
Synthesis and Characterization of Ion Pairs between Alkaline Metal Ions and Anionic Anti-Aromatic and Aromatic Hydrocarbons with π-Conjugated Central Seven- and Eight-Membered Rings

 , , , and
, , , and
Abstract
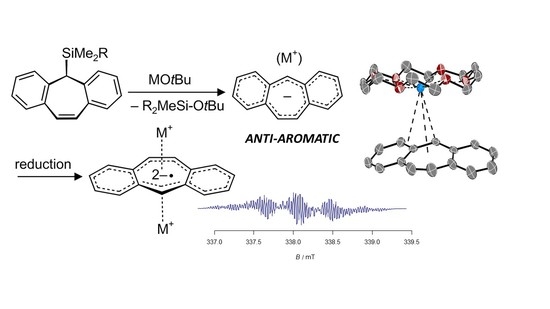
1. Introduction
2. Results
2.1. Characterization by NMR, EPR and X-ray Diffraction Methods
2.2. Evaluation of Anti-Aromaticity and Aromaticity by Calculation of NICS Values
2.3. Electrochemistry
3. Materials and Methods
3.1. General Comments
3.2. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thomaier, J.; Boulmaâz, S.; Schönberg, H.; Rüegger, H.; Currao, A.; Grützmacher, H.; Hillebrecht, H.; Pritzkow, H. Dibenzotropylidene Phosphanes (TROPPs): Synthesis and Coinage Metal Complexes. New J. Chem. 1998, 22, 947–958. [Google Scholar] [CrossRef]
- Schönberg, H.; Boulmaâz, S.; Wörle, M.; Liesum, L.; Schweiger, A.; Grützmacher, H. A Monomeric d9-Rhodium(0) Complex. Angew. Chem. Int. Ed. 1998, 37, 1423–1426. [Google Scholar] [CrossRef]
- Ostendorf, D.; Landis, C.; Grützmacher, H. Trigonal Pyramids: Alternative Ground-State Structures for Sixteen-Electron Complexes. Angew. Chem. Int. Ed. 2006, 45, 5169–5173. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, C.; Viciu, L.; Adelhardt, M.; Sutter, J.; Meyer, K.; de Bruin, B.; Grützmacher, H. Low-Valent Iron(I) Amido Olefin Complexes as Promotors for Dehydrogenation Reactions. Angew. Chem. Int. Ed. 2015, 54, 5766–5771. [Google Scholar] [CrossRef]
- Grützmacher, H. Cooperating Ligands in Catalysis. Angew. Chem. Int. Ed. 2008, 47, 1814–1818. [Google Scholar] [CrossRef]
- Maire, P.; Büttner, T.; Breher, F.; Le Floch, P.; Grützmacher, H. Heterolytic Splitting of Hydrogen with Rhodium(I) Amides. Angew. Chem. Int. Ed. 2005, 44, 6318–6323. [Google Scholar] [CrossRef]
- Zweifel, T.; Naubron, J.-V.; Büttner, T.; Ott, T.; Grützmacher, H. Ethanol as Hydrogen Donor: Highly Efficient Transfer Hydrogenations with Rhodium(I) Amides. Angew. Chem. Int. Ed. 2008, 47, 3245–3249. [Google Scholar] [CrossRef]
- Zweifel, T.; Naubron, J.-V.; Grützmacher, H. Catalyzed Dehydrogenative Coupling of Primary Alcohols with Water, Methanol, or Amines. Angew. Chem. Int. Ed. 2009, 48, 559–563. [Google Scholar] [CrossRef]
- Gianetti, T.L.; Annen, S.P.; Santiso-Quinones, G.; Reiher, M.; Driess, M.; Grützmacher, H. Nitrous Oxide as a Hydrogen Acceptor for the Dehydrogenative Coupling of Alcohols. Angew. Chem. Int. Ed. 2016, 55, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grützmacher, H. A Homogeneous Transition Metal Complex for Clean Hydrogen Production From Methanol–Water Mixtures. Nat. Chem. 2013, 5, 342. [Google Scholar] [CrossRef]
- Trincado, M.; Sinha, V.; Rodriguez-Lugo, R.E.; Pribanic, B.; de Bruin, B.; Grützmacher, H. Homogeneously Catalysed Conversion of Aqueous Formaldehyde to H2 and Carbonate. Nat. Commun. 2017, 8, 14990. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Pribanic, B.; de Bruin, B.; Trincado, M.; Grützmacher, H. Ligand- and Metal-Based Reactivity of a Neutral Ruthenium Diolefin Diazadiene Complex: The Innocent, the Guilty and the Suspicious. Chem. Eur. J. 2018, 24, 5513–5521. [Google Scholar] [CrossRef] [PubMed]
- Defieber, C.; Grützmacher, H.; Carreira, E.M. Chiral Olefins as Steering Ligands in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2008, 47, 4482–4502. [Google Scholar] [CrossRef] [PubMed]
- Harder, S.; Martin, J.; Langer, J.; Wiesinger, M.; Elsen, H. Dibenzotropylidene Substituted Ligands for Early Main Group Metal-Alkene Bonding. Eur. J. Inorg. Chem. 2020, 2020, 2582–2595. [Google Scholar] [CrossRef]
- Bock, H.; Ruppert, K.; Näther, C.; Havlas, Z.; Herrmann, H.-F.; Arad, C.; Göbel, I.; John, A.; Meuret, J.; Nick, S.; et al. Distorted Molecules: Perturbation Design, Preparation and Structures. Angew. Chem. Int. Ed. 1992, 31, 550–581. [Google Scholar] [CrossRef]
- Bock, H.; Gharagozloo-Hubmann, K.; Sievert, M.; Prisner, T.; Havlas, Z. Single Crystals of an Ionic Anthracene Aggregate with a Triplet Ground State. Nature 2000, 404, 267–269. [Google Scholar] [CrossRef]
- Müllen, K. Reduction and Oxidation of Annulenes. Chem. Rev. 1984, 84, 603–646. [Google Scholar] [CrossRef]
- Vos, H.W.; Bakker, Y.W.; Maclean, C.; Velthorst, N.H. Paramagnetic Ring Currents in the Carbanion of 5H-dibenzo[a,d]cycloheptene and the Nitranion of 5H-dibenz[b,f]azepine. Chem. Phys. Lett. 1974, 25, 80–83. [Google Scholar] [CrossRef]
- von Schleyer, P.R.; Jiao, H. What is Aromaticity? Pure Appl. Chem. 1996, 68, 209–218. [Google Scholar] [CrossRef]
- McAuley, I.; Krogh, E.; Wan, P. Carbanion Intermediates in the Photodecarboxylation of Benzannelated Acetic Acids in Aqueous Solution. J. Am. Chem. Soc. 1988, 110, 600–602. [Google Scholar] [CrossRef]
- Krogh, E.; Wan, P. Photodecarboxylation of Diarylacetic Acids in Aqueous Solution: Enhanced Photogeneration of Cyclically Conjugated Eight π Electron Carbanions. J. Am. Chem. Soc. 1992, 114, 705–712. [Google Scholar] [CrossRef]
- Wan, P.; Krogh, E.; Chak, B. Enhanced Formation of 8π(4n) Conjugated Cyclic Carbanions in the Excited State: First Example of Photochemical C–H Bond Heterolysis in Photoexcited Suberene. J. Am. Chem. Soc. 1988, 110, 4073–4074. [Google Scholar] [CrossRef]
- Budac, D.; Wan, P. Excited-State Carbon Acids. Facile Benzylic Carbon-Hydrogen Bond Heterolysis of Suberene on Photolysis in Aqueous Solution: A Photogenerated Cyclically Conjugated Eight π Electron Carbanion. J. Org. Chem. 1992, 57, 887–894. [Google Scholar] [CrossRef]
- Bauld, N.L.; Brown, M.S. Dianion Radicals. II. Tropenide Systems. J. Am. Chem. Soc. 1967, 89, 5417–5421. [Google Scholar] [CrossRef]
- Ceccon, A.; Gambaro, A.; Pizzato, L.; Romanin, A.; Venzo, A. Metal Stabilized Carbanions. Kinetic Acidity and 1H NMR Spectrum of the π-(Tricarbonylchromium)-5H-dibenzo[a,d]cycloheptenyl Anion. J. Chem. Soc. Chem. Commun. 1982, 16, 907–908. [Google Scholar] [CrossRef]
- Ceccon, A.; Gambaro, A.; Venzo, A. Metal-Stabilized Carbanions. J. Organomet. Chem. 1984, 275, 209–222. [Google Scholar] [CrossRef]
- Irngartinger, H.; Reibel, W.R.K. Structures of Dibenzo[a,e]cyclooctatetraene and Tetrabenzo[a,c,e,g]cyclooctatetraene (o-Tetraphenylene). Acta Cryst. B 1981, 37, 1724–1728. [Google Scholar] [CrossRef]
- Katz, T.J.; Yoshida, M.; Siew, L.C. The sym-Dibenzcyclooctatetraene Anion Radical and Dianion. J. Am. Chem. Soc. 1965, 87, 4516–4520. [Google Scholar] [CrossRef]
- Kojima, H.; Bard, A.J.; Wong, H.N.C.; Sondheimer, F. Electrochemical Reduction of sym-Dibenzocyclooctatetraene, sym-Dibenzo-1,5-cyclooctadiene-3,7-diyne, and sym-Dibenzo-1,3,5-cyclooctatrien-7-yne. J. Am. Chem. Soc. 1976, 98, 5560–5565. [Google Scholar] [CrossRef]
- Gerson, F.; Martin, W.B., Jr.; Plattner, G.; Sondheimer, F.; Wong, H.N.C. ESR. Spectra and Structures of Radical Anions in the Dibenzo[a,e]cyclooctene Series. Helv. Chim. Acta 1976, 59, 2038–2048. [Google Scholar] [CrossRef]
- Sygula, A.; Fronczek, F.R.; Rabideau, P.W. The First Example of η8 Coordination of Lithium Cations with a Cyclooctatetraene Dianion: Crystal Structure of Li2(dibenzo[a,e]cyclooctatetraene)(TMEDA)2. J. Organomet. Chem. 1996, 526, 389–391. [Google Scholar] [CrossRef]
- Franck, G.; Brill, M.; Helmchen, G. Dibenzo[a,e]cyclooctene: Multi-gram Synthesis of a Bidentate Ligand. Org. Synth. 2012, 89, 55–65. [Google Scholar]
- Berti, G. Communications - Dibenzo[a,e]tropylium and 5-Phenyldibenzo[a,e]tropylium Cations. J. Org. Chem. 1957, 22, 230. [Google Scholar] [CrossRef]
- Ball, R.G.; Edelmann, F.; Kiel, G.Y.; Takats, J.; Drews, R. Cycloheptatrienyl-Bridged Heterobimetallic Complexes: Facile Phosphine Substitution Reactions of (μ-C7H7)Fe(CO)3Rh(CO)2. Organometallics 1986, 5, 829–839. [Google Scholar] [CrossRef]
- Dickson, R.S.; Jenkins, S.M.; Skelton, B.W.; White, A.H. The Addition of Small Molecules to (η-C5H5)2Rh2(CO)(CF3C2CF3)—IX. C–H Bond Activation in the Reactions with Dienes, Polyenes and Arenes; the Crystal and Molecular Structure of [(η-C5H5)2Rh2{C(CF3)=C(CF3)H}]2(C6H4). Polyhedron 1988, 7, 859–870. [Google Scholar] [CrossRef]
- Oro, L.A.; Valderrama, M.; Cifuentes, P.; Foces-Foces, C.; Cano, F.H. Azulene as a Ligand in Cationic Rhodium and Iridium Complexes. Crystal Structure of [Rh(TFB)(az)]PF6. J. Organomet. Chem. 1984, 276, 67–77. [Google Scholar] [CrossRef]
- Kunkely, H.; Vogler, A. Ligand-to-Ligand Charge Transfer in [(η-C5Me5)RhIII(η-C7H7)]3+: Absorption and Emission. Inorg. Chem. Commun. 2004, 7, 650–653. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in 1997, 78, 1396–1396. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- von Schleyer, P.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; von Schleyer, P.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, C.; Prokopchuk, D.E.; Adelhardt, M.; Viciu, L.; Meyer, K.; Grützmacher, H. Reactivity of an All-Ferrous Iron–Nitrogen Heterocubane under Reductive and Oxidative Conditions. Chem. Eur. J. 2015, 21, 15797–15805. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.M.; Rawal, V.H. 1,3-Pentadiene. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: New York, NY, USA, 2001. [Google Scholar]
- Ernst, R.D. Structural and Reactivity Patterns in Transition-Metal-Pentadienyl Chemistry. Chem. Rev. 1988, 88, 1255–1291. [Google Scholar] [CrossRef]
- Cordoneanu, A.; Drewitt, M.J.; Bavarian, N.; Baird, M.C. Synthesis and Characterization of Weakly Coordinating Anion Salts of a New, Stable Carbocationic Reagent, the Dibenzosuberenyl (Dibenzotropylium) ion. New J. Chem. 2008, 32, 1890–1898. [Google Scholar] [CrossRef]
- Wu, J.I.C.; Mo, Y.; Evangelista, F.A.; von Ragué Schleyer, P. Is Cyclobutadiene really highly Destabilized by Antiaromaticity? Chem. Commun. 2012, 48, 8437–8439. [Google Scholar] [CrossRef]
- Bahl, J.J.; Bates, R.B.; Beavers, W.A.; Launer, C.R. Cycloheptatrienyl and Heptatrienyl Trianions. J. Am. Chem. Soc. 1977, 99, 6126–6127. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Salcedo, R.; Sansores, L.E.; Fomina, L. Theoretical Study about the Radicals and Anions of [8]annulenes. J. Mol. Struct. 1997, 397, 159–166. [Google Scholar] [CrossRef]
- Viculis, L.M.; Mack, J.J.; Mayer, O.M.; Hahn, H.T.; Kaner, R.B. Intercalation and Exfoliation Routes to Graphite Nanoplatelets. J. Mater. Chem. 2005, 15, 974–978. [Google Scholar] [CrossRef]
- Giordano, G.; Crabtree, R.H.; Heintz, R.M.; Forster, D.; Morris, D.E. Di-μ-Chloro-Bis(η4-1,5-Cyclooctadiene)-Dirhodium(I). In Inorganic Syntheses; Angelici, R.J., Ed.; John Wiley & Sons, Ltd.: New York NY, USA, 2007. [Google Scholar]
- IUPAC. Section A -Hydrocarbons. In Nomenclature of Organic Chemistry; Pergamon Press: Oxford, UK, 1979. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds tropCl, 1a, 1b, and dbcot are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| δ 1Hol | δ 13Col | δ 1Hbz | δ 13Cbz | 1JHC | |
|---|---|---|---|---|---|
| trop2 | 6.90 | 131.43 | 4.90 | 52.18 | |
| Litrop | 2.64 | 139.03 | 0.50 | 83.67 | 139.8 |
| Natrop | 2.60 | 138.79 | 0.56 | 81.56 | 142.9 |
| Ktrop | 2.00 | 139.26 | 0.06 | 89.44 | 144.5 |
| [Rh(trop)(cod)] | 5.16 | 97.52 | 2.88 | 55.84 | 144.6 |
| dbcot [32] | 6.72 | 126.76 | |||
| Li2dbcot [28] | 7.08 | ||||
| Na2dbcot | 7.17 | 93.37 | |||
| K2dbcot | 7.17 | 95.78 |
| Lithftrop | Lidmetrop a | Nadmetrop | Kthftrop | K18c6trop | [Rh(trop)(cod)] | ||
|---|---|---|---|---|---|---|---|
| C1–C2 | 1.440(2) | 1.430(15) | 1.420(16) | 1.4265(19) | 1.428(3) | 1.429(3) | 1.470(2) |
| C2–C3 | 1.423(2) | 1.422(14) | 1.435(17) | 1.4342(19) | 1.433(2) | 1.430(3) | 1.435(2) |
| C3–C4 | 1.468(2) | 1.40(2) | 1.54(2) | 1.455(3) | 1.463(3) | 1.461(3) | 1.458(3) |
| C4–C5 | 1.338(3) | 1.325(14) | 1.324(14) | 1.308(3) | 1.332(3) | 1.339(3) | 1.380(3) |
| C5–C6 | 1.464(3) | 1.47(3) | 1.46(3) | 1.414(3) | 1.467(3) | 1.464(3) | 1.482(3) |
| C6–C7 | 1.423(2) | 1.422(16) | 1.412(17) | 1.419(2) | 1.430(3) | 1.434(3) | 1.398(2) |
| C7–C1 | 1.434(2) | 1.422(13) | 1.414(15) | 1.419(2) | 1.425(3) | 1.431(3) | 1.492(2) |
| C1–M1 | 2.279(3) | 6.788(17) | 7.91(2) | 5.3528(15) | 3.0977(17) b | 3.0213(18) | 2.0912(16) |
| C4–M1 | 3.715(4) | 7.63(2) | 7.50(2) | 5.4943(17) | 3.4072(19) | 4.937(3) | 2.2397(17) |
| C5–M1 | 3.783(3) | 8.35(2) | 6.64(2) | 5.5599(19) | 3.346(2) | 4.694(2) | 2.4044(18) |
| M1–Cnt1 | 7.282(17) | 7.34(2) | 5.2530(15) | 2.8223(4) c | 3.8117(18) | 2.2600(16) | |
| M1–Cnt2 | 2.9708(4) d | 3.303(2) | 2.2186(18) | ||||
| M1–Cnt3 | 1.9682(19) | ||||||
| M1–Cnt4 | 2.1139(19) | ||||||
| Central Ring | Trop Angles | Dbcot Angles | ||||
|---|---|---|---|---|---|---|
| Compound | ||||||
| trop2 [42] | 1.4451(7) | 0.0802(10) | 49.37(12) | 22.46(9) | ||
| trop+ [45],a | 1.4134(11) | 0.0283(15) | 1.6(2) | 3.44(14) | ||
| Lithftrop | 1.4271(9) | 0.0537(12) | 24.16(19) | 14.13(14) | ||
| Lidmetrop | 1.421(10) | 0.060(14) | 6(4) | 2.9(25) | ||
| Natrop | 1.4108(9) | 0.0420(13) | 6.49(19) | 4.66(12) | ||
| Kthftrop | 1.4253(11) | 0.0491(15) | 1.7(2) | 2.37(14) | ||
| K18c6trop | 1.4269(11) | 0.0449(16) | 12.4(3) | 12.35(17) | ||
| K2trop | 1.4346(10) | 0.0337(14) | 1.3(2) | 2.30(13) | ||
| [Rh(trop)(cod)] | 1.4450(9) | 0.0626(13) | 55.49(18) | 32.29(13) | ||
| Li2dbcot [31] | 1.421(2) | 0.024(2) | 0 b,c | |||
| Na2dbcot | 1.4281(5) | 0.0234(7) | 5.38(3) | 4.88(3) | ||
| K2dbcot | 1.4279(7) | 0.0263(10) | 1.40(12) b | |||
| K2trop | Na2dbcot | K2dbcot a,b | ||
|---|---|---|---|---|
| C1–C2 | 1.426(3) | 1.4121(15) | 1.409(2) | 1.409(2) |
| C2–C3 | 1.463(2) | 1.4254(14) | 1.426(2) | 1.418(2) |
| C3–C4 | 1.434(3) | 1.4555(14) | 1.460(2) | 1.463(2) |
| C4–C5 | 1.385(3) | 1.4223(14) | ||
| C5–C6 | 1.440(3) | 1.4094(15) | ||
| C6–C7 | 1.466(2) | 1.4209(14) | ||
| C7–C8 | 1.4596(14) | |||
| C4–C1 | 1.420(2) | 1.418(2) | ||
| C7–C1 | 1.428(2) | |||
| C8–C1 | 1.4199(14) | |||
| C1–M1 | 3.2455(18) | 2.6792(11) | 3.0482(15) | 3.0905(14) |
| C2–M1 | 2.7258(11) | 3.0093(15) | 3.0728(14) | |
| C4–M1 | 3.2607(18) | |||
| C5–M1 | 3.1092(17) | 2.6971(11) | ||
| C6–M1 | 2.8896(11) | |||
| C1–M2 | 3.0902(17) | 2.7507(11) | ||
| C2–M2 | 2.6557(11) | |||
| C4–M2 | 3.3042(18) | |||
| C5–M2 | 3.1789(18) | 2.9216(11) | ||
| C6–M2 | 2.7689(11) | |||
| M1–Cnt1 | 2.7876(18) | 2.0924(11) | 2.4669(15) | 2.5385(14) |
| M2–Cnt1 | 2.7495(17) | 2.1348(11) | ||
| K1-Cnt1-K2 | 166.507(12) | 174.05(4) | 180.00(5) | 180.00(4) |
 |  | |||||
|---|---|---|---|---|---|---|
| A | B | C | ||||
| trop+ | NICS(0) | −8.87 | −4.71 | −8.87 | 0.00 | 0.00 |
| NICS(1) | −11.76 | −7.56 | −11.76 | |||
| trop– | NICS(0) | 21.05 | 45.00 | 21.02 | 0.01 | 0.01 |
| NICS(1) | 15.12 | 34.54 | 15.10 | |||
| Litropm | NICS(0) | 6.41 | 25.23 | 6.66 | 26.19 | 13.54 |
| NICS(1) a | 3.85 | 18.82 | 3.89 | |||
| NICS(1) b | 0.95 | 17.36 | 1.67 | |||
| Natropm | NICS(0) | 10.23 | 30.78 | 10.23 | 19.99 | 11.00 |
| NICS(1) a | 7.00 | 23.32 | 7.00 | |||
| NICS(1) b | 4.40 | 22.04 | 4.41 | |||
| Ktropm | NICS(0) | 17.02 | 42.39 | 17.02 | 7.41 | 1.77 |
| NICS(1) a | 12.03 | 32.38 | 12.02 | |||
| NICS(1) b | 11.18 | 32.86 | 11.18 | |||
| [Rh(trop)(cod)] | NICS(0) | −6.04 | −4.33 | −5.79 | 58.20 | 34.54 |
| NICS(1) a | −7.41 | −1.38 | −7.90 | |||
| NICS(1) b | −8.47 | −26.89 | −8.53 | |||
| K2tropm | NICS(0) | 4.77 | 12.87 | 4.77 | 0.02 | 0.02 |
| NICS(1) | 1.93 | 8.78 | 1.93 | |||
| CBD | NICS(0) | 27.49 | ||||
| NICS(1) | 18.03 | |||||
 |  | |||||
|---|---|---|---|---|---|---|
| A | B | C | ||||
| dbcot2- | NICS(0) | −6.60 | −17.20 | −6.60 | 0.07 | 0.07 |
| NICS(1) | −7.39 | −15.13 | −7.39 | |||
| Li2dbcotm | NICS(0) | −4.42 | −15.27 | −6.76 | 3.73 | 2.50 |
| NICS(1) | −6.51 | −13.49 | −8.45 | |||
| NICS(1) | −5.31 | −15.04 | −7.77 | |||
| Na2dbcotm | NICS(0) | −5.59 | −14.57 | −5.59 | 0.03 | 0.05 |
| NICS(1) | −7.06 | −14.15 | −7.06 | |||
| K2dbcotm | NICS(0) | −5.41 | −17.48 | −5.42 | 0.03 | 0.04 |
| NICS(1) | −6.93 | −16.96 | −6.94 | |||
| Benzene | NICS(0) | −7.68 | ||||
| NICS(1) | −10.09 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bloch, J.; Kradolfer, S.; L. Gianetti, T.; Ostendorf, D.; Dey, S.; Mougel, V.; Grützmacher, H. Synthesis and Characterization of Ion Pairs between Alkaline Metal Ions and Anionic Anti-Aromatic and Aromatic Hydrocarbons with π-Conjugated Central Seven- and Eight-Membered Rings. Molecules 2020, 25, 4742. https://doi.org/10.3390/molecules25204742
Bloch J, Kradolfer S, L. Gianetti T, Ostendorf D, Dey S, Mougel V, Grützmacher H. Synthesis and Characterization of Ion Pairs between Alkaline Metal Ions and Anionic Anti-Aromatic and Aromatic Hydrocarbons with π-Conjugated Central Seven- and Eight-Membered Rings. Molecules. 2020; 25(20):4742. https://doi.org/10.3390/molecules25204742
Chicago/Turabian StyleBloch, Jan, Stefan Kradolfer, Thomas L. Gianetti, Detlev Ostendorf, Subal Dey, Victor Mougel, and Hansjörg Grützmacher. 2020. "Synthesis and Characterization of Ion Pairs between Alkaline Metal Ions and Anionic Anti-Aromatic and Aromatic Hydrocarbons with π-Conjugated Central Seven- and Eight-Membered Rings" Molecules 25, no. 20: 4742. https://doi.org/10.3390/molecules25204742
APA StyleBloch, J., Kradolfer, S., L. Gianetti, T., Ostendorf, D., Dey, S., Mougel, V., & Grützmacher, H. (2020). Synthesis and Characterization of Ion Pairs between Alkaline Metal Ions and Anionic Anti-Aromatic and Aromatic Hydrocarbons with π-Conjugated Central Seven- and Eight-Membered Rings. Molecules, 25(20), 4742. https://doi.org/10.3390/molecules25204742