Energy Relationships: A Diffusion Clock for Determining Carbocation Reactivities. Angew. Chem. Int. Ed. Engl. 1995, 34, 2250–2252. [Google Scholar] [CrossRef]
- Roy, R.K.; Krishnamurti, S.; Geerlings, P.; Pal, S. Local Softness and Hardness Based Reactivity Descriptors for Predicting Intra- and Intermolecular Reactivity Sequences: Carbonyl Compounds. J. Phys. Chem. A 1998, 102, 3746–3755. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B. Reactivity Dynamics in Atom−Field Interactions: A Quantum Fluid Density Functional Study. J. Phys. Chem. A 2001, 105, 169–183. [Google Scholar] [CrossRef]
- Ormazabal-Toledo, R.; Contreras, R. Philicity and Fugality Scales for Organic Reactions. Adv. Chem. 2014, 2014, 13. [Google Scholar] [CrossRef]
- Błażej, S.; Mąkosza, M. Substituent Effects on the Electrophilic Activity of Nitroarenes in Reactions with Carbanions. Chem. Eur. J. 2008, 14, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Persson, J.; Matsson, O. Use of Fluorine Kinetic Isotope Effects in the Study of Steric Effects in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 1998, 63, 9348–9350. [Google Scholar] [CrossRef]
- Matsson, O.; Persson, J.; Axelsson, B.S.; Laangstroem, B. Fluorine Kinetic Isotope Effects. J. Am. Chem. Soc. 1993, 115, 5288–5289. [Google Scholar] [CrossRef]
- Mąkosza, M.; Lemek, T.; Kwast, A.; Terrier, F. Elucidation of the Vicarious Nucleophilic Substitution of Hydrogen Mechanism Via Studies of Competition between Substitution of Hydrogen, Deuterium, and Fluorine. J. Org. Chem. 2002, 67, 394–400. [Google Scholar] [CrossRef]
- Makosza, M.; Staliski, K. Oxidative Nucleophilic Substitution of Hydrogen with 2-Phenylpropanenitrile Carbanion in Heterocyclic Nitroarenes. Synthesis 1998, 11, 1631–1634. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules 2020, 25, 4819. https://doi.org/10.3390/molecules25204819
Błaziak K, Danikiewicz W, Mąkosza M. How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules. 2020; 25(20):4819. https://doi.org/10.3390/molecules25204819
Chicago/Turabian StyleBłaziak, Kacper, Witold Danikiewicz, and Mieczysław Mąkosza. 2020. "How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices" Molecules 25, no. 20: 4819. https://doi.org/10.3390/molecules25204819
APA StyleBłaziak, K., Danikiewicz, W., & Mąkosza, M. (2020). How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules, 25(20), 4819. https://doi.org/10.3390/molecules25204819