
Regulatory Impact of the C-Terminal Tail on Charge Transfer Pathways in Drosophila Cryptochrome



Abstract
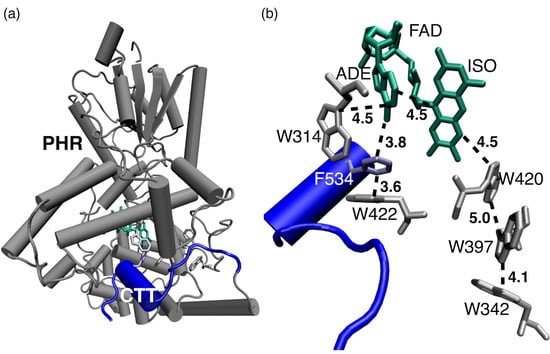
1. Introduction
2. Results
2.1. Nanosecond Structural Fluctuations
2.2. Driving Force of Charge Transfer Reactions
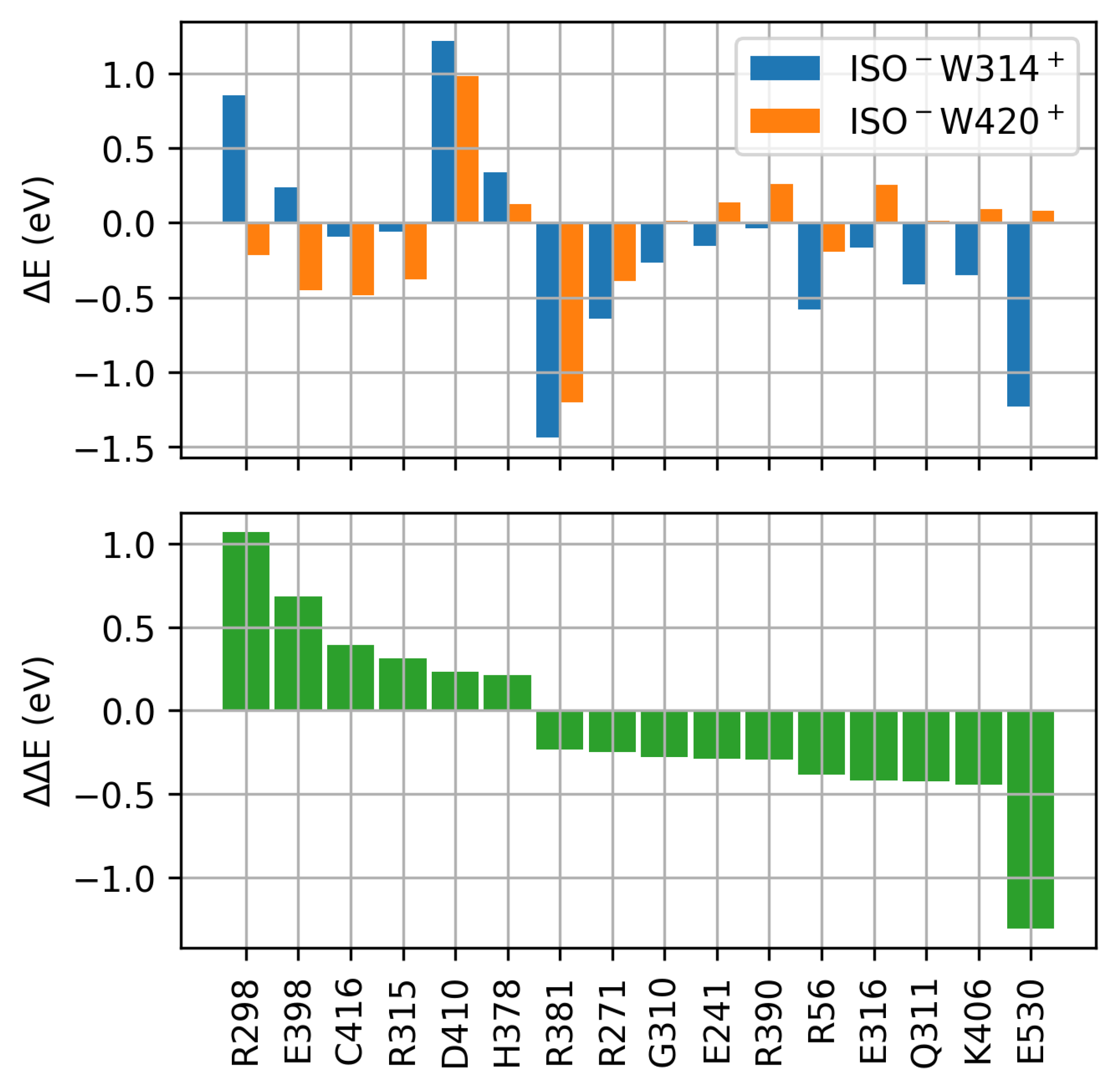
2.3. Protein Electrostatic Environment
2.4. Charge Transfer Dynamics
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics (MD)
4.2. QM/MM Simulations
4.3. Reorganization Energies and Driving Forces of CT Reactions:
4.4. Model Hamiltonian
4.5. MACGIC-QUAPI Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sancar, A. Structure and Function of DNA Photolyase and Cryptochrome Blue-Light Photoreceptors. Chem. Rev. 2003, 103, 2203–2238. [Google Scholar] [CrossRef]
- Ozturk, N.; Selby, C.P.; Zhong, D.; Sancar, A. Mechanism of Photosignaling by Drosophila Cryptochrome: Role of the Redox Status of the Flavin Chromophore. J. Biol. Chem. 2013, 289, 4634–4642. [Google Scholar] [CrossRef]
- Vechtomova, Y.L.; Telegina, T.A.; Kritsky, M.S. Evolution of Proteins of the DNA Photolyase/Cryptochrome Family. Biochemistry 2020, 85, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.L.; Sancar, A. Photolyase/cryptochrome blue-light photoreceptors use photon energy to repair DNA and reset the circadian clock. Oncogene 2002, 21, 9043–9056. [Google Scholar] [CrossRef] [PubMed]
- Merbitz-Zahradnik, T.; Wolf, E. How is the inner circadian clock controlled by interactive clock proteins?: Structural analysis of clock proteins elucidates their physiological role. FEBS Lett. 2015, 589, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- Crane, B.R.; Young, M.W. Interactive Features of Proteins Composing Eukaryotic Circadian Clocks. Annu. Rev. Biochem. 2014, 83, 191–219. [Google Scholar] [CrossRef]
- Thieulin-Pardo, G.; Avilan, L.; Kojadinovic, M.; Gontero, B. Fairy “tails”: Flexibility and function of intrinsically disordered extensions in the photosynthetic world. Front. Mol. Biosci. 2015, 2, 23. [Google Scholar] [CrossRef]
- Michael, A.K.; Fribourgh, J.L.; Van Gelder, R.N.; Partch, C.L. Animal Cryptochromes: Divergent Roles in Light Perception, Circadian Timekeeping and Beyond. Photochem. Photobiol. 2016, 93, 128–140. [Google Scholar] [CrossRef]
- Zoltowski, B.D.; Vaidya, A.T.; Top, D.; Widom, J.; Young, M.W.; Crane, B.R. Structure of full-length Drosophila cryptochrome. Nature 2011, 480, 396–399. [Google Scholar] [CrossRef]
- Levy, C.; Zoltowski, B.D.; Jones, A.R.; Vaidya, A.T.; Top, D.; Widom, J.; Young, M.W.; Scrutton, N.S.; Crane, B.R.; Leys, D. Updated structure of Drosophila cryptochrome. Nature 2013, 495, E3–E4. [Google Scholar] [CrossRef]
- Czarna, A.; Berndt, A.; Singh, H.R.; Grudziecki, A.; Ladurner, A.G.; Timinszky, G.; Kramer, A.; Wolf, E. Structures of Drosophila Cryptochrome and Mouse Cryptochrome1 Provide Insight into Circadian Function. Cell 2013, 153, 1394–1405. [Google Scholar] [CrossRef]
- Partch, C.L.; Sancar, A. Photochemistry and Photobiology of Cryptochrome Blue-light Photopigments: The Search for a Photocycle. Photochem. Photobiol. 2005, 81, 1291–1304. [Google Scholar] [CrossRef]
- Ozturk, N. Phylogenetic and Functional Classification of the Photolyase/Cryptochrome Family. Photochem. Photobiol. 2017, 93, 104–111. [Google Scholar] [CrossRef]
- Dissel, S.; Codd, V.; Fedic, R.; Garner, K.J.; Costa, R.; Kyriacou, C.P.; Rosato, E. A constitutively active cryptochrome in Drosophila melanogaster. Nat. Neurosci. 2004, 7, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Busza, A.; Emery-Le, M.; Rosbash, M.; Emery, P. Roles of the Two Drosophila CRYPTOCHROME Structural Domains in Circadian Photoreception. Science 2004, 304, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, N.; Selby, C.P.; Annayev, Y.; Zhong, D.; Sancar, A. Reaction mechanism of Drosophila cryptochrome. Proc. Natl. Acad. Sci. USA 2011, 108, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.; Schleicher, E.; Kacprzak, S.; Bouly, J.P.; Picot, M.; Wu, W.; Berndt, A.; Wolf, E.; Bittl, R.; Ahmad, M. Human and Drosophila cryptochromes are light activated by flavin photoreduction in living cells. PLoS Biol. 2008, 6, 1559–1569. [Google Scholar]
- Vaidya, A.T.; Top, D.; Manahan, C.C.; Tokuda, J.M.; Zhang, S.; Pollack, L.; Young, M.W.; Crane, B.R. Flavin reduction activates Drosophila cryptochrome. Proc. Natl. Acad. Sci. USA 2013, 110, 20455–20460. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Manahan, C.C.; Top, D.; Yee, E.F.; Lin, C.; Young, M.W.; Thiel, W.; Crane, B.R. Changes in active site histidine hydrogen bonding trigger cryptochrome activation. Proc. Natl. Acad. Sci. USA 2016, 113, 10073–10078. [Google Scholar] [CrossRef]
- Aubert, C.; Vos, M.H.; Mathis, P.; Eker, A.M.; Brettel, K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 2000, 405, 586–590. [Google Scholar] [CrossRef]
- Giovani, B.; Byrdin, M.; Ahmad, M.; Brettel, K. Light-induced electron transfer in a cryptochrome blue-light photoreceptor. Nat. Struct. Mol. Biol. 2003, 10, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Solov’yov, I.A.; Domratcheva, T.; Moughal Shahi, A.R.; Schulten, K. Decrypting Cryptochrome: Revealing the Molecular Identity of the Photoactivation Reaction. J. Am. Chem. Soc. 2012, 134, 18046–18052. [Google Scholar] [CrossRef] [PubMed]
- Cailliez, F.; Müller, P.; Gallois, M.; de la Lande, A. ATP Binding and Aspartate Protonation Enhance Photoinduced Electron Transfer in Plant Cryptochrome. J. Am. Chem. Soc. 2014, 136, 12974–12986. [Google Scholar] [CrossRef] [PubMed]
- Lüdemann, G.; Solov’yov, I.A.; Kubař, T.; Elstner, M. Solvent Driving Force Ensures Fast Formation of a Persistent and Well-Separated Radical Pair in Plant Cryptochrome. J. Am. Chem. Soc. 2015, 137, 1147–1156. [Google Scholar] [CrossRef]
- Lin, C.; Top, D.; Manahan, C.C.; Young, M.W.; Crane, B.R. Circadian clock activity of cryptochrome relies on tryptophan-mediated photoreduction. Proc. Natl. Acad. Sci. USA 2018, 115, 3822–3827. [Google Scholar] [CrossRef]
- Ahmad, M. Photocycle and signaling mechanisms of plant cryptochromes. Curr. Opin. Plant. Biol. 2016, 33, 108–115. [Google Scholar] [CrossRef]
- Gegear, R.J.; Foley, L.E.; Casselman, A.; Reppert, S.M. Animal cryptochromes mediate magnetoreception by an unconventional photochemical mechanism. Nature 2010, 463, 804–807. [Google Scholar] [CrossRef]
- Li, X.; Wang, Q.; Yu, X.; Liu, H.; Yang, H.; Zhao, C.; Liu, X.; Tan, C.; Klejnot, J.; Zhong, D.; et al. Arabidopsis cryptochrome 2 (CRY2) functions by the photoactivation mechanism distinct from the tryptophan (trp) triad-dependent photoreduction. Proc. Natl. Acad. Sci. USA 2011, 108, 20844–20849. [Google Scholar] [CrossRef]
- Gao, J.; Wang, X.; Zhang, M.; Bian, M.; Deng, W.; Zuo, Z.; Yang, Z.; Zhong, D.; Lin, C. Trp triad-dependent rapid photoreduction is not required for the function of Arabidopsis CRY1. Proc. Natl. Acad. Sci. USA 2015, 112, 9135–9140. [Google Scholar] [CrossRef]
- Biskup, T.; Paulus, B.; Okafuji, A.; Hitomi, K.; Getzoff, E.D.; Weber, S.; Schleicher, E. Variable Electron-Transfer Pathways in an Amphibian Cryptochrome: Tryptophan versus Tyrosine-Based Radical Pairs. J. Biol. Chem. 2013, 288, 9249–9260. [Google Scholar] [CrossRef]
- Parson, W.W.; Chu, Z.T.; Warshel, A. Reorganization Energy of the Initial Electron-Transfer Step in Photosynthetic Bacterial Reaction Centers. Biophys. J. 1998, 74, 182–191. [Google Scholar] [CrossRef]
- Richter, M.; Fingerhut, B.P. Coarse-grained representation of the quasi adiabatic propagator path integral for the treatment of non-Markovian long-time bath memory. J. Chem. Phys. 2017, 146, 214101. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.T.; Tan, C.; Song, S.H.; Öztürk, N.; Li, J.; Wang, L.; Sancar, A.; Zhong, D. Ultrafast Dynamics and Anionic Active States of the Flavin Cofactor in Cryptochrome and Photolyase. J. Am. Chem. Soc. 2008, 130, 7695–7701. [Google Scholar] [CrossRef]
- Solov’yov, I.A.; Domratcheva, T.; Schulten, K. Separation of photo-induced radical pair in cryptochrome to a functionally critical distance. Sci Rep. 2014, 4, 3845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, L.; Shu, S.; Sancar, A.; Zhong, D. Bifurcating electron-transfer pathways in DNA photolyases determine the repair quantum yield. Science 2016, 354, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, B.P.; Zinth, W.; de Vivie-Riedle, R. Design criteria for optimal photosynthetic energy conversion. Chem. Phys. Lett. 2008, 466, 209–213. [Google Scholar] [CrossRef]
- Langenbacher, T.; Immeln, D.; Dick, B.; Kottke, T. Microsecond Light-Induced Proton Transfer to Flavin in the Blue Light Sensor Plant Cryptochrome. J. Am. Chem. Soc. 2009, 131, 14274–14280. [Google Scholar] [CrossRef]
- Muller, P.; Ignatz, E.; Kiontke, S.; Brettel, K.; Essen, L.O. Sub-nanosecond tryptophan radical deprotonation mediated by a protein-bound water cluster in class II DNA photolyases. Chem. Sci. 2018, 9, 1200–1212. [Google Scholar] [CrossRef]
- Hemsley, M.J.; Mazzotta, G.M.; Mason, M.; Dissel, S.; Toppo, S.; Pagano, M.A.; Sandrelli, F.; Meggio, F.; Rosato, E.; Costa, R.; et al. Linear motifs in the C-terminus of D. melanogaster cryptochrome. Biochem. Biophys. Res. Commun. 2007, 355, 531–537. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinf. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Baer, R.; Neuhauser, D. Density Functional Theory with Correct Long-Range Asymptotic Behavior. Phys. Rev. Lett. 2005, 94, 043002. [Google Scholar] [CrossRef] [PubMed]
- Voityuk, A.A.; Rösch, N. Fragment charge difference method for estimating donor–acceptor electronic coupling: Application to DNA π-stacks. J. Chem. Phys. 2002, 117, 5607–5616. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Kats, D.; Korona, T.; Schütz, M. Local CC2 electronic excitation energies for large molecules with density fitting. J. Chem. Phys. 2006, 125, 104106. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO; Version 2015.1; A Package of Ab Initio Programs; University of Cardiff Chemistry Consultants (UC3): Cardiff, Wales, UK, 2015. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general–purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Firmino, T.; Mangaud, E.; Cailliez, F.; Devolder, A.; Mendive-Tapia, D.; Gatti, F.; Meier, C.; Desouter-Lecomte, M.; de la Lande, A. Quantum effects in ultrafast electron transfers within cryptochromes. Phys. Chem. Chem. Phys. 2016, 18, 21442–21457. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Fingerhut, B.P. Coupled excitation energy and charge transfer dynamics in reaction centre inspired model systems. Faraday Discuss. 2019, 216, 72–93. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | ||||||
|---|---|---|---|---|---|---|
| 0.25 | — | — | — | — | — | |
| Trp triad: | ||||||
| ISOW420 | 1.10 | 1.14 | 1.14 | +0.13 | −0.16 (0.15) | −0.16 |
| ISOW397 | 2.02 | 2.03 | 2.69 | −0.44 | −0.70 (0.31) | −1.35 |
| CTT: | ||||||
| ISOADE | 1.20 | 1.28 | — | +0.24 | −0.10 (0.19) | — |
| ISOW314 | 1.90 | 1.98 | 2.92 | −0.40 | −0.75 (0.22) | −1.66 |
| ISOW422 | 1.55 | 1.64 | 1.98 | −0.16 | −0.52 (0.26) | −0.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, M.; Fingerhut, B.P. Regulatory Impact of the C-Terminal Tail on Charge Transfer Pathways in Drosophila Cryptochrome. Molecules 2020, 25, 4810. https://doi.org/10.3390/molecules25204810
Richter M, Fingerhut BP. Regulatory Impact of the C-Terminal Tail on Charge Transfer Pathways in Drosophila Cryptochrome. Molecules. 2020; 25(20):4810. https://doi.org/10.3390/molecules25204810
Chicago/Turabian StyleRichter, Martin, and Benjamin P. Fingerhut. 2020. "Regulatory Impact of the C-Terminal Tail on Charge Transfer Pathways in Drosophila Cryptochrome" Molecules 25, no. 20: 4810. https://doi.org/10.3390/molecules25204810
APA StyleRichter, M., & Fingerhut, B. P. (2020). Regulatory Impact of the C-Terminal Tail on Charge Transfer Pathways in Drosophila Cryptochrome. Molecules, 25(20), 4810. https://doi.org/10.3390/molecules25204810