Determination of Lekethromycin, a Novel Macrolide Lactone, in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study

 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Stock Solutions, Quality Control Samples and Standard Curves
2.3. LC-MS/MS Analysis
2.4. Animals
2.5. Sample Preparation
2.6. Method Validation
2.7. Application to a Pharmacokinetic Study
3. Results and Discussion
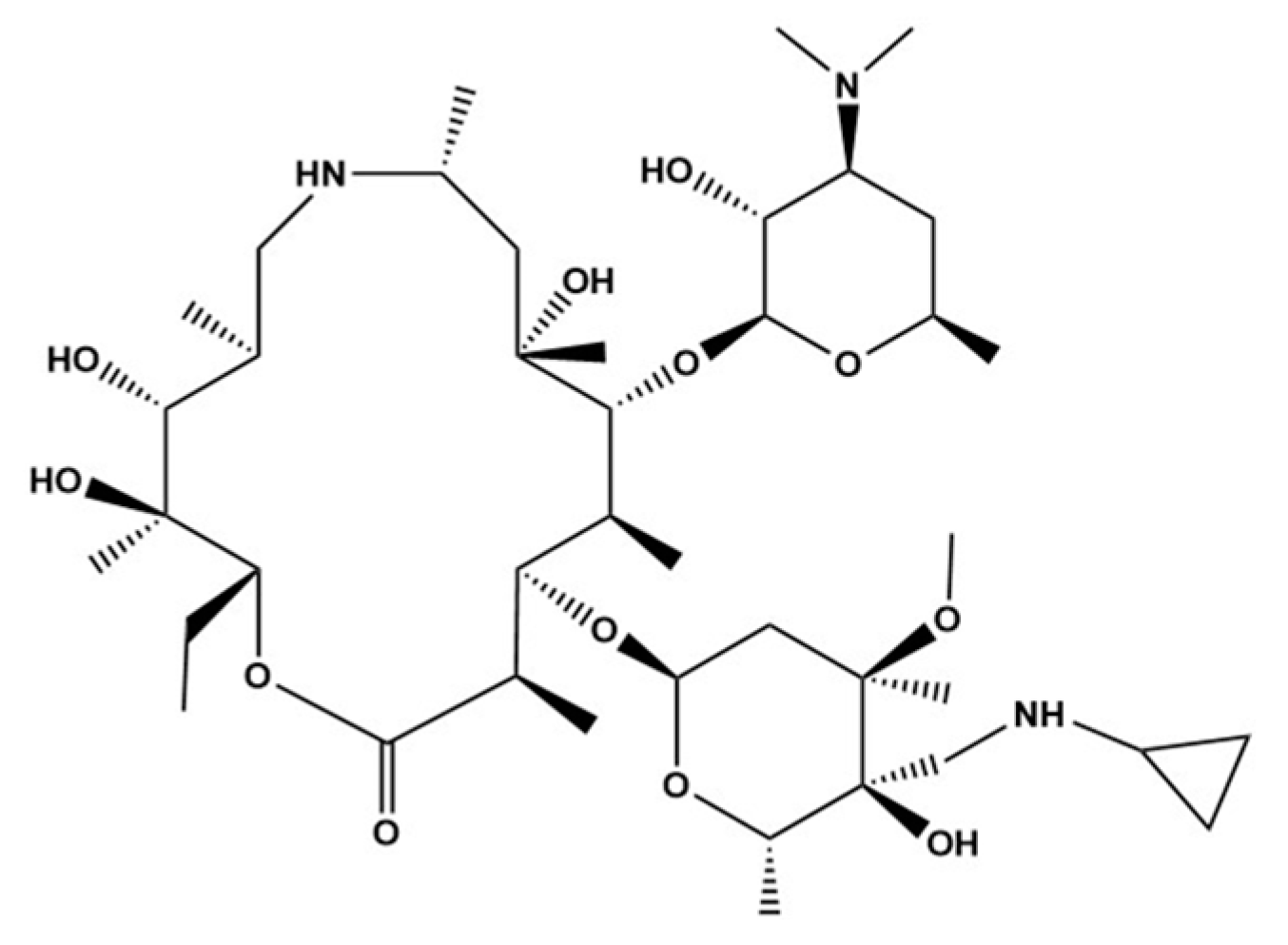
3.1. UPLC-Mass Spectrometry Optimization
3.2. Method Validation
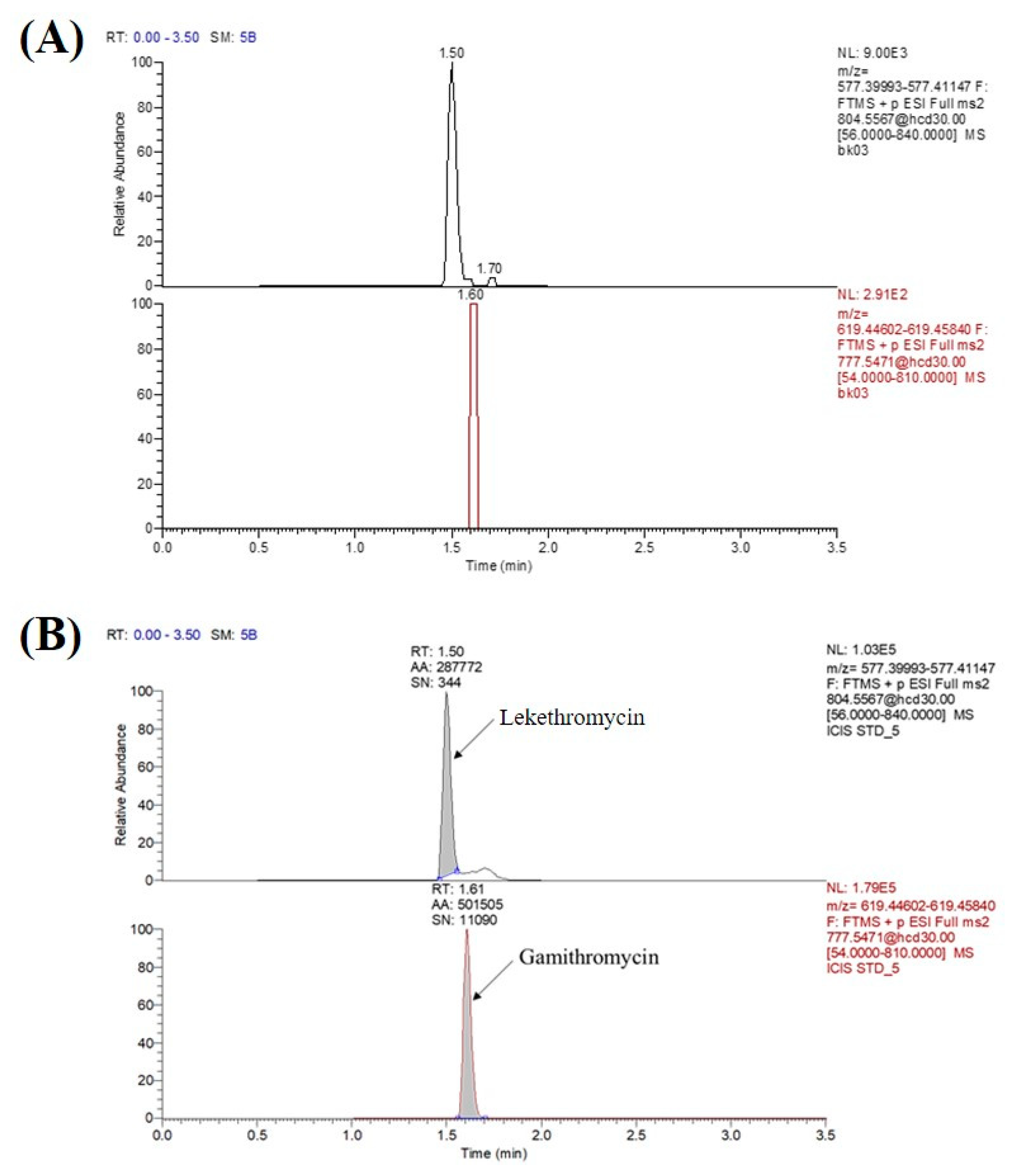
3.2.1. Specificity and Sensitivity
3.2.2. Linearity and Calibration Curve
3.2.3. Precision and Accuracy
3.2.4. Recovery and Matrix Effects
3.2.5. Dilution Integrity
3.2.6. Stability
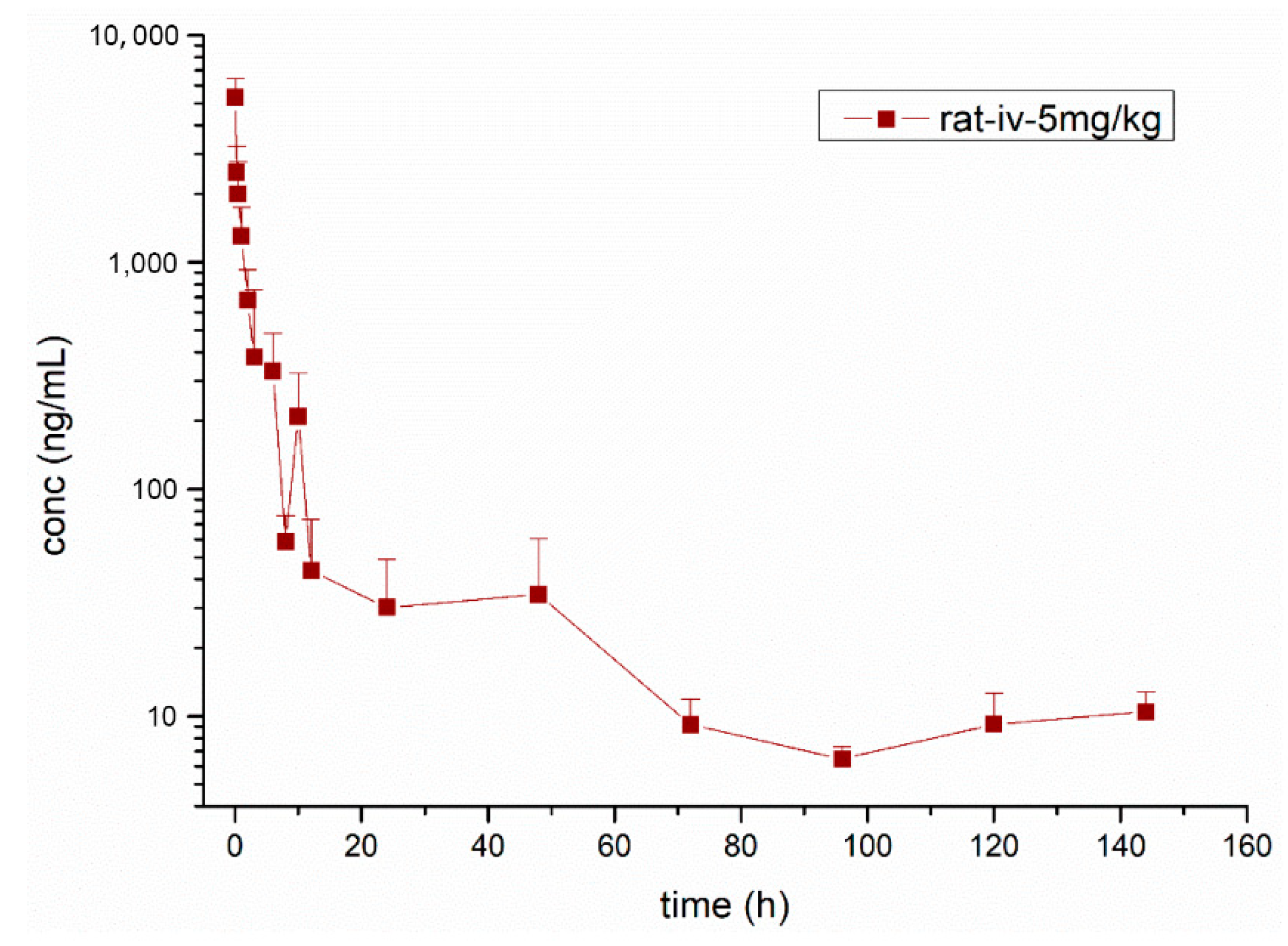
3.3. Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murphy, D.M.; Forrest, I.A.; Curran, D.; Ward, C. Macrolide antibiotics and the airway: Antibiotic or non-antibiotic effects. Expert Opin. Investig. Drugs 2010, 19, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Carlson, E.E. Structure Elucidation of Macrolide Antibiotics Using MS(n) Analysis and Deuterium Labelling. J. Am. Soc. Mass. Spectrom. 2019, 30, 1464–1480. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, B.; Maslinska, M. Macrolide therapy in chronic inflammatory diseases. Mediat. Inflamm. 2012, 2012, 636157. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, I.; Skinner, M.F.; Walker, R.B. Analysis of macrolide antibiotics. J. Chromatogr. A 1998, 812, 255–286. [Google Scholar] [CrossRef]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef]
- Retsema, J.; Fu, W. Macrolides: Structures and microbial targets. Int. J. Antimicrob. Agents 2001, 18 (Suppl. 1), S3–S10. [Google Scholar] [CrossRef]
- Janas, A.; Przybylski, P. 14- and 15-membered lactone macrolides and their analogues and hybrids: Structure, molecular mechanism of action and biological activity. Eur. J. Med. Chem. 2019, 182, 111662. [Google Scholar] [CrossRef]
- Bebear, C.M.; Renaudin, H.; Aydin, M.D.; Chantot, J.F.; Bebear, C. In-vitro activity of ketolides against mycoplasmas. J. Antimicrob. Chemother. 1997, 39, 669–670. [Google Scholar] [CrossRef]
- Doucet-Populaire, F.; Capobianco, J.O.; Zakula, D.; Jarlier, V.; Goldman, R.C. Molecular basis of clarithromycin activity against Mycobacterium avium and Mycobacterium smegmatis. J. Antimicrob. Chemother. 1998, 41, 179–187. [Google Scholar] [CrossRef]
- Alekshun, M.N. New advances in antibiotic development and discovery. Expert Opin. Investig. Drugs 2005, 14, 117–134. [Google Scholar] [CrossRef]
- Wilson, D.N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, S.; Pulido, S.A.; Tutz, S.; Wagner, G.E.; Kriechbaum, M.; Gubensak, N.; Trifunovic, J.; Dorn, M.; Fabian, W.M.; Novak, P.; et al. Structural and functional implications of the interaction between macrolide antibiotics and bile acids. Chemistry 2015, 21, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Mankin, A.S. Macrolide myths. Curr. Opin. Microbiol. 2008, 11, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Arsic, B.; Barber, J.; Cikos, A.; Mladenovic, M.; Stankovic, N.; Novak, P. 16-membered macrolide antibiotics: A review. Int. J. Antimicrob. Agents 2018, 51, 283–298. [Google Scholar] [CrossRef]
- Park, J.W.; Yoon, Y.J. Recent advances in the discovery and combinatorial biosynthesis of microbial 14-membered macrolides and macrolactones. J. Ind. Microbiol. Biotechnol. 2019, 46, 445–458. [Google Scholar] [CrossRef]
- Suzaki, H.; Asano, K.; Ohki, S.; Kanai, K.; Mizutani, T.; Hisamitsu, T. Suppressive activity of a macrolide antibiotic, roxithromycin, on pro-inflammatory cytokine production in vitro and in vivo. Mediat. Inflamm. 1999, 8, 199–204. [Google Scholar] [CrossRef]
- Shimane, T.; Asano, K.; Suzuki, M.; Hisamitsu, T.; Suzaki, H. Influence of a macrolide antibiotic, roxithromycin, on mast cell growth and activation in vitro. Mediat. Inflamm. 2001, 10, 323–332. [Google Scholar] [CrossRef]
- Schultz, M.J.; Speelman, P.; Hack, C.E.; Buurman, W.A.; van Deventer, S.J.; van der Poll, T. Intravenous infusion of erythromycin inhibits CXC chemokine production, but augments neutrophil degranulation in whole blood stimulated with Streptococcus pneumoniae. J. Antimicrob. Chemother. 2000, 46, 235–240. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Wada, H.; Rossios, C.; Takagi, D.; Higaki, M.; Mikura, S.; Goto, H.; Barnes, P.J.; Ito, K. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-kappaB inhibition. J. Pharmacol. Exp. Ther. 2013, 345, 76–84. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E.; Pereira, D. Nature nurtures the design of new semi-synthetic macrolide antibiotics. J. Antibiot. 2017, 70, 527–533. [Google Scholar] [CrossRef]
- Asano, K.; Tryka, E.; Cho, J.S.; Keicho, N. Macrolide therapy in chronic inflammatory diseases. Mediat. Inflamm. 2012, 2012, 692352. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. Prevalence of macrolide resistance in Treponema pallidum is associated with macrolide consumption. J. Med. Microbiol. 2019, 68, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Smajs, D.; Pospisilova, P. Macrolide resistance in yaws. Lancet 2018, 391, 1555–1556. [Google Scholar] [CrossRef]
- Smajs, D.; Pastekova, L.; Grillova, L. Macrolide Resistance in the Syphilis Spirochete, Treponema pallidum ssp. pallidum: Can We Also Expect Macrolide-Resistant Yaws Strains. Am. J. Trop. Med. Hyg. 2015, 93, 678–683. [Google Scholar] [CrossRef]
- Wang, N.; Zhou, Y.; Zhang, H.; Liu, Y. In vitro activities of acetylmidecamycin and other antimicrobials against human macrolide-resistant Mycoplasma pneumoniae isolates. J. Antimicrob. Chemother. 2020, 75, 1513–1517. [Google Scholar] [CrossRef]
- Southon, S.B.; Beres, S.B.; Kachroo, P.; Saavedra, M.O.; Erlendsdottir, H.; Haraldsson, G.; Yerramilli, P.; Pruitt, L.; Zhu, L.; Musser, J.M.; et al. Population genomic molecular epidemiological study of macrolide resistant Streptococcus pyogenes in Iceland, 1995–2016: Identification of a large clonal population with a pbp2x mutation conferring reduced in vitro beta-lactam susceptibility. J. Clin. Microbiol. 2020, 58, 1–13. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X. A Kind of Macrolides Compound, China Patent CN 103965273 B, 25 May 2016. [Google Scholar]
- Yi, X.; Lan, X. LC-MS/MS method for the determination of erianin in rat plasma: Application to a pharmacokinetic study. Biomed. Chromatogr. 2020, 34, e4826. [Google Scholar] [CrossRef]
- Guo, N.; Zhang, A.; Zhuang, H.; Zhang, C. LC-MS/MS assay for the quantification of foretinib in rat plasma and its application to preclinical pharmacokinetic study. Biomed. Chromatogr. 2020, 34, 1–8. [Google Scholar] [CrossRef]
- Sun, X.; Niu, L.; Li, X.; Lu, X.; Li, F. Characterization of metabolic profile of mosapride citrate in rat and identification of two new metabolites: Mosapride N-oxide and morpholine ring-opened mosapride by UPLC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2009, 50, 27–34. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry on Bioanalytical Method Validation, in: C.f.D.E.a.R. Food and Drug Administration (Ed.). Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 2 May 2020).
- EMA. Guideline onBioanalyticalMethodValidation, in: E.M. Agency (Ed.). Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 2 May 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (ng/mL) | Intra-day (n = 6) | Inter-day (n = 18) | ||
|---|---|---|---|---|
| Accuracy (%) | Precision (RSD%) | Accuracy (%) | Precision (RSD%) | |
| 15 | 94 | 5.5 | 97 | 5.1 |
| 200 | 95. | 3.9 | 97 | 3.5 |
| 800 | 89. | 7.3 | 93 | 6.3 |
| Concentration (ng/mL) | Recovery (n = 6) | Matrix Effects (n = 6) | ||
|---|---|---|---|---|
| Accuracy (%) | Precision (RSD%) | Mean (%) | Precision (RSD%) | |
| 15 | 98 | 3.3 | 0.96 | 5.6 |
| 200 | 98 | 2.9 | 0.97 | 2.2 |
| 800 | 95 | 5.1 | 1.0 | 1.2 |
| Dilution Factor | Nominal Conc (ng/mL) | Measured Conc (ng/mL) | Accuracy (%) | Precision (RSD%) |
|---|---|---|---|---|
| 100 | 20 | 20 | 99 | 2.0 |
| 10 | 200 | 2.2 × 102 | 1.1 × 102 | 1.7 |
| 2 | 1000 | 9.7 × 102 | 97 | 3.2 |
| Stability | Nominal Conc (ng/mL) | Measured Conc (ng/mL) | Accuracy (%) | Precision (RSD%) |
|---|---|---|---|---|
| Short-term (24 h) | 15 | 15 | 1.0 × 102 | 5.6 |
| 200 | 2.1 × 102 | 1.0 × 102 | 3.3 | |
| 800 | 7.7 × 102 | 96 | 7.7 | |
| Long-term (2 months) | 15 | 15 | 1.0 × 102 | 3.8 |
| 200 | 2.2 × 102 | 1.1 × 102 | 8.2 | |
| 800 | 9.1 × 102 | 1.1 × 102 | 11.7 | |
| Thaw and freeze (3 cycles) | 15 | 15 | 97 | 7.4 |
| 200 | 2.0 × 102 | 1.0 × 102 | 5.3 | |
| 800 | 7.4 × 102 | 93 | 7.0 | |
| Auto-samples (8 h) | 15 | 15 | 98 | 5.2 |
| 200 | 2.0 × 102 | 100 | 3.1 | |
| 800 | 7.3 × 102 | 92 | 4.9 | |
| Stock solution (3 months) | 100 | 97 | 98 | 2.8 |
| Parameters | Unit | Mean ± SD |
|---|---|---|
| λz | H−1 | 0.02 ± 0.01 |
| T1/2λz | h | 32.33 ± 14.63 |
| Cmax | ng·mL−1 | 5735.97 ± 1395.96 |
| AUClast | ng·h·mL−1 | 8710.69 ± 2318.88 |
| AUCINF_obs | ng·h·mL−1 | 9133.46 ± 2372.71 |
| VZ_obs | L·kg−1 | 25.56 ± 7.93 |
| CL_obs | L·h−1·kg−1 | 0.58 ± 0.17 |
| MRTlast | h | 17.38 ± 7.71 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.; Sun, P.; Qiu, J.; Wang, J.; Yan, L.; Zhang, S.; Cao, X. Determination of Lekethromycin, a Novel Macrolide Lactone, in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study. Molecules 2020, 25, 4676. https://doi.org/10.3390/molecules25204676
Xiao H, Sun P, Qiu J, Wang J, Yan L, Zhang S, Cao X. Determination of Lekethromycin, a Novel Macrolide Lactone, in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study. Molecules. 2020; 25(20):4676. https://doi.org/10.3390/molecules25204676
Chicago/Turabian StyleXiao, Hongzhi, Pan Sun, Jicheng Qiu, Jianzhong Wang, Lei Yan, Suxia Zhang, and Xingyuan Cao. 2020. "Determination of Lekethromycin, a Novel Macrolide Lactone, in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study" Molecules 25, no. 20: 4676. https://doi.org/10.3390/molecules25204676
APA StyleXiao, H., Sun, P., Qiu, J., Wang, J., Yan, L., Zhang, S., & Cao, X. (2020). Determination of Lekethromycin, a Novel Macrolide Lactone, in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study. Molecules, 25(20), 4676. https://doi.org/10.3390/molecules25204676