
Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches


Abstract
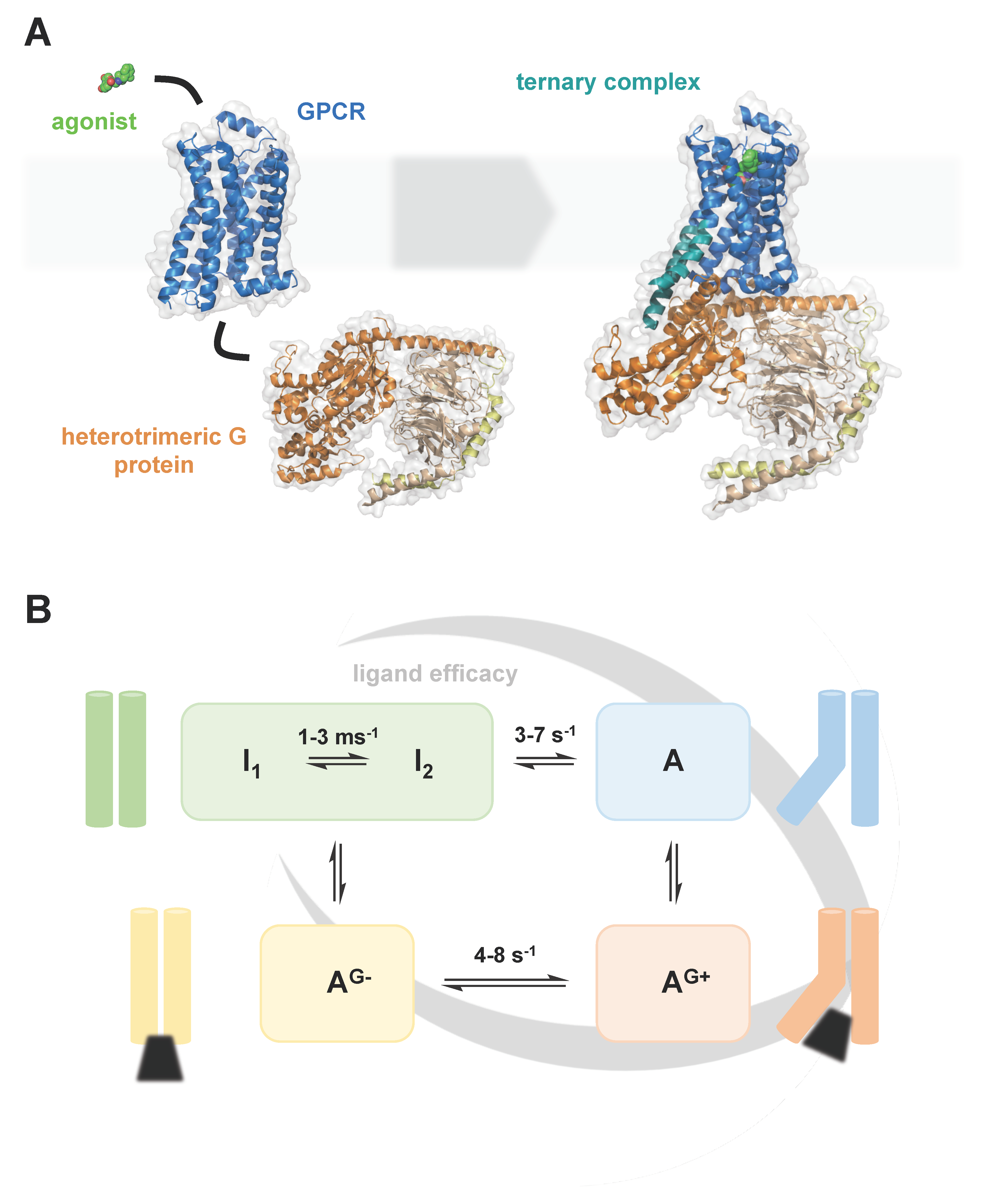
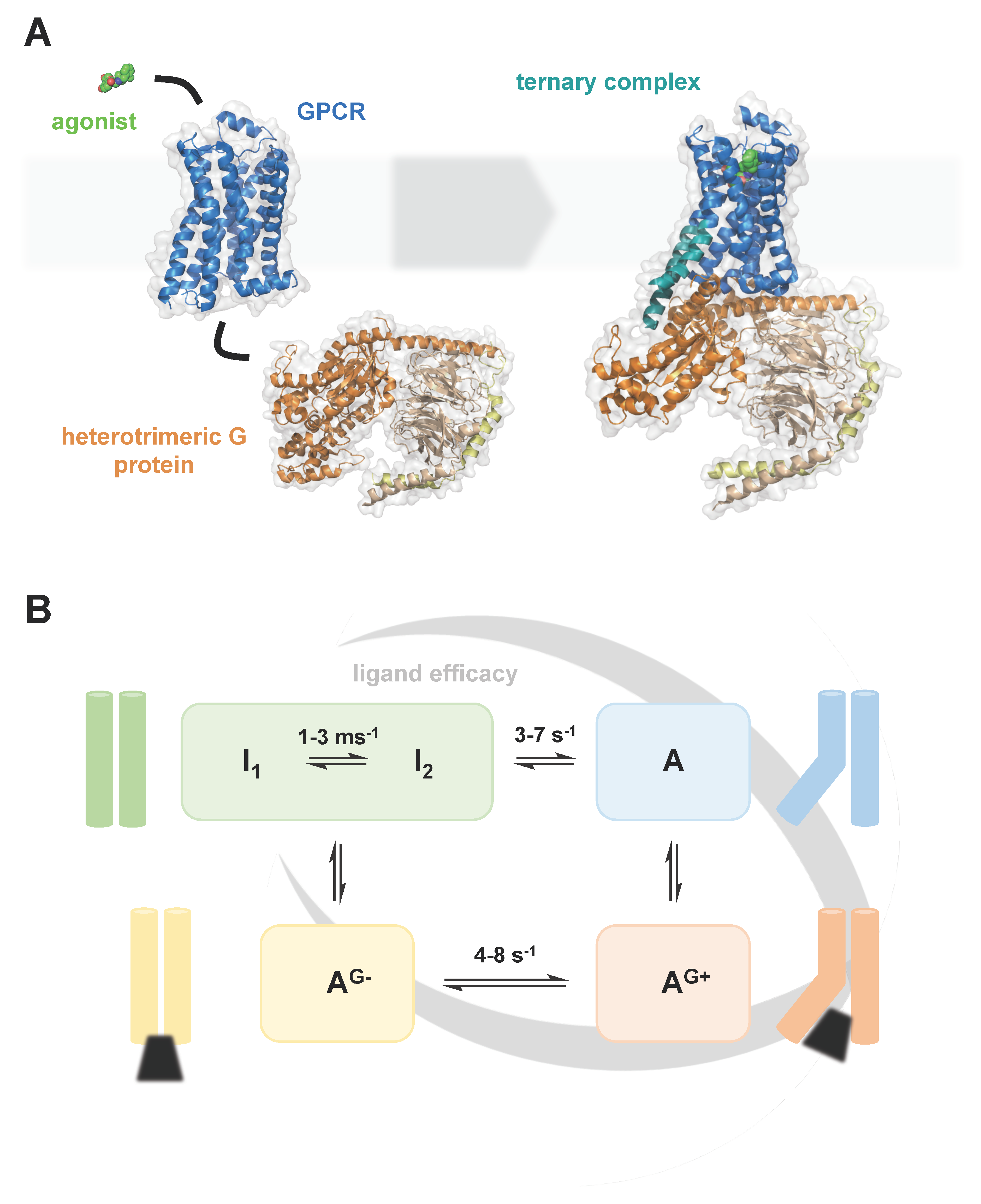
:1. Introduction
2. Cellular Trafficking of Membrane Lipids and GPCRs
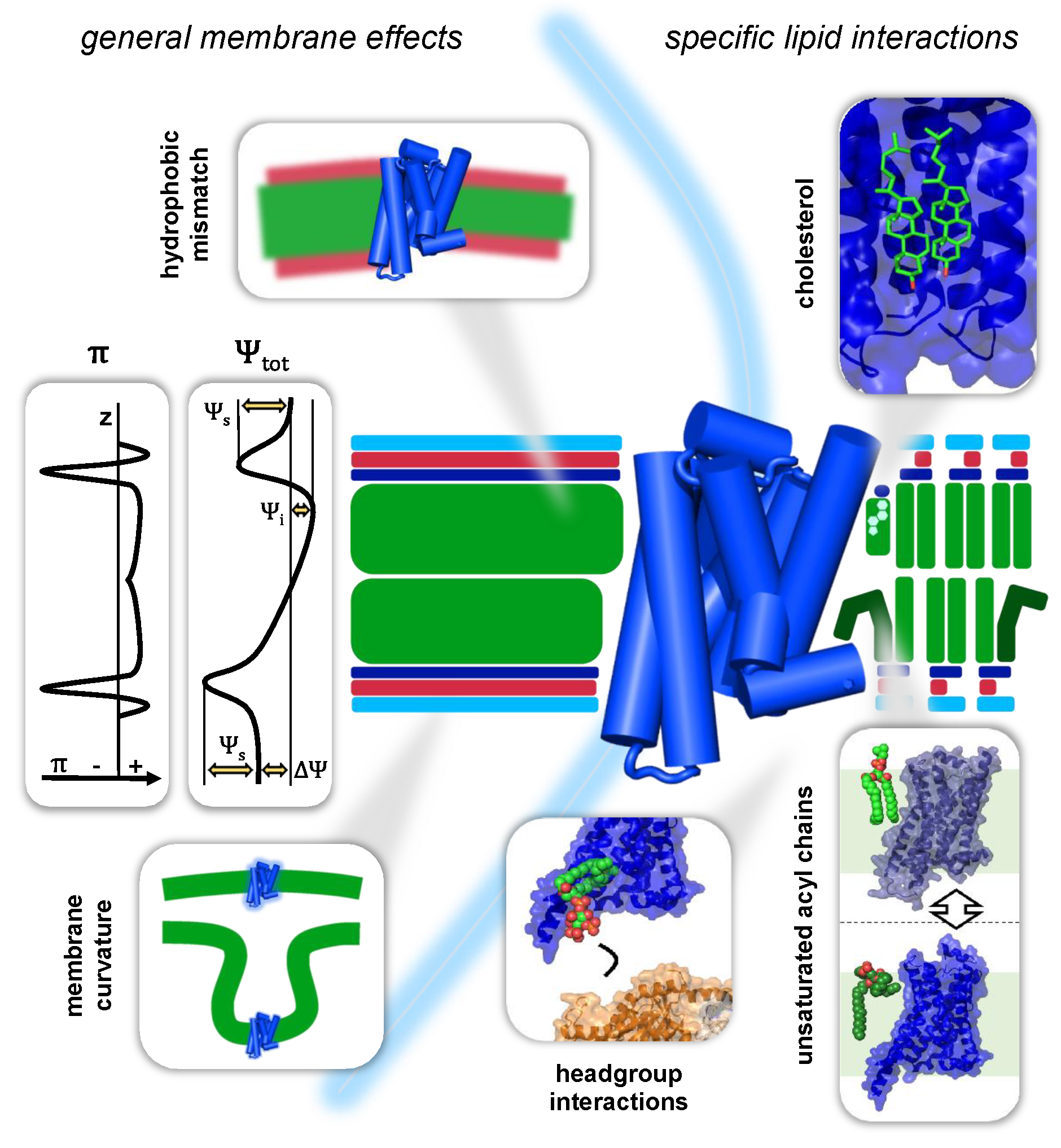
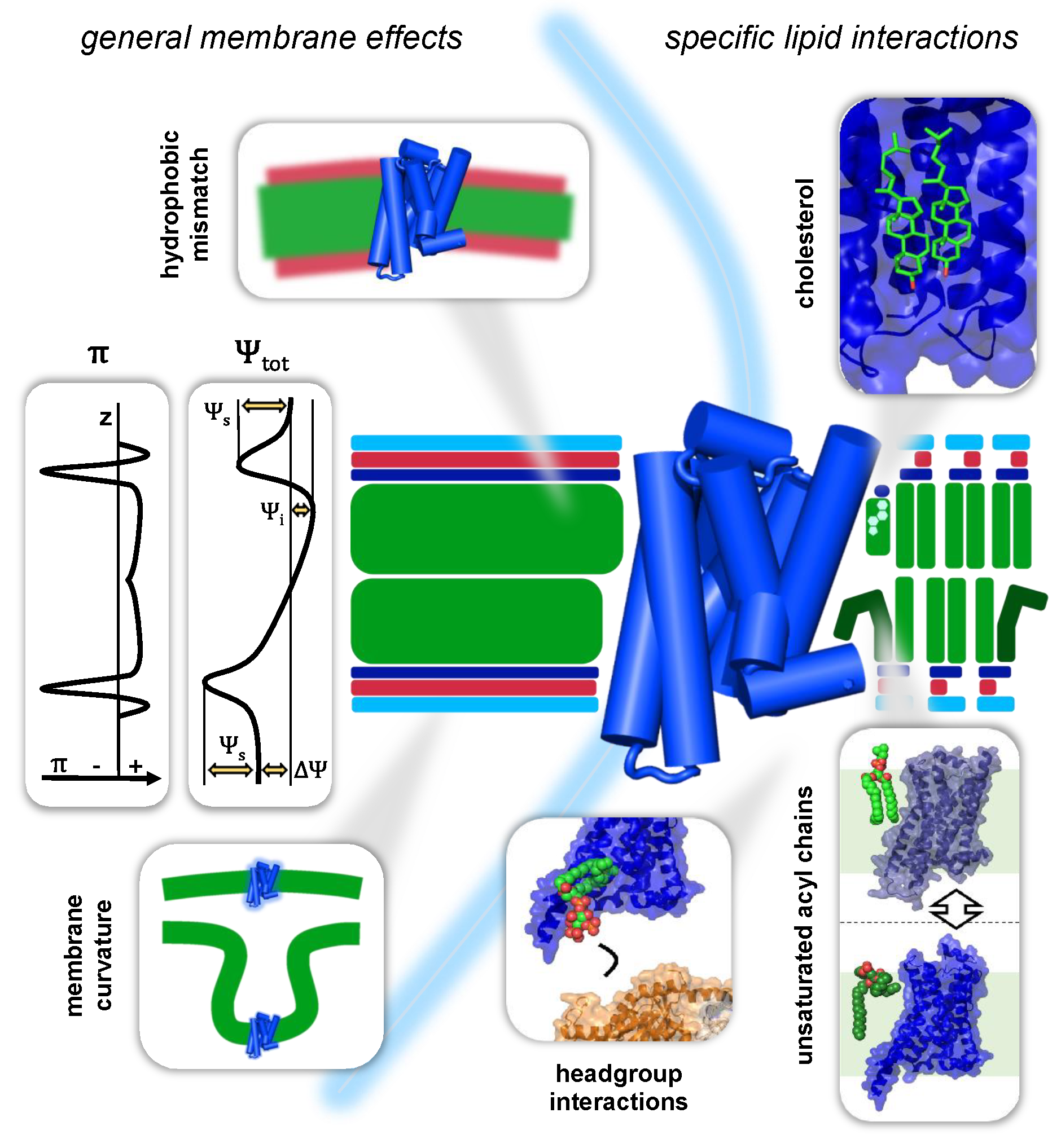
3. Interactions of GPCRs with Their Membrane Environment
3.1. General Membrane Effects on GPCRs
3.1.1. Mechanical Forces in the Bilayer
3.1.2. Membrane Curvature
3.1.3. GPCRs and the Hydrophobic Mismatch
3.1.4. Electrostatic Membrane Potentials and GPCR Function
3.1.5. External Factors Governing Membrane Properties
The Aqueous Phases Shape Membrane Properties In Vivo and In Vitro
Temperature
3.2. Specific Lipid–GPCR Interactions
3.2.1. Cholesterol
3.2.2. Anionic Lipids
3.2.3. Sphingolipids
3.2.4. Unsaturated Acyl Chains
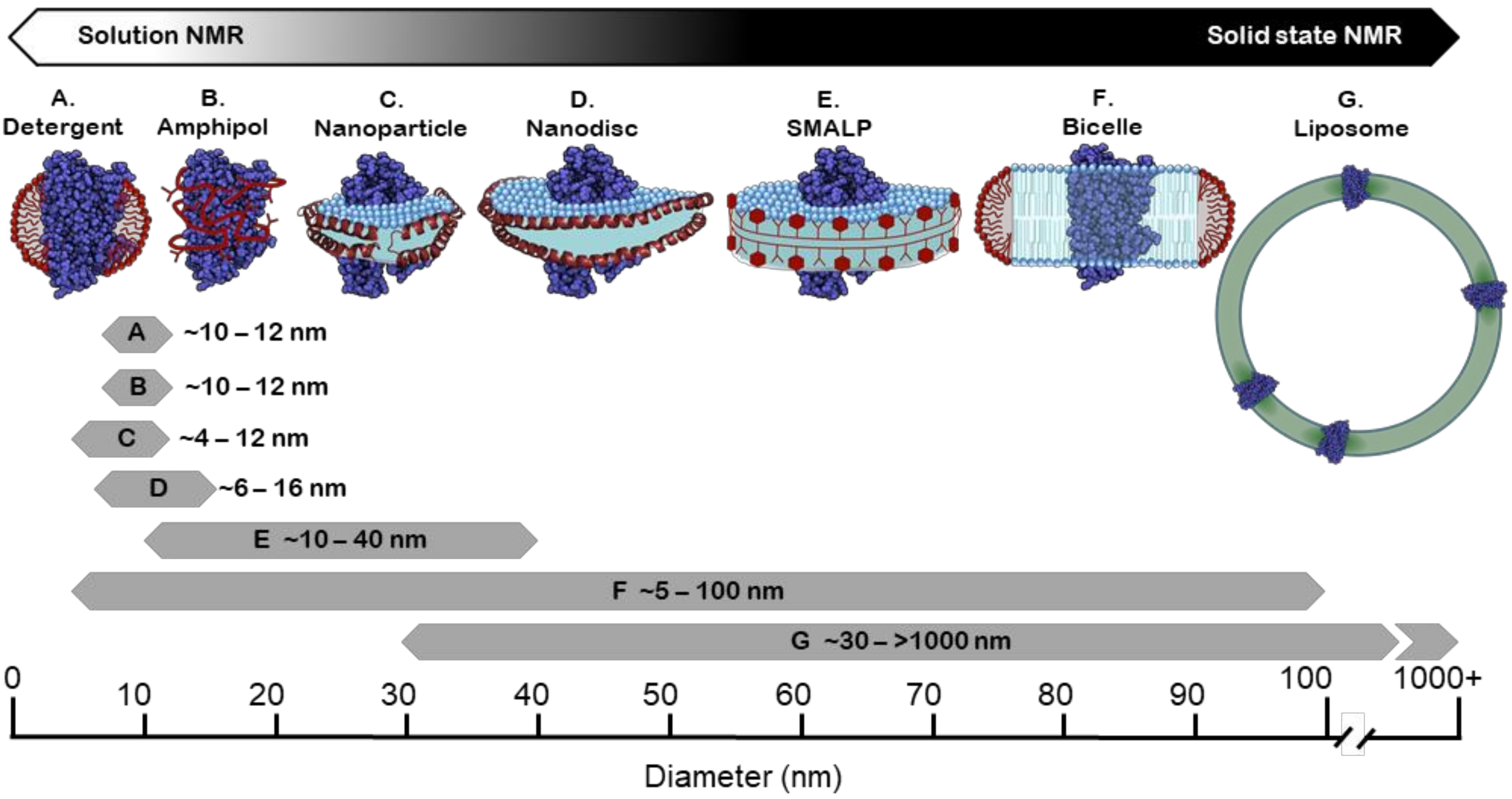
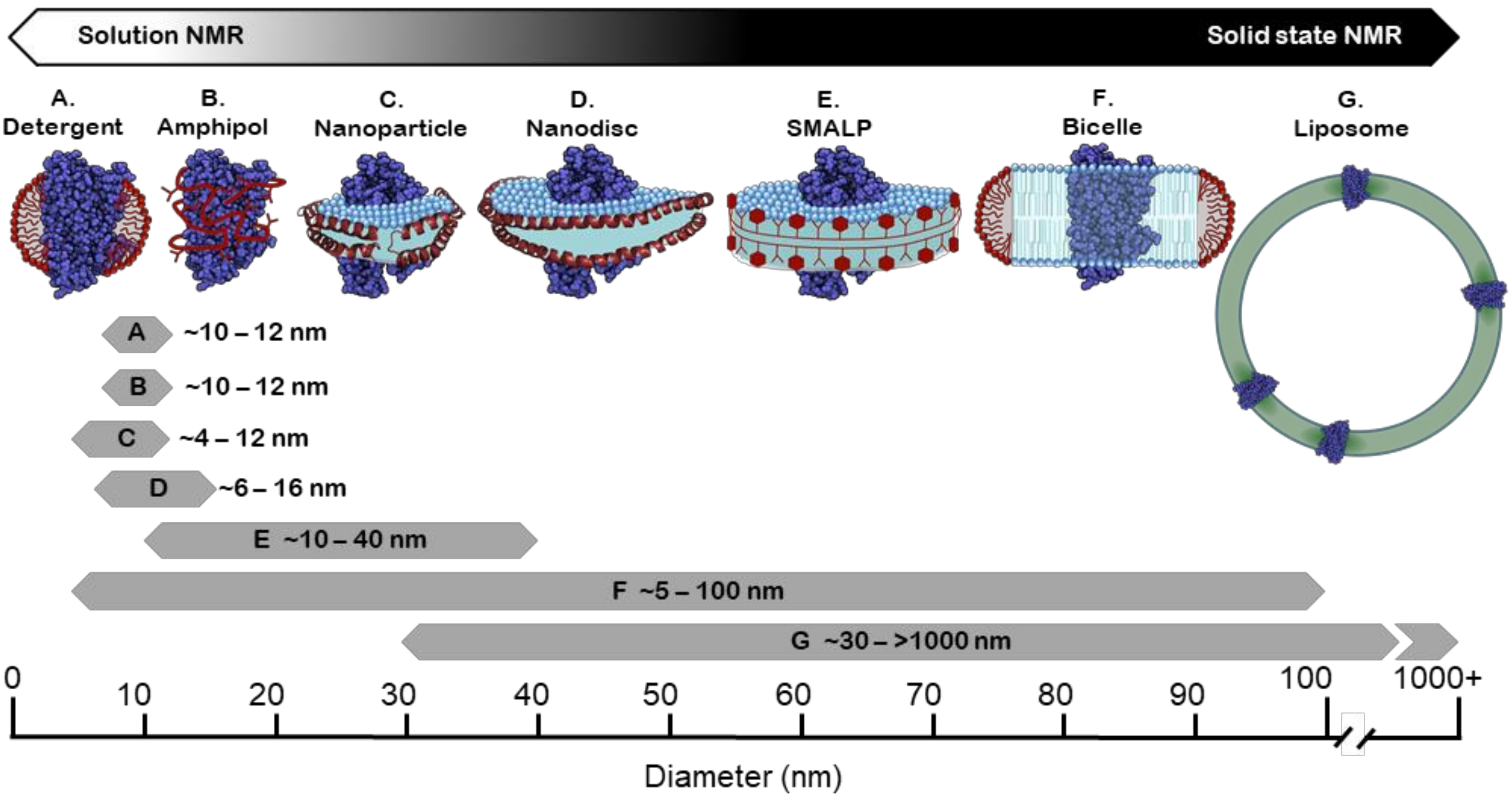
4. Membrane Mimetic Systems for Structural and Functional Studies
4.1. Detergents
4.2. Amphipols
4.3. Bicelles
4.4. Liposomes
4.5. Membrane Scaffold Protein (MSP) Nanodiscs
4.6. Styrene Maleic Acid (SMA) Copolymer and Copolymer Variants
4.7. Saposin Nanoparticles
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Yokomizo, T.; Kofuku, Y.; Shiraishi, Y.; Ueda, T.; Shimada, I. Structural equilibrium underlying ligand-dependent activation of β2-adrenoreceptor. Nat. Chem. Biol. 2020, 16, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, R.; Liu, J.J.; White, K.L.; Katritch, V.; Stevens, R.C.; Wüthrich, K.; Millar, D.P. Biased Signaling of the G-Protein-Coupled Receptor β2AR Is Governed by Conformational Exchange Kinetics. Structure 2020, 28, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Duc, N.M.; Rasmussen, S.G.F.; Hilger, D.; Kubiak, X.; Wang, L.; Bohon, J.; Kim, H.R.; Wegrecki, M.; Asuru, A.; et al. Assembly of a GPCR-G Protein Complex. Cell 2019, 177, 1232–1242.e11. [Google Scholar] [CrossRef] [PubMed]
- Solt, A.S.; Bostock, M.J.; Shrestha, B.; Kumar, P.; Warne, T.; Tate, C.G.; Nietlispach, D. Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound β1-adrenergic receptor. Nat. Commun. 2017, 8, 1795. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.N.; Broadhurst, R.W.; Bostock, M.J.; Solt, A.; Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Shrestha, B.; Nietlispach, D. Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19F NMR of the β1-adrenergic receptor. Nat. Commun. 2020, 11, 669. [Google Scholar] [CrossRef]
- Bischof, H.; Burgstaller, S.; Waldeck-Weiermair, M.; Rauter, T.; Schinagl, M.; Ramadani-Muja, J.; Graier, W.F.; Malli, R. Live-Cell Imaging of Physiologically Relevant Metal Ions Using Genetically Encoded FRET-Based Probes. Cells 2019, 8, 492. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Van Meer, G. Cellular lipidomics. EMBO J. 2005, 24, 3159–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L.; Walch, L.; Verbavatz, J.M. Lipids and Their Trafficking: An Integral Part of Cellular Organization. Dev. Cell 2016, 39, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, H.; Emmerstorfer-Augustin, A. Modification of membrane lipid compositions in single-celled organisms – From basics to applications. Methods 2018, 147, 50–65. [Google Scholar] [CrossRef]
- Sunshine, H.; Iuela-Arispe, L. Membrane Lipids and Cell Signaling Hannah. Physiol. Behav. 2016, 176, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Filipeanu, C.M.; Duvernay, M.T.; Wu, G. Regulation of G protein-coupled receptor export trafficking. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 853–870. [Google Scholar] [CrossRef] [Green Version]
- Hurt, C.M.; Ho, V.K.; Angelotti, T. Systematic and quantitative analysis of G protein-coupled receptor trafficking motifs. Methods Enzymol. 2013, 521, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Hegde, R.S. Membrane Protein Insertion at the Endoplasmic Reticulum. Adapt. Opt. Anal. Methods Syst. AO 2015, 2015, 289. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Hegde, R.S. Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol. 2011, 27, 25–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, G. Mechanisms of the anterograde trafficking of GPCRs: Regulation of AT1R transport by interacting proteins and motifs. Traffic 2019, 20, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef]
- Dores, M.R.; Trejo, J. Endo-lysosomal sorting of G-protein-coupled receptors by ubiquitin: Diverse pathways for G-protein-coupled receptor destruction and beyond. Traffic 2019, 20, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.; Xu, H.; Guo, D.; Shi, H.; Li, Y.; Zhang, W.; Gu, Y. 5-HTR3 and 5-HTR4 located on the mitochondrial membrane and functionally regulated mitochondrial functions. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sergin, I.; Jong, Y.J.I.; Harmon, S.K.; Kumar, V.; O’Malley, K.L. Sequences within the C terminus of the metabotropic glutamate receptor 5 (mGluR5) are responsible for inner nuclear membrane localization. J. Biol. Chem. 2017, 292, 3637–3655. [Google Scholar] [CrossRef] [Green Version]
- Jong, Y.J.I.; Harmon, S.K.; O’Malley, K.L. GPCR signalling from within the cell. Br. J. Pharmacol. 2018, 175, 4026–4035. [Google Scholar] [CrossRef] [Green Version]
- White, S.H.; Ladokhin, A.S.; Jayasinghe, S.; Hristova, K. How Membranes Shape Protein Structure. J. Biol. Chem. 2001, 276, 32395–32398. [Google Scholar] [CrossRef] [Green Version]
- van Meer, G. Dynamic transbilayer lipid asymmetry. Cold Spring Harb. Perspect. Biol. 2011, 3, a004671. [Google Scholar] [CrossRef] [Green Version]
- Fadeel, B.; Xue, D. The ins and outs of phospholipid asymmetry in the plasma membrane: Roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. [Google Scholar] [CrossRef]
- Honig, B.H.; Hubbell, W.L.; Flewelling, R.F. Electrostatic interactions in membranes and proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 163–193. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W.; Wu, D.; Heine, P.; Warne, T.; Lee, Y.; et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559, 423–427. [Google Scholar] [CrossRef]
- Cantor, R.S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999, 76, 2625–2639. [Google Scholar] [CrossRef] [Green Version]
- Ollila, S. Lateral Pressure in Lipid Membranes and Its Role in Function of Membrane Proteins. Ph.D. Thesis, Tampere University of Technology, Tampere, Finland, November 2010. [Google Scholar]
- Reddy, B.; Bavi, N.; Lu, A.; Park, Y.; Perozo, E. Molecular basis of force-from-lipids gating in the mechanosensitive channel mscs. Elife 2019, 8, 1–24. [Google Scholar] [CrossRef]
- Ollila, S.; Hyvönen, M.T.; Vattulainen, I. Polyunsaturation in lipid membranes: Dynamic properties and lateral pressure profiles. J. Phys. Chem. B 2007, 111, 3139–3150. [Google Scholar] [CrossRef]
- Findlay, H.E.; Booth, P.J. The biological significance of lipid-protein interactions. J. Phys. Condens. Matter 2006, 18. [Google Scholar] [CrossRef]
- Harris, N.J.; Charalambous, K.; Findlay, H.E.; Booth, P.J. Lipids modulate the insertion and folding of the nascent chains of alpha helical membrane proteins. Biochem. Soc. Trans. 2018, 46, 1355–1366. [Google Scholar] [CrossRef] [Green Version]
- Patra, M. Lateral pressure profiles in cholesterol-DPPC bilayers. Eur. Biophys. J. 2005, 35, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.R.; Findlay, H.E.; Booth, P.J. Lipid bilayer composition modulates the unfolding free energy of a knotted α-helical membrane protein. Proc. Natl. Acad. Sci. USA 2018, 115, E1709–E1808. [Google Scholar] [CrossRef] [Green Version]
- Sreekumari, A.; Lipowsky, R. Lipids with bulky head groups generate large membrane curvatures by small compositional asymmetries. J. Chem. Phys. 2018, 149, 084901. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Ridone, P.; Grage, S.L.; Patkunarajah, A.; Battle, A.R.; Ulrich, A.S.; Martinac, B. “Force-from-lipids” gating of mechanosensitive channels modulated by PUFAs. J. Mech. Behav. Biomed. Mater. 2018, 79, 158–167. [Google Scholar] [CrossRef]
- Ermakov, Y.A.; Kamaraju, K.; Sengupta, K.; Sukharev, S. Gadolinium ions block mechanosensitive channels by altering the packing and lateral pressure of anionic lipids. Biophys. J. 2010, 98, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Charalambous, K.; Miller, D.; Curnow, P.; Booth, P.J. Lipid bilayer composition influences small multidrug transporters. BMC Biochem. 2008, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.; Charalambous, K.; Rotem, D.; Schuldiner, S.; Curnow, P.; Booth, P.J. In vitro Unfolding and Refolding of the Small Multidrug Transporter EmrE. J. Mol. Biol. 2009, 393, 815–832. [Google Scholar] [CrossRef]
- Rosholm, K.R.; Leijnse, N.; Mantsiou, A.; Tkach, V.; Pedersen, S.L.; Wirth, V.F.; Oddershede, L.B.; Jensen, K.J.; Martinez, K.L.; Hatzakis, N.S.; et al. Membrane curvature regulates ligand-specific membrane sorting of GPCRs in living cells. Nat. Chem. Biol. 2017, 13, 724–729. [Google Scholar] [CrossRef]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zhang, Y.L.; Frangos, J.A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.F. Soft Matter in Lipid-Protein Interactions. Annu. Rev. Biophys. 2017, 46, 379–410. [Google Scholar] [CrossRef] [Green Version]
- Erlandson, S.C.; McMahon, C.; Kruse, A.C. Structural Basis for G Protein–Coupled Receptor Signaling. Annu. Rev. Biophys. 2018, 47, 1–18. [Google Scholar] [CrossRef]
- Weinberg, Z.Y.; Puthenveedu, M.A. Regulation of G protein-coupled receptor signaling by plasma membrane organization and endocytosis. Traffic 2019, 20, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubias, O.; Teague, W.E.; Hines, K.G.; Gawrisch, K. The role of membrane curvature elastic stress for function of rhodopsin-like G protein-coupled receptors. Biochimie 2015, 107, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerasinghe, N.; Fried, S.D.; Perera, S.M.D.C.; Eitel, A.R.; Chawla, U.; Molugu, T.R.; Struts, A.V.; Brown, M.F. G-Protein-Coupled Receptor Activation through Membrane Deformation. Biophys. J. 2018, 114, 274a. [Google Scholar] [CrossRef]
- Von Heijne, G. Formation of transmembrane helices in vivo - Is hydrophobicity all that matters? J. Gen. Physiol. 2007, 129, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Koehler Leman, J.; Bonneau, R.; Ulmschneider, M.B. Statistically derived asymmetric membrane potentials from α-helical and β-barrel membrane proteins. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- De Jesus, A.J.; Allen, T.W. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbaye, M.N.; Hou, Q.; Basu, S.; Teheux, F.; Pucci, F.; Rooman, M. A comprehensive computational study of amino acid interactions in membrane proteins. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gleason, N.J.; Vostrikov, V.V.; Greathouse, D.V.; Koeppe, R.E. Buried lysine, but not arginine, titrates and alters transmembrane helix tilt. Proc. Natl. Acad. Sci. USA 2013, 110, 1692–1695. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Schmidt, T.; Cho, E.-G.; Ye, F.; Ulmer, T.S.; Ginsberg, M.H. Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 2011, 481, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Andersen, O.S.; Koeppe, R.E. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [Green Version]
- Heimburg, T. Thermal Biophysics of Membranes; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527404711. [Google Scholar]
- Gahbauer, S.; Böckmann, R.A. Membrane-mediated oligomerization of G protein coupled receptors and its implications for GPCR function. Front. Physiol. 2016, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, B.; Javanainen, M.; García-Murria, M.J.; Kulig, W.; Vattulainen, I.; Mingarro, I.; Martínez-Gil, L. The role of hydrophobic matching on transmembrane helix packing in cells. Cell Stress 2017, 1, 90–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubias, O.; Teague, W.E.; Hines, K.G.; Gawrisch, K. Rhodopsin/lipid hydrophobic matching - Rhodopsin oligomerization and function. Biophys. J. 2015, 108, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K.; Ubarretxena-Belandia, I.; Taguchi, T.; Warren, G.; Engelman, D.M. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. USA 2004, 101, 4083–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayermann, S.P.; Rayermann, G.E.; Cornell, C.E.; Merz, A.J.; Keller, S.L. Hallmarks of Reversible Separation of Living, Unperturbed Cell Membranes into Two Liquid Phases. Biophys. J. 2017, 113, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Levental, I. Lipid Rafts come of Age. Nat. Rev. Mol. Cell Biol. 2020, 21, 420. [Google Scholar] [CrossRef]
- Periole, X.; Huber, T.; Marrink, S.J.; Sakmar, T.P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 2007, 129, 10126–10132. [Google Scholar] [CrossRef] [Green Version]
- Botelho, A.V.; Huber, T.; Sakmar, T.P.; Brown, M.F. Curvature and Hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006, 91, 4464–4477. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rohrer, B.; Levental, K.R.; Levental, I. Rafting through traffic: Membrane domains in cellular logistics. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Sodt, A.J.; Sandar, M.L.; Gawrisch, K.; Pastor, R.W.; Lyman, E. The molecular structure of the liquid-ordered phase of lipid bilayers. J. Am. Chem. Soc. 2014, 136, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Sodt, A.J.; Pastor, R.W.; Lyman, E. Hexagonal Substructure and Hydrogen Bonding in Liquid-Ordered Phases Containing Palmitoyl Sphingomyelin. Biophys. J. 2015, 109, 948–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javanainen, M.; Martinez-Seara, H.; Vattulainen, I. Nanoscale Membrane Domain Formation Driven by Cholesterol. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risselada, H.J.; Marrink, S.J. The molecular face of lipid rafts in model membranes. Proc. Natl. Acad. Sci. 2008, 105, 17367–17372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosetti, C.; Pastorino, C. Comparison of ternary bilayer mixtures with asymmetric or symmetric unsaturated phosphatidylcholine lipids by coarse grained molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 3525–3537. [Google Scholar] [CrossRef]
- Baoukina, S.; Mendez-Villuendas, E.; Bennett, W.F.D.; Tieleman, D.P. Computer simulations of the phase separation in model membranes. Faraday Discuss. 2012, 161, 63–75. [Google Scholar] [CrossRef]
- Davis, R.S.; Sunil Kumar, P.B.; Sperotto, M.M.; Laradji, M. Predictions of phase separation in three-component lipid membranes by the MARTINI force field. J. Phys. Chem. B 2013, 117, 4072–4080. [Google Scholar] [CrossRef]
- Lin, X.; Lorent, J.H.; Skinkle, A.D.; Levental, K.R.; Waxham, M.N.; Gorfe, A.A.; Levental, I. Domain stability in biomimetic membranes driven by lipid polyunsaturation. J. Phys. Chem. B 2016, 120, 11930–11941. [Google Scholar] [CrossRef] [PubMed]
- Destainville, N.; Manghi, M.; Cornet, J. A rationale for mesoscopic domain formation in biomembranes. Biomolecules 2018, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.A.; Engelman, D.M. Bacteriorhodopsin remains dispersed in fluid phospholipid bilayers over a wide range of bilayer thicknesses. J. Mol. Biol. 1983, 166, 203–210. [Google Scholar] [CrossRef]
- Gumbart, J.; Khalili-Araghi, F.; Sotomayor, M.; Roux, B. Constant electric field simulations of the membrane potential illustrated with simple systems. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L.; Kashiwaya, Y.; King, M.T. The resting membrane potential of cells are measures of electrical work, not of ionic currents. Integr. Physiol. Behav. Sci. 1995, 30, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Anishkin, A.; Loukin, S.H.; Teng, J.; Kung, C. Feeling the hidden mechanical forces in lipid bilayer is an original sense. Proc. Natl. Acad. Sci. USA 2014, 111, 7898–7905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, H.B.; Nicolini, A.M.; Arrowood, C.A.; Chvatal, S.A.; Wolfson, D.W.; Cho, H.C.; Sullivan, D.D.; Chal, J.; Fermini, B.; Clements, M.; et al. Novel method for action potential measurements from intact cardiac monolayers with multiwell microelectrode array technology. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- Abdulkader, F.; Arcisio-Miranda, M.; Curi, R.; Procopio, J. Surface potential determination in planar lipid bilayers: A simplification of the conductance-ratio method. J. Biochem. Biophys. Methods 2007, 70, 515–518. [Google Scholar] [CrossRef]
- Wadsäter, M.; Maric, S.; Simonsen, J.B.; Mortensen, K.; Cardenas, M. The effect of using binary mixtures of zwitterionic and charged lipids on nanodisc formation and stability. Soft Matter 2013, 9, 2329–2337. [Google Scholar] [CrossRef]
- Strohman, M.J.; Maeda, S.; Hilger, D.; Masureel, M.; Du, Y.; Kobilka, B.K. Local membrane charge regulates β2 adrenergic receptor coupling to Gi3. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Pearlstein, R.A.; Dickson, C.J.; Hornak, V. Contributions of the membrane dipole potential to the function of voltage-gated cation channels and modulation by small molecule potentiators. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 177–194. [Google Scholar] [CrossRef]
- Zheng, C.; Vanderkooi, G. Molecular origin of the internal dipole potential in lipid bilayers: Calculation of the electrostatic potential. Biophys. J. 1992, 63, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Lenaeus, M.J.; Gamal El-Din, T.M.; Ing, C.; Ramanadane, K.; Pomès, R.; Zheng, N.; Catterall, W.A. Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2017, 114, E3051–E3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Mahaut-Smith, M.P.; Martinez-Pinna, J.; Gurung, I.S. A role for membrane potential in regulating GPCRs? Trends Pharmacol. Sci. 2008, 29, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinna, J.; Tolhurst, G.; Gurung, I.S.; Vandenberg, J.I.; Mahaut-Smith, M.P. Sensitivity limits for voltage control of P2Y receptor-evoked Ca2+ mobilization in the rat megakaryocyte. J. Physiol. 2004, 555, 61–70. [Google Scholar] [CrossRef]
- Vickery, O.N.; Machtens, J.P.; Tamburrino, G.; Seeliger, D.; Zachariae, U. Structural Mechanisms of Voltage Sensing in G Protein-Coupled Receptors. Structure 2016, 24, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Rinne, A.; Mobarec, J.C.; Mahaut-Smith, M.; Kolb, P.; Bünemann, M. The mode of agonist binding to a G protein-coupled receptor switches the effect that voltage changes have on signaling. Sci. Signal. 2015, 8, 1–9. [Google Scholar] [CrossRef]
- Böckmann, R.A.; Hac, A.; Heimburg, T.; Grubmüller, H. Effect of sodium chloride on a lipid bilayer. Biophys. J. 2003, 85, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Morata, L.; Giannotti, M.I.; Sanz, F. Structural impact of cations on lipid bilayer models: Nanomechanical properties by AFM-force spectroscopy. Mol. Membr. Biol. 2014, 31, 17–28. [Google Scholar] [CrossRef]
- Deplazes, E.; Poger, D.; Cornell, B.; Cranfield, C.G. The effect of hydronium ions on the structure of phospholipid membranes. Phys. Chem. Chem. Phys. 2017, 20, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Raphael, R.M. Solution pH alters mechanical and electrical properties of phosphatidylcholine membranes: Relation between interfacial electrostatics, intramembrane potential, and bending elasticity. Biophys. J. 2007, 92, 2451–2462. [Google Scholar] [CrossRef] [Green Version]
- Seeger, H. Kinetics of Domain Formation; University of Goettingen: Göttingen, Germany, 2006. [Google Scholar]
- Peters, G.H.; Wang, C.; Cruys-Bagger, N.; Velardez, G.F.; Madsen, J.J.; Westh, P. Binding of serotonin to lipid membranes. J. Am. Chem. Soc. 2013, 135, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Isidoro, R.; Sierra-Valdez, F.J.; Ruiz-Suárez, J.C. Anesthetic diffusion through lipid membranes depends on the protonation rate. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, K.F.; Sanford, R.L.; Lee, W.; Andersen, O.S.; Hemmings, H.C. Clinical concentrations of chemically diverse general anesthetics minimally affect lipid bilayer properties. Proc. Natl. Acad. Sci. USA 2017, 114, 3109–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippov, A.; Orädd, G.; Lindblom, G. Lipid Lateral Diffusion in Ordered and Disordered Phases in Raft Mixtures. Biophys. J. 2004, 86, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Siliakus, M.F.; van der Oost, J.; Kengen, S.W.M. Adaptations of archaeal and bacterial membranes to variations in temperature, pH and pressure. Extremophiles 2017, 21, 651–670. [Google Scholar] [CrossRef]
- Pucadyil, T.J.; Chattopadhyay, A. Role of cholesterol in the function and organization of G-protein coupled receptors. Prog. Lipid Res. 2006, 45, 295–333. [Google Scholar] [CrossRef]
- Oates, J.; Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807. [Google Scholar] [CrossRef]
- Jafurulla, M.; Chattopadhyay, A. Sphingolipids in the function of G protein-coupled receptors. Eur. J. Pharmacol. 2015, 763, 241–246. [Google Scholar] [CrossRef]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife 2016, 5. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; Van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef]
- Inagaki, S.; Ghirlando, R.; White, J.F.; Gvozdenovic-Jeremic, J.; Northup, J.K.; Grisshammer, R. Modulation of the interaction between neurotensin receptor NTS1 and Gq protein by lipid. J. Mol. Biol. 2012, 417, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Kirilovsky, J.; Schramm, M. Delipidation of a beta-adrenergic receptor preparation and reconstitution by specific lipids. J. Biol. Chem. 1983, 258, 6841–6849. [Google Scholar]
- Alemany, R.; Perona, J.S.; Sánchez-Dominguez, J.M.; Montero, E.; Cañizares, J.; Bressani, R.; Escribá, P.V.; Ruiz-Gutierrez, V. G protein-coupled receptor systems and their lipid environment in health disorders during aging. Biochim. Biophys. Acta 2007, 1768, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Paila, Y.D.; Tiwari, S.; Chattopadhyay, A. Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta 2009, 1788, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.G. Biological membranes: The importance of molecular detail. Trends Biochem. Sci. 2011, 36, 493–500. [Google Scholar] [CrossRef]
- Jones, O.T.; Mcnamee, M.G. Annular and Nonannular Binding Sites for Cholesterol Associated with the Nicotinic Acetylcholine Receptor. Biochemistry 1988, 27, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Vaidehi, N. Differences in allosteric communication pipelines in the inactive and active states of a GPCR. Biophys. J. 2014, 107, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [Green Version]
- Casiraghi, M.; Damian, M.; Lescop, E.; Point, E.; Moncoq, K.; Morellet, N.; Levy, D.; Marie, J.; Guittet, E.; Banères, J.-L.; et al. Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J. Am. Chem. Soc. 2016, 138, 11170–11175. [Google Scholar] [CrossRef]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Chattopadhyay, A. Molecular dynamics simulations of GPCR-cholesterol interaction: An emerging paradigm. Biochim. Biophys. Acta 2015, 1848, 1775–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Manna, M.; Niemelä, M.; Tynkkynen, J.; Javanainen, M.; Kulig, W.; Müller, D.J.; Rog, T.; Vattulainen, I. Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. Elife 2016, 5. [Google Scholar] [CrossRef]
- Muth, S.; Fries, A.; Gimpl, G. Cholesterol-induced conformational changes in the oxytocin receptor. Biochem. J. 2011, 437, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Jafurulla, M.; Rao, B.D.; Sreedevi, S.; Ruysschaert, J.M.; Covey, D.F.; Chattopadhyay, A. Stereospecific requirement of cholesterol in the function of the serotonin1A receptor. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 158–163. [Google Scholar] [CrossRef]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.; Chattopadhyay, A. Cholesterol interaction motifs in G protein-coupled receptors: Slippery hot spots? Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1481. [Google Scholar] [CrossRef]
- Sengupta, D. Cholesterol modulates the structure, binding modes, and energetics of caveolin-membrane interactions. J. Phys. Chem. B 2012, 116, 14556–14564. [Google Scholar] [CrossRef]
- Jafurulla, M.; Chattopadhyay, A. Membrane lipids in the function of serotonin and adrenergic receptors. Curr. Med. Chem. 2013, 20, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, X.; Chattopadhyay, A.; Sengupta, D. Cholesterol modulates the dimer interface of the β2- adrenergic receptor via cholesterol occupancy sites. Biophys. J. 2014, 106, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Zocher, M.; Zhang, C.; Rasmussen, S.G.F.; Kobilka, B.K.; Müller, D.J. Cholesterol increases kinetic, energetic, and mechanical stability of the human β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [Green Version]
- Gater, D.L.; Saurel, O.; Iordanov, I.; Liu, W.; Cherezov, V.; Milon, A. Two classes of cholesterol binding sites for the β2AR revealed by thermostability and NMR. Biophys. J. 2014, 107, 2305–2312. [Google Scholar] [CrossRef] [Green Version]
- Cang, X.; Du, Y.; Mao, Y.; Wang, Y.; Yang, H.; Jiang, H. Mapping the functional binding sites of cholesterol in β2- adrenergic receptor by long-time molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 1085–1094. [Google Scholar] [CrossRef]
- Song, W.; Yen, H.Y.; Robinson, C.V.; Sansom, M.S.P. State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 2019, 27, 392–403.e3. [Google Scholar] [CrossRef] [Green Version]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehmé, R.; Edwards, P.C.; Leslie, A.G.W.; Schertler, G.F.X.; Tate, C.G. The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature 2011, 469, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, K.; Gao, Z.G.; Paoletta, S.; Zhang, D.; Han, G.W.; Li, T.; Ma, L.; Zhang, W.; Müller, C.E.; et al. Agonist-bound structure of the human P2Y12 receptor. Nature 2014, 508, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, J.; Gao, Z.G.; Zhang, D.; Zhu, L.; Han, G.W.; Moss, S.M.; Paoletta, S.; Kiselev, E.; Lu, W.; et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 2014, 508, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Fu, Z.; Frangaj, A.; Liu, J.; Mosyak, L.; Shen, T.; Slavkovich, V.N.; Ray, K.M.; Taura, J.; Cao, B.; et al. Structure of human GABAB receptor in an inactive state. Nature 2020, 584, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Zhang, Y.; Li, Y.; Li, G. Computer Simulations to Explore Membrane Organization and Transport. In Membrane Biophysics; Springer: Singapore, 2018; pp. 355–392. [Google Scholar]
- Marius, P.; Zagnoni, M.; Sandison, M.E.; Malcolm East, J.; Morgan, H.; Lee, A.G. Binding of anionic lipids to at least three nonannular sites on the potassium channel KcsA is required for channel opening. Biophys. J. 2008, 94, 1689–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorobyov, I.; Allen, T.W. On the role of anionic lipids in charged protein interactions with membranes. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; pp. 366–428. [Google Scholar]
- Neale, C.; Herce, H.D.; Pomès, R.; García, A.E. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor? Biophys. J. 2015, 109, 1652–1662. [Google Scholar] [CrossRef] [Green Version]
- Holthuis, J.C.; Pomorski, T.; Raggers, R.J.; Sprong, H.; Van Meer, G. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 2001, 81, 1689–1723. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Chattopadhyay, A. Removal of sphingomyelin headgroup inhibits the ligand binding function of hippocampal serotonin1A receptors. Biochem. Biophys. Res. Commun. 2012, 419, 321–325. [Google Scholar] [CrossRef]
- Jafurulla, M.; Bandari, S.; Pucadyil, T.J.; Chattopadhyay, A. Sphingolipids modulate the function of human serotonin1A receptors: Insights from sphingolipid-deficient cells. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Garmy, N.; Yahi, N. Prediction of glycolipid-binding domains from the amino acid sequence of lipid raft-associated proteins: Application to HpaA, a protein involved in the adhesion of Helicobacter pylori to gastrointestinal cells. Biochemistry 2006, 45, 10957–10962. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Paila, Y.D.; Shrivastava, S.; Tiwari, S.; Singh, P.; Fantini, J. Sphingolipid-binding domain in the serotonin(1A) receptor. Adv. Exp. Med. Biol. 2012, 749, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Jafurulla, M.; Tiwari, S.; Chattopadhyay, A. Identification of Sphingolipid-binding Motif in G Protein-coupled Receptors. Adv. Exp. Med. Biol. 2018, 1112, 141–149. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.C.; McIntosh, T.; Needham, D.; Evans, E.A. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Antonny, B.; Vanni, S.; Shindou, H.; Ferreira, T. From zero to six double bonds: Phospholipid unsaturation and organelle function. Trends Cell Biol. 2015, 25, 427–436. [Google Scholar] [CrossRef]
- Innis, S.M. Dietary (n-3) fatty acids and brain development. J. Nutr. 2007, 137, 855–859. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Huang, B.X.; Spector, A.A. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 2014, 56, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Singh, M. Essential fatty acids, DHA and human brain. Indian J. Pediatr. 2005, 72, 239–242. [Google Scholar] [CrossRef]
- Choe, H.W.; Kim, Y.J.; Park, J.H.; Morizumi, T.; Pai, E.F.; Krau, N.; Hofmann, K.P.; Scheerer, P.; Ernst, O.P. Crystal structure of metarhodopsin II. Nature 2011, 471, 651–655. [Google Scholar] [CrossRef]
- Mizumura, T.; Kondo, K.; Kurita, M.; Kofuku, Y.; Natsume, M.; Imai, S.; Shiraishi, Y.; Ueda, T.; Shimada, I. Activation of adenosine A2A receptor by lipids from docosahexaenoic acid revealed by NMR. Sci. Adv. 2020, 6, 8544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javanainen, M.; Enkavi, G.; Guixà-Gonzaléz, R.; Kulig, W.; Martinez-Seara, H.; Levental, I.; Vattulainen, I. Reduced level of docosahexaenoic acid shifts GPCR neuroreceptors to less ordered membrane regions. PLoS Comput. Biol. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guixà-González, R.; Javanainen, M.; Gómez-Soler, M.; Cordobilla, B.; Domingo, J.C.; Sanz, F.; Pastor, M.; Ciruela, F.; Martinez-Seara, H.; Selent, J. Membrane omega-3 fatty acids modulate the oligomerisation kinetics of adenosine A2A and dopamine D2 receptors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ximenes da Silva, A.; Lavialle, F.; Gendrot, G.; Guesnet, P.; Alessandri, J.-M.; Lavialle, M. Glucose transport and utilization are altered in the brain of rats deficient in n-3 polyunsaturated fatty acids. J. Neurochem. 2002, 81, 1328–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anson, M.L. The denaturation of proteins by detergents and bile salts. Science 1939, 90, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Brooks, M.M. Comparative studies on respiration: XV. The effect of bile salts and of saponin upon respiration. J. Gen. Physiol. 1921, 3, 527–532. [Google Scholar] [CrossRef]
- Linke, D. Detergents: An overview. Methods Enzymol. 2009, 463, 603–617. [Google Scholar] [CrossRef]
- Garavito, R.M.; Ferguson-Miller, S. Detergents as tools in membrane biochemistry. J. Biol. Chem. 2001, 276, 32403–32406. [Google Scholar] [CrossRef] [Green Version]
- Maibaum, L.; Dinner, A.R.; Chandler, D. Micelle Formation and the Hydrophobic Effect. J. Phys. Chem. B 2004, 108, 6778–6781. [Google Scholar] [CrossRef] [Green Version]
- Kragh-Hansen, U.; le Maire, M.; Møller, J. V The mechanism of detergent solubilization of liposomes and protein-containing membranes. Biophys. J. 1998, 75, 2932–2946. [Google Scholar] [CrossRef] [Green Version]
- Popot, J.-L. Amphipols, nanodiscs, and fluorinated surfactants: Three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu. Rev. Biochem. 2010, 79, 737–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidehi, N.; Grisshammer, R.; Tate, C.G. How Can Mutations Thermostabilize G-Protein-Coupled Receptors? Trends Pharmacol. Sci. 2016, 37, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulumello, D.V.; Deber, C.M. Efficiency of detergents at maintaining membrane protein structures in their biologically relevant forms. Biochim. Biophys. Acta 2012, 1818, 1351–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Urner, L.H.; Liko, I.; Yen, H.-Y.; Hoi, K.-K.; Bolla, J.R.; Gault, J.; Almeida, F.G.; Schweder, M.-P.; Shutin, D.; Ehrmann, S.; et al. Modular detergents tailor the purification and structural analysis of membrane proteins including G-protein coupled receptors. Nat. Commun. 2020, 11, 564. [Google Scholar] [CrossRef]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [Green Version]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Ghosh, S.; Jana, S.; Robertson, N.; Tate, C.G.; Vaidehi, N. How Do Branched Detergents Stabilize GPCRs in Micelles? Biochemistry 2020, 59, 2125–2134. [Google Scholar] [CrossRef]
- Chung, K.Y.; Kim, T.H.; Manglik, A.; Alvares, R.; Kobilka, B.K.; Prosser, R.S. Role of detergents in conformational exchange of a G protein-coupled receptor. J. Biol. Chem. 2012, 287, 36305–36311. [Google Scholar] [CrossRef] [Green Version]
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G protein-coupled receptors: The evolution of structural insight. AIMS Biophys. 2017, 4, 491–527. [Google Scholar] [CrossRef]
- Bostock, M.J.; Solt, A.S.; Nietlispach, D. The role of NMR spectroscopy in mapping the conformational landscape of GPCRs. Curr. Opin. Struct. Biol. 2019, 57, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, M.A.; Helgeson, M.E.; Wagner, N.J.; Robinson, A.S. The morphology and composition of cholesterol-rich micellar nanostructures determine transmembrane protein (GPCR) activity. Biophys. J. 2011, 100, L11–L13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sounier, R.; Mas, C.; Steyaert, J.; Laeremans, T.; Manglik, A.; Huang, W.; Kobilka, B.K.; Déméné, H.; Granier, S. Propagation of conformational changes during μ-opioid receptor activation. Nature 2015, 524, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Bokoch, M.P.; Zou, Y.; Rasmussen, S.G.F.; Liu, C.W.; Nygaard, R.; Rosenbaum, D.M.; Fung, J.J.; Choi, H.-J.; Thian, F.S.; Kobilka, T.S.; et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 2010, 463, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Maeda, M.; Tsujishita, H.; Shimada, I. Efficacy of the β₂-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 2012, 3, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Chung, K.Y.; Manglik, A.; Hansen, A.L.; Dror, R.O.; Mildorf, T.J.; Shaw, D.E.; Kobilka, B.K.; Prosser, R.S. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J. Am. Chem. Soc. 2013, 135, 9465–9474. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β(2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Horst, R.; Liu, J.J.; Stevens, R.C.; Wüthrich, K. β₂-adrenergic receptor activation by agonists studied with 19F NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 10762–10765. [Google Scholar] [CrossRef] [Green Version]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Mizumura, T.; Suzuki, S.; Shimada, I. Functional dynamics of deuterated β2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2014, 53, 13376–13379. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew. Chem. Int. Ed. Engl. 2015, 54, 15771–15776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, C.; Isogai, S.; Grzesiek, S. An economic approach to efficient isotope labeling in insect cells using homemade 15N-, 13C- and 2H-labeled yeast extracts. J. Biomol. NMR 2015, 62, 373–385. [Google Scholar] [CrossRef]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.-J.; Brueckner, F.; Schertler, G.F.X.; Veprintsev, D.B.; Grzesiek, S. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 2016, 530, 237–241. [Google Scholar] [CrossRef]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef]
- Clark, L.D.; Dikiy, I.; Chapman, K.; Rödström, K.E.; Aramini, J.; LeVine, M.V.; Khelashvili, G.; Rasmussen, S.G.; Gardner, K.H.; Rosenbaum, D.M. Ligand modulation of sidechain dynamics in a wild-type human GPCR. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Nasr, M.L.; Baptista, D.; Strauss, M.; Sun, Z.-Y.J.; Grigoriu, S.; Huser, S.; Plückthun, A.; Hagn, F.; Walz, T.; Hogle, J.M.; et al. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nat. Methods 2017, 14, 49–52. [Google Scholar] [CrossRef]
- Eddy, M.T.; Gao, Z.-G.; Mannes, P.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Extrinsic Tryptophans as NMR Probes of Allosteric Coupling in Membrane Proteins: Application to the A2A Adenosine Receptor. J. Am. Chem. Soc. 2018, 140, 8228–8235. [Google Scholar] [CrossRef]
- Franke, B.; Opitz, C.; Isogai, S.; Grahl, A.; Delgado, L.; Gossert, A.D.; Grzesiek, S. Production of isotope-labeled proteins in insect cells for NMR. J. Biomol. NMR 2018, 71, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Bumbak, F.; Keen, A.C.; Gunn, N.J.; Gooley, P.R.; Bathgate, R.A.D.; Scott, D.J. Optimization and 13CH3 methionine labeling of a signaling competent neurotensin receptor 1 variant for NMR studies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Natsume, M.; Kofuku, Y.; Imai, S.; Nakata, K.; Mizukoshi, T.; Ueda, T.; Iwaï, H.; Shimada, I. Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat. Commun. 2018, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofuku, Y.; Yokomizo, T.; Imai, S.; Shiraishi, Y.; Natsume, M.; Itoh, H.; Inoue, M.; Nakata, K.; Igarashi, S.; Yamaguchi, H.; et al. Deuteration and selective labeling of alanine methyl groups of β2-adrenergic receptor expressed in a baculovirus-insect cell expression system. J. Biomol. NMR 2018, 71, 185–192. [Google Scholar] [CrossRef]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomès, R.; García, A.E.; Ernst, O.P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef]
- Sušac, L.; Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 2018, 115, 12733–12738. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Hu, Y.; Kaindl, J.; Risel, P.; Hübner, H.; Maeda, S.; Niu, X.; Li, H.; Gmeiner, P.; Jin, C.; et al. Conformational Complexity and Dynamics in a Muscarinic Receptor Revealed by NMR Spectroscopy. Mol. Cell 2019, 75, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Wingler, L.M.; Pichugin, D.; Prosser, R.S.; Lefkowitz, R.J. Detergent- and phospholipid-based reconstitution systems have differential effects on constitutive activity of G-protein-coupled receptors. J. Biol. Chem. 2019, 294, 13218–13223. [Google Scholar] [CrossRef]
- Abiko, L.A.; Grahl, A.; Grzesiek, S. High Pressure Shifts the β1-Adrenergic Receptor to the Active Conformation in the Absence of G Protein. J. Am. Chem. Soc. 2019, 141, 16663–16670. [Google Scholar] [CrossRef]
- Wu, F.-J.; Williams, L.M.; Abdul-Ridha, A.; Gunatilaka, A.; Vaid, T.M.; Kocan, M.; Whitehead, A.R.; Griffin, M.D.W.; Bathgate, R.A.D.; Scott, D.J.; et al. Probing the correlation between ligand efficacy and conformational diversity at the α1A-adrenoreceptor reveals allosteric coupling of its microswitches. J. Biol. Chem. 2020, 295, 7404–7417. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-X.; Cross, T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013, 42, 361–392. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Mao, A.; Bhattacharya, S.; Robertson, N.; Grisshammer, R.; Tate, C.G.; Vaidehi, N. How Do Short Chain Nonionic Detergents Destabilize G-Protein-Coupled Receptors? J. Am. Chem. Soc. 2016, 138, 15425–15433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaptal, V.; Delolme, F.; Kilburg, A.; Magnard, S.; Montigny, C.; Picard, M.; Prier, C.; Monticelli, L.; Bornert, O.; Agez, M.; et al. Quantification of Detergents Complexed with Membrane Proteins. Sci. Rep. 2017, 7, 41751. [Google Scholar] [CrossRef] [PubMed]
- Gohon, Y.; Dahmane, T.; Ruigrok, R.W.H.; Schuck, P.; Charvolin, D.; Rappaport, F.; Timmins, P.; Engelman, D.M.; Tribet, C.; Popot, J.-L.; et al. Bacteriorhodopsin/amphipol complexes: Structural and functional properties. Biophys. J. 2008, 94, 3523–3537. [Google Scholar] [CrossRef] [Green Version]
- Rahmeh, R.; Damian, M.; Cottet, M.; Orcel, H.; Mendre, C.; Durroux, T.; Sharma, K.S.; Durand, G.; Pucci, B.; Trinquet, E.; et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 6733–6738. [Google Scholar] [CrossRef] [Green Version]
- Tribet, C.; Audebert, R.; Popot, J.L. Amphipols: Polymers that keep membrane proteins soluble in aqueous solutions. Proc. Natl. Acad. Sci. USA 1996, 93, 15047–15050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoonens, M.; Popot, J.-L. Amphipols for each season. J. Membr. Biol. 2014, 247, 759–796. [Google Scholar] [CrossRef] [Green Version]
- Catoire, L.J.; Warnet, X.L.; Warschawski, D.E. Micelles, Bicelles, Amphipols, Nanodiscs, Liposomes, or Intact Cells: The Hitchhiker’s Guide to the Study of Membrane Proteins by NMR. In Membrane Proteins Production for Structural Analysis; Springer: New York, NY, USA, 2014; pp. 315–345. [Google Scholar]
- Ferré, G.; Louet, M.; Saurel, O.; Delort, B.; Czaplicki, G.; M’Kadmi, C.; Damian, M.; Renault, P.; Cantel, S.; Gavara, L.; et al. Structure and dynamics of G protein-coupled receptor-bound ghrelin reveal the critical role of the octanoyl chain. Proc. Natl. Acad. Sci. USA 2019, 116, 17525–17530. [Google Scholar] [CrossRef] [Green Version]
- Dahmane, T.; Damian, M.; Mary, S.; Popot, J.-L.; Banères, J.-L. Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 2009, 48, 6516–6521. [Google Scholar] [CrossRef]
- Autzen, H.E.; Julius, D.; Cheng, Y. Membrane mimetic systems in CryoEM: Keeping membrane proteins in their native environment. Curr. Opin. Struct. Biol. 2019, 58, 259–268. [Google Scholar] [CrossRef]
- Popot, J.L.; Althoff, T.; Bagnard, D.; Banères, J.L.; Bazzacco, P.; Billon-Denis, E.; Catoire, L.J.; Champeil, P.; Charvolin, D.; Cocco, M.J.; et al. Amphipols from A to Z. Annu. Rev. Biophys. 2011, 40, 379–408. [Google Scholar] [CrossRef]
- Zubcevic, L.; Hsu, A.L.; Borgnia, M.J.; Lee, S.-Y. Symmetry transitions during gating of the TRPV2 ion channel in lipid membranes. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Mary, S.; Damian, M.; Rahmeh, R.; Mouillac, B.; Marie, J.; Granier, S.; Banères, J.-L. Amphipols in G protein-coupled receptor pharmacology: What are they good for? J. Membr. Biol. 2014, 247, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Planchard, N.; Point, É.; Dahmane, T.; Giusti, F.; Renault, M.; Le Bon, C.; Durand, G.; Milon, A.; Guittet, É.; Zoonens, M.; et al. The use of amphipols for solution NMR studies of membrane proteins: Advantages and constraints as compared to other solubilizing media. J. Membr. Biol. 2014, 247, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Catoire, L.J.; Damian, M.; Giusti, F.; Martin, A.; van Heijenoort, C.; Popot, J.-L.; Guittet, E.; Banères, J.-L. Structure of a GPCR ligand in its receptor-bound state: Leukotriene B4 adopts a highly constrained conformation when associated to human BLT2. J. Am. Chem. Soc. 2010, 132, 9049–9057. [Google Scholar] [CrossRef] [PubMed]
- Giusti, F.; Casiraghi, M.; Point, E.; Damian, M.; Rieger, J.; Le Bon, C.; Pozza, A.; Moncoq, K.; Banères, J.-L.; Catoire, L.J. Structure of the agonist 12-HHT in its BLT2 receptor-bound state. Sci. Rep. 2020, 10, 2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzkorn, M.; Raschle, T.; Hagn, F.; Gelev, V.; Rice, A.J.; Walz, T.; Wagner, G. Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: Biophysical properties and NMR accessibility. Structure 2013, 21, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elter, S.; Raschle, T.; Arens, S.; Viegas, A.; Gelev, V.; Etzkorn, M.; Wagner, G. The use of amphipols for NMR structural characterization of 7-TM proteins. J. Membr. Biol. 2014, 247, 957–964. [Google Scholar] [CrossRef] [Green Version]
- Bosco, M.; Damian, M.; Chauhan, V.; Roche, M.; Guillet, P.; Fehrentz, J.-A.; Bonneté, F.; Polidori, A.; Banères, J.-L.; Durand, G. Biotinylated non-ionic amphipols for GPCR ligands screening. Methods 2020. [Google Scholar] [CrossRef]
- Dürr, U.H.N.; Soong, R.; Ramamoorthy, A. When detergent meets bilayer: Birth and coming of age of lipid bicelles. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 69, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Morrison, E.A.; Henzler-Wildman, K.A. Reconstitution of integral membrane proteins into isotropic bicelles with improved sample stability and expanded lipid composition profile. Biochim. Biophys. Acta 2012, 1818, 814–820. [Google Scholar] [CrossRef] [Green Version]
- Puthenveetil, R.; Vinogradova, O. Solution NMR: A powerful tool for structural and functional studies of membrane proteins in reconstituted environments. J. Biol. Chem. 2019, 294, 15914–15931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Prytulla, S.; De Angelis, A.A.; Brown, J.M.; Kiefer, H.; Opella, S.J. High-resolution NMR spectroscopy of a GPCR in aligned bicelles. J. Am. Chem. Soc. 2006, 128, 7402–7403. [Google Scholar] [CrossRef] [Green Version]
- Mineev, K.S.; Nadezhdin, K.D.; Goncharuk, S.A.; Arseniev, A.S. Characterization of Small Isotropic Bicelles with Various Compositions. Langmuir 2016, 32, 6624–6637. [Google Scholar] [CrossRef]
- Piai, A.; Fu, Q.; Dev, J.; Chou, J.J. Optimal Bicelle Size q for Solution NMR Studies of the Protein Transmembrane Partition. Chemistry 2017, 23, 1361–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, T.A.; Baoukina, S.; Brock, A.T.; Oliver, R.C.; Root, K.T.; Krueger, J.K.; Glover, K.J.; Tieleman, D.P.; Columbus, L. Low- q Bicelles Are Mixed Micelles. J. Phys. Chem. Lett. 2018, 9, 4469–4473. [Google Scholar] [CrossRef] [PubMed]
- Glover, K.J.; Whiles, J.A.; Wu, G.; Yu, N.; Deems, R.; Struppe, J.O.; Stark, R.E.; Komives, E.A.; Vold, R.R. Structural evaluation of phospholipid bicelles for solution-state studies of membrane-associated biomolecules. Biophys. J. 2001, 81, 2163–2171. [Google Scholar] [CrossRef] [Green Version]
- Björnerås, J.; Nilsson, M.; Mäler, L. Analysing DHPC/DMPC bicelles by diffusion NMR and multivariate decomposition. Biochim. Biophys. Acta 2015, 1848, 2910–2917. [Google Scholar] [CrossRef]
- Schmidt, P.; Bender, B.J.; Kaiser, A.; Gulati, K.; Scheidt, H.A.; Hamm, H.E.; Meiler, J.; Beck-Sickinger, A.G.; Huster, D. Improved in Vitro Folding of the Y2 G Protein-Coupled Receptor into Bicelles. Front. Mol. Biosci. 2017, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.A.; Liu, J.J.; Chun, E.; Wacker, D.; Wu, H.; Cherezov, V.; Stevens, R.C. GPCR stabilization using the bicelle-like architecture of mixed sterol-detergent micelles. Methods 2011, 55, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Schrottke, S.; Kaiser, A.; Vortmeier, G.; Els-Heindl, S.; Worm, D.; Bosse, M.; Schmidt, P.; Scheidt, H.A.; Beck-Sickinger, A.G.; Huster, D. Expression, Functional Characterization, and Solid-State NMR Investigation of the G Protein-Coupled GHS Receptor in Bilayer Membranes. Sci. Rep. 2017, 7, 46128. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, J.M.; Shih, K.-C.; Scheidt, H.A.; Fantin, S.M.; Parson, K.F.; Pantelopulos, G.A.; Harrington, H.R.; Mittendorf, K.F.; Qian, S.; Stein, R.A.; et al. Bicelles Rich in both Sphingolipids and Cholesterol and Their Use in Studies of Membrane Proteins. J. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Duc, N.M.; Du, Y.; Zhang, C.; Lee, S.Y.; Thorsen, T.S.; Kobilka, B.K.; Chung, K.Y. Effective application of bicelles for conformational analysis of G protein-coupled receptors by hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 808–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, Y.; Racker, E. Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation XXV. Reconstitution of vesicles catalyzing 32pi—Adenosine triphosphate exchange. J. Biochem. 1971, 246, 5477–5487. [Google Scholar]
- Sessa, G.; Weissmann, G. Phospholipid spherules (liposomes) as a model for biological membranes. J. Lipid Res. 1968, 9, 310–318. [Google Scholar] [PubMed]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv. 2011, 2011, 591325. [Google Scholar] [CrossRef] [Green Version]
- Siontorou, C.G.; Nikoleli, G.-P.; Nikolelis, D.P.; Karapetis, S.K. Artificial Lipid Membranes: Past, Present, and Future. Membranes 2017, 7, 38. [Google Scholar] [CrossRef]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, J.; Shigematsu, H.; Xu, W.; Shih, W.M.; Rothman, J.E.; Lin, C. Self-assembly of size-controlled liposomes on DNA nanotemplates. Nat. Chem. 2016, 8, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Ogasawara, T.; Tanaka, Y.; Takeda, H.; Sawasaki, T.; Mogi, M.; Liu, S.; Maeyama, K. Functional G-Protein-Coupled Receptor (GPCR) Synthesis: The Pharmacological Analysis of Human Histamine H1 Receptor (HRH1) Synthesized by a Wheat Germ Cell-Free Protein Synthesis System Combined with Asolectin Glycerosomes. Front. Pharmacol. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Yeliseev, A. Expression and Preparation of a G-Protein-Coupled Cannabinoid Receptor CB2 for NMR Structural Studies. Curr. Protoc. Protein Sci. 2019, 96, e83. [Google Scholar] [CrossRef]
- Redka, D.S.; Morizumi, T.; Elmslie, G.; Paranthaman, P.; Shivnaraine, R.V.; Ellis, J.; Ernst, O.P.; Wells, J.W. Coupling of G proteins to reconstituted monomers and tetramers of the M2 muscarinic receptor. J. Biol. Chem. 2014, 289, 24347–24365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, J.; Faust, B.; Attrill, H.; Harding, P.; Orwick, M.; Watts, A. The role of cholesterol on the activity and stability of neurotensin receptor 1. Biochim. Biophys. Acta 2012, 1818, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.J.Y.; Blaza, J.N.; Bridges, H.R.; May, B.; Moore, A.L.; Hirst, J. A Self-Assembled Respiratory Chain that Catalyzes NADH Oxidation by Ubiquinone-10 Cycling between Complex I and the Alternative Oxidase. Angew. Chem. Int. Ed. Engl. 2016, 55, 728–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berstein, G.; Blank, J.L.; Smrcka, A.V.; Higashijima, T.; Sternweis, P.C.; Exton, J.H.; Ross, E.M. Reconstitution of agonist-stimulated phosphatidylinositol 4,5-bisphosphate hydrolysis using purified m1 muscarinic receptor, Gq/11, and phospholipase C-beta 1. J. Biol. Chem. 1992, 267, 8081–8088. [Google Scholar] [PubMed]
- Biner, O.; Fedor, J.G.; Yin, Z.; Hirst, J. Bottom-Up Construction of a Minimal System for Cellular Respiration and Energy Regeneration. ACS Synth. Biol. 2020, 9, 1450–1459. [Google Scholar] [CrossRef]
- Mouritsen, O.G. Model answers to lipid membrane questions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004622. [Google Scholar] [CrossRef] [PubMed]
- Gallier, S.; Laubscher, A.; Jiménez-Flores, R. The Milk Fat Globule Membrane. In Food Structures, Digestion and Health; Elsevier: Amsterdam, The Netherlands, 2014; pp. 107–142. [Google Scholar]
- Goddard, A.D.; Dijkman, P.M.; Adamson, R.J.; Watts, A. Lipid-dependent GPCR dimerization. Methods Cell Biol. 2013, 117, 341–357. [Google Scholar] [CrossRef]
- Conner, M.; Hicks, M.R.; Dafforn, T.; Knowles, T.J.; Ludwig, C.; Staddon, S.; Overduin, M.; Günther, U.L.; Thome, J.; Wheatley, M.; et al. Functional and biophysical analysis of the C-terminus of the CGRP-receptor; a family B GPCR. Biochemistry 2008, 47, 8434–8444. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Das, B.B.; Casagrande, F.; Tian, Y.; Nothnagel, H.J.; Chu, M.; Kiefer, H.; Maier, K.; De Angelis, A.A.; Marassi, F.M.; et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 2012, 491, 779–783. [Google Scholar] [CrossRef]
- Luca, S.; White, J.F.; Sohal, A.K.; Filippov, D.V.; van Boom, J.H.; Grisshammer, R.; Baldus, M. The conformation of neurotensin bound to its G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 10706–10711. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Li, C.; Gao, L.; Wang, A. High-density lipoprotein synthesis and metabolism (Review). Mol. Med. Rep. 2015, 12, 4015–4021. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Grinkova, Y.V.; Sligar, S.G. Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- Hagn, F.; Etzkorn, M.; Raschle, T.; Wagner, G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisov, I.G.; Grinkova, Y.V.; Lazarides, A.A.; Sligar, S.G. Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J. Am. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef]
- McLean, M.A.; Gregory, M.C.; Sligar, S.G. Nanodiscs: A Controlled Bilayer Surface for the Study of Membrane Proteins. Annu. Rev. Biophys. 2018, 47, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef] [PubMed]
- Bibow, S.; Polyhach, Y.; Eichmann, C.; Chi, C.N.; Kowal, J.; Albiez, S.; McLeod, R.A.; Stahlberg, H.; Jeschke, G.; Güntert, P.; et al. Solution structure of discoidal high-density lipoprotein particles with a shortened apolipoprotein A-I. Nat. Struct. Mol. Biol. 2017, 24, 187–193. [Google Scholar] [CrossRef]
- Grinkova, Y.V.; Denisov, I.G.; Sligar, S.G. Engineering extended membrane scaffold proteins for self-assembly of soluble nanoscale lipid bilayers. Protein Eng. Des. Sel. 2010, 23, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Frauenfeld, J.; Gumbart, J.; van der Sluis, E.O.; Funes, S.; Gartmann, M.; Beatrix, B.; Mielke, T.; Berninghausen, O.; Becker, T.; Schulten, K.; et al. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol. 2011, 18, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Denisov, I.G.; McLean, M.A.; Shaw, A.W.; Grinkova, Y.V.; Sligar, S.G. Thermotropic phase transition in soluble nanoscale lipid bilayers. J. Phys. Chem. B 2005, 109, 15580–15588. [Google Scholar] [CrossRef] [Green Version]
- Padmanabha Das, K.M.; Shih, W.M.; Wagner, G.; Nasr, M.L. Large Nanodiscs: A Potential Game Changer in Structural Biology of Membrane Protein Complexes and Virus Entry. Front. Bioeng. Biotechnol. 2020, 8, 539. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, P.M.; Watts, A. Lipid modulation of early G protein-coupled receptor signalling events. Biochim. Biophys. Acta 2015, 1848, 2889–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Klenk, C.; Merklinger, L.; Morstein, L.; Hagn, F.; Plückthun, A.; Gui, M.; Wang, Z.-F.; Gorgulla, C.; Yu, J.J.; et al. Cryo-EM structure of an activated GPCR-G protein complex in lipid nanodiscs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lee, S.C.; Knowles, T.J.; Postis, V.L.G.; Jamshad, M.; Parslow, R.A.; Lin, Y.-P.; Goldman, A.; Sridhar, P.; Overduin, M.; Muench, S.P.; et al. A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat. Protoc. 2016, 11, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Hothersall, J.D.; Jones, A.Y.; Dafforn, T.R.; Perrior, T.; Chapman, K.L. Releasing the technical “shackles” on GPCR drug discovery: Opportunities enabled by detergent-free polymer lipid particle (PoLiPa) purification. Drug Discov. Today 2020. [Google Scholar] [CrossRef]
- Xue, M.; Cheng, L.; Faustino, I.; Guo, W.; Marrink, S.J. Molecular Mechanism of Lipid Nanodisk Formation by Styrene-Maleic Acid Copolymers. Biophys. J. 2018, 115, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, P.S.; Bozdaganyan, M.E.; Voskoboynikova, N.; Mulkidjanian, A.Y.; Steinhoff, H.-J.; Shaitan, K.V. Styrene/Maleic Acid Copolymers Form SMALPs by Pulling Lipid Patches out of the Lipid Bilayer. Langmuir 2019, 35, 3748–3758. [Google Scholar] [CrossRef]
- Park, S.H.; Wu, J.; Yao, Y.; Singh, C.; Tian, Y.; Marassi, F.M.; Opella, S.J. Membrane proteins in magnetically aligned phospholipid polymer discs for solid-state NMR spectroscopy. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183333. [Google Scholar] [CrossRef]
- Fiori, M.C.; Jiang, Y.; Altenberg, G.A.; Liang, H. Polymer-encased nanodiscs with improved buffer compatibility. Sci. Rep. 2017, 7, 7432. [Google Scholar] [CrossRef]
- Pollock, N.L.; Lee, S.C.; Patel, J.H.; Gulamhussein, A.A.; Rothnie, A.J. Structure and function of membrane proteins encapsulated in a polymer-bound lipid bilayer. Biochim. Biophys. Acta Biomembr. 2018, 1860, 809–817. [Google Scholar] [CrossRef]
- Jamshad, M.; Grimard, V.; Idini, I.; Knowles, T.J.; Dowle, M.R.; Schofield, N.; Sridhar, P.; Lin, Y.-P.; Finka, R.; Wheatley, M.; et al. Structural analysis of a nanoparticle containing a lipid bilayer used for detergent-free extraction of membrane proteins. Nano Res. 2015, 8, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.C.K.; Lee, S.C.; Pollock, N.L.; Stroud, Z.; Hall, S.; Thakker, A.; Pitt, A.R.; Dafforn, T.R.; Spickett, C.M.; Roper, D.I. Analysis of SMALP co-extracted phospholipids shows distinct membrane environments for three classes of bacterial membrane protein. Sci. Rep. 2019, 9, 1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van’t Klooster, J.S.; Cheng, T.-Y.; Sikkema, H.R.; Jeucken, A.; Moody, B.; Poolman, B. Periprotein lipidomes of Saccharomyces cerevisiae provide a flexible environment for conformational changes of membrane proteins. Elife 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Fu, Z.; Xu, G.G.; Grassucci, R.A.; Zhang, Y.; Frank, J.; Hendrickson, W.A.; Guo, Y. Structure and activity of lipid bilayer within a membrane-protein transporter. Proc. Natl. Acad. Sci. USA 2018, 115, 12985–12990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Benlekbir, S.; Venkatakrishnan, P.; Wang, Y.; Hong, S.; Hosler, J.; Tajkhorshid, E.; Rubinstein, J.L.; Gennis, R.B. Structure of the alternative complex III in a supercomplex with cytochrome oxidase. Nature 2018, 557, 123–126. [Google Scholar] [CrossRef]
- Broecker, J.; Eger, B.T.; Ernst, O.P. Crystallogenesis of Membrane Proteins Mediated by Polymer-Bounded Lipid Nanodiscs. Structure 2017, 25, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logez, C.; Damian, M.; Legros, C.; Dupré, C.; Guéry, M.; Mary, S.; Wagner, R.; M’Kadmi, C.; Nosjean, O.; Fould, B.; et al. Detergent-free Isolation of Functional G Protein-Coupled Receptors into Nanometric Lipid Particles. Biochemistry 2016, 55, 38–48. [Google Scholar] [CrossRef]
- Routledge, S.J.; Jamshad, M.; Little, H.A.; Lin, Y.-P.; Simms, J.; Thakker, A.; Spickett, C.M.; Bill, R.M.; Dafforn, T.R.; Poyner, D.R.; et al. Ligand-induced conformational changes in a SMALP-encapsulated GPCR. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183235. [Google Scholar] [CrossRef] [PubMed]
- Radoicic, J.; Park, S.H.; Opella, S.J. Macrodiscs Comprising SMALPs for Oriented Sample Solid-State NMR Spectroscopy of Membrane Proteins. Biophys. J. 2018, 115, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.H.; Cook, G.M.; Spratley, S.J.; Fawke, S.; Graham, S.C.; Deane, J.E. The mechanism of glycosphingolipid degradation revealed by a GALC-SapA complex structure. Nat. Commun. 2018, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Popovic, K.; Holyoake, J.; Pomès, R.; Privé, G.G. Structure of saposin A lipoprotein discs. Proc. Natl. Acad. Sci. USA. 2012, 109, 2908–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfeld, J.; Löving, R.; Armache, J.-P.; Sonnen, A.F.-P.; Guettou, F.; Moberg, P.; Zhu, L.; Jegerschöld, C.; Flayhan, A.; Briggs, J.A.G.; et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 2016, 13, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Richards, M.R.; Bagal, D.; Campuzano, I.D.G.; Kitova, E.N.; Xiong, Z.J.; Privé, G.G.; Klassen, J.S. Characterizing the Size and Composition of Saposin A Lipoprotein Picodiscs. Anal. Chem. 2016, 88, 9524–9531. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-T.H.; Helfinger, L.R.; Bostock, M.J.; Solt, A.; Tan, Y.L.; Nietlispach, D. An Adaptable Phospholipid Membrane Mimetic System for Solution NMR Studies of Membrane Proteins. J. Am. Chem. Soc. 2017, 139, 14829–14832. [Google Scholar] [CrossRef]
- Flayhan, A.; Mertens, H.D.T.; Ural-Blimke, Y.; Martinez Molledo, M.; Svergun, D.I.; Löw, C. Saposin Lipid Nanoparticles: A Highly Versatile and Modular Tool for Membrane Protein Research. Structure 2018, 26, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Neuberger, A.; Orr, M.W.; Newman, C.E.; Hsu, P.-C.; Samsudin, F.; Szewczak-Harris, A.; Ramos, L.M.; Debela, M.; Khalid, S.; et al. Interactions of a Bacterial RND Transporter with a Transmembrane Small Protein in a Lipid Environment. Structure 2020, 28, 625–634.e6. [Google Scholar] [CrossRef]
- Nguyen, N.X.; Armache, J.-P.; Lee, C.; Yang, Y.; Zeng, W.; Mootha, V.K.; Cheng, Y.; Bai, X.-C.; Jiang, Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 2018, 559, 570–574. [Google Scholar] [CrossRef]
- Lloris-Garcerá, P.; Klinter, S.; Chen, L.; Skynner, M.J.; Löving, R.; Frauenfeld, J. DirectMX—One-Step Reconstitution of Membrane Proteins from Crude Cell Membranes into Salipro Nanoparticles. Front. Bioeng. Biotechnol. 2020, 8, 215. [Google Scholar] [CrossRef]
- Yao, X.; Fan, X.; Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proc. Natl. Acad. Sci. USA 2020, 117, 18497–18503. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
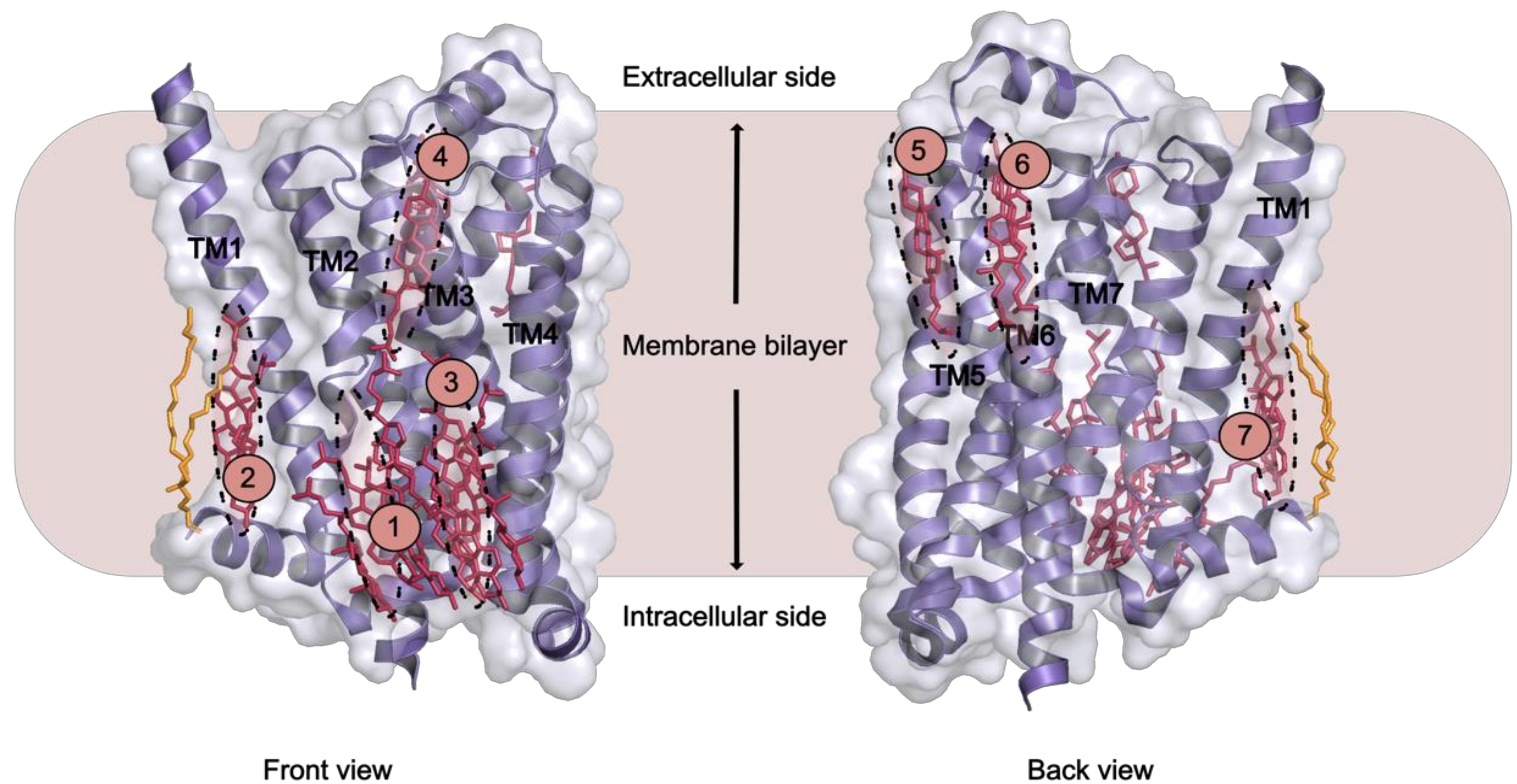
| Site | GPCR | PDB IDs | Number of Bound Lipids | Binding Interface | Crystal Contacts (C) or Dimer Interface (D) |
|---|---|---|---|---|---|
| 1 | β2AR | 2RH1, 3D4S | 2 × C | IC; helices I–IV | C |
| 1 | β1AR | 2Y00 | 1 × CHS | IC; helices II–IV, ICL1 | C |
| 1 | P2Y12 | 4PXZ | 1 × C | IC; helices II–IV, ICL1 | C |
| 2 | β2AR | 2RH1 | 1 × P | IC; helices I, VIII, palmitate | D |
| 2 | 5-HT2B | 4IB4 | 1 × P | IC; helices I, VIII, palmitate | D |
| 3 | β1AR | 2Y00 | 1 × CHS | IC; helices III–V | C |
| 3 | P2Y12 GABAB | 4NTJ 6WIV | 1 × C 2 × C | IC; helices III–V IC; helices III–V | C D |
| 4 | A2AAR | 4EIY | 1 × C | EC; helices V–VI | C |
| 4 | β1AR | 2Y00 | 1 × CHS | EC; helix V | C |
| 5 | A2AAR µ-OR | 4EIY 4DKL | 11 × C | EC; helices VI–VII, ECL3 EC; helices VI–VII, ECL3 | |
| 6 | P2Y12 | 4NTJ | 1 × C | EC; helices VII, I | |
| 7 | A2AAR | 4EIY | 1 × C | EC; helices II–III, ECL1 | C |
| 7 | mGlu1 | 4OR2 | 3 × C | EC; N-term, helices I–III, ECL1 | D |
| Receptor | Expression System | Membrane Mimetic | Labelling | NMR Experiment | Reference |
|---|---|---|---|---|---|
| β2AR | Sf9 | DDM | 13CH3-Lys (reductive methylation) | STD-filtered 1H,13C HMQC; 1H,13C HSQC | [191] |
| β2AR | Sf9 | DDM/CHS | 19F-TET | 19F; 1D | [143] |
| β2AR | expressSF+ | DDM | 13CH3-Met or α,β,β-2H3,13CH3-Met | 1H,13C-HMQC | [192] |
| β2AR | Sf9 | DDM/CHS or LMNG | 19F-BTFA | 19F; 1D, T1, T2 | [184] |
| β2AR | Sf9 | LMNG | 19F-BTFA | 19F; 1D, T1, T2 | [193] |
| β2AR | Sf9 | DDM | 13CH3-Met | 1H,13C HSQC | [194] |
| β2AR | Sf9 | DDM/CHS | 19F-TET | 19F; 1D, 2D EXSY | [195] |
| β2AR | expressSF+ | POPC/POPG nanodiscs | b2AR [2H-9AA, abg 2H,13CH3-Met] | 1H,13C-HMQC; 1H,15N | [196] |
| β2AR | Sf9 | LMNG | 19F-BTFA | 19F; 1D, CPMG, STD | [197] |
| mOR | Sf9 | LMNG/CHS | 2H-8AA, ab-2H-13CH3-Met | 1H,13C-HMQC | [198] |
| mOR | Sf9 | LMNG/CHS | 13CH3-Lys (reductive methylation) | 1H,13C-HMQC | [189] |
| b1AR | Sf9 | DDM | u-2H,15N | 1H,15N TROSY | [199] |
| b1AR | High five | DM | 15N-Val | 1H,15N HSQC | [200] |
| A2AAR | P. pastoris | LMNG | 19F-BTFMA | 19F; 1D, STD | [201] |
| BLT2 | E. coli | DMPC/CHS nanodiscs | U-2H, 13CH3-δ1-Ile, 13CH3-ϵ-Met | 1H,13C-HMQC | [123] |
| b1AR | Sf9 or Sf21 | LMNG | 13CH3-Met | 1H,13C-HMQC | [6] |
| A2AAR | P. pastoris | DDM | 13CH3 Ile d1/2H | 1H,13C HMQC, 3Q-relaxation | [202] |
| NTR1 | E. coli | DMPC/DMPC nanodiscs | U-2H, 13CH3-δ1-Ile,13CH3-ϵ-Met] | 1H,13C-HMQC | [203] |
| A2AAR | P. pastoris | LMNG/CHS | U-15N, 70% 2H | 1H,15N TROSY | [204] |
| A2AAR | P. pastoris | LMNG/CHS | U-15N, 70% 2H | 1H,15N TROSY | [190] |
| CCR5 | Sf9 | DDM | U-2H,15N | 1H,15N TROSY | [205] |
| NTR1 | E. coli | DDM | 13CH3-Met | 1H,13C-HMQC | [206] |
| β2AR | b2AR: expressSF +C-terminal tail: E. coli | POPC/POPG nanodiscs | b2AR [2H-9AA, abg2H,13CH3-Met] C-tail: U-[2H, 13C, 15N] or 13CH3 Thr g2 and Ile d1 | 1H,13C-HMQC; 1H,15N HSQC; cross-saturation | [207] |
| β2AR | expressSF+ | β-DDM or POPC/POPG nanodiscs | α-2H,13CH3-Ala; αβγ-2H,13CH3-Met; 13C-Ile; 13C-Leu; 13C-Thr; <80% 2H | 1H,13C HSQC; 1H, 13C TROSY | [208] |
| A2AAR | P. pastoris | LMNG | 19F-BTFMA; metal ions | 19F, 23Na+, 25Mg+; 1D, CPMG | [209] |
| A2AAR | Sf9 | DDM/CHS | 19F-TET (in membrane labelling) | 19F; 1D, 2D EXSY | [210] |
| M2R | Sf9 | LMNG/CHS | 13 CH3-ε-Met | 1H,13C HSQC | [211] |
| β2AR | Sf9 | LMNG/CHS or POPC/POPG nanodiscs | 19F-BTFMA | 19F; 1D | [212] |
| β1AR | Sf9 | DM | 15N-Val | 1H,15N TROSY | [213] |
| β2AR | Sf9 | LMNG | [2,3,3-2 H, 15N]-Leu, MSTL | 1H,15N TROSY, PRE | [3] |
| β1AR | Sf9 | LMNG | 19F-TET | 19F; 1D, CPMG, STD | [7] |
| A2AR | P. pastoris | nanodiscs (POPC/POPG and/or SAPC or SDPC) | α,β,β-2H,13CH3] Met, u-2H | 1H,13C-HMQC; 1H 1D; 31P 1D; solution PRE | [166] |
| α1AR | E. coli | DDM/CHS | 13CH3-ϵ-Met | 1H,13C-HMQC | [214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. https://doi.org/10.3390/molecules25204729
Jones AJY, Gabriel F, Tandale A, Nietlispach D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules. 2020; 25(20):4729. https://doi.org/10.3390/molecules25204729
Chicago/Turabian StyleJones, Andrew J. Y., Florian Gabriel, Aditi Tandale, and Daniel Nietlispach. 2020. "Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches" Molecules 25, no. 20: 4729. https://doi.org/10.3390/molecules25204729