2.3. A-Complexes of Pyridines.OCS
The A complexes are planar or nearly so. The N…S distances are shorter than the sum of the van der Waals radii of N and S (3.35 Å) and are in the range between 3.021 and 3.068 Å. The N…SC chalcogen bond is not linear, and the N…SC angle varies between 167° and 169°, suggesting an interaction between the C1H bond and the S atom. This is in agreement with the intermolecular H…S distances which range between 2.989 Å and 2.964 Å, thus shorter than the sum of the van der Waals distances of H and S (3.0 Å). Let us observe that these distances are decreasing on going from NH2-pyridine to NO2-pyridine.
The binding energies are rather low, between −12.24 and −9.58 kJ/mol. The
A-complex of pyr.OCS systems is slightly more stable than the corresponding pyr.CS
2 complex as expected from the higher V
s max value of the S atom in OCS. Similarly, for complexes between pyridines and CS
2 [
46], these energies depend on the proton affinity and the ionization potential of the pyridines (correlation equations are given in
S.I.1 of Supplementary Information). The binding energies are also related to the V
s,min of the pyridines (both in kJ/mol):
For the pyridines–CS
2 interaction, a slope of −0.029 was calculated. The results of the present work show that the
A-complexes of pyr.OCS systems are slightly stronger than the corresponding pyr.CS
2 complex [
46], as expected from the higher V
s,max value of the S atom in OCS. This is in agreement with results on the interaction with NH
3 and H
2O which is somewhat stronger for the OCS than for the CS
2 interaction; for the weak complexes with PH
3 and H
2S, they are of the same order of magnitude [
33]. However, for the strong interaction with the Cl
− anion, the CS
2 complexes seem to be stronger than the OCS ones [
32]. Contrary to the expectation from the much greater V
s,max value of the S atom in OCS compared to CS
2, the difference of binding energies between pyr.OCS and pyr.CS
2 complexes is very small. The CH…S H–bonding interaction is weaker for the pyr.OCS complex because of the presence of electronegative O-atom in OCS. In fact, AIM results do not reveal BCPs for CH…S H–bonding in pyr.OCS complexes except for the NO
2-pyr.OCS complex.
Let us compare the binding energies of the
A-pyridines.OCS and
A-pyridines.CS
2 complexes with the electrostatic potential of the interacting atoms. The binding energies of the NH
2-pyr.OCS and NO
2-pyr.OCS complexes are equal to −12.24 and −9.58 kJ/mol and the difference between the V
s,min values is about 70 kJ/mol (
Table 1). The NO
2-pyr.OCS and NO
2-pyr.CS
2 systems have binding energies of −9.58 and −9.34 kJ/mol, the difference between the electrostatic potential of the S atom being about 16 kJ/mol. From these data, it can be concluded that, in the present cases, the electrostatic potential of the electron donor is more important than the electrostatic potential of the electron acceptor in determining the binding energies. Results on other interactions with PH
3, H
2S, [
33] and Cl
− [
32] are in agreement with this conclusion.
Table 3 reports the different parameters obtained from an AIM analysis. Let us observe that the AIM results have been questioned for weak interactions [
52]. The results of the present work show that the AIM parameters are well correlated with other parameters describing the nature of the interaction (see further discussion).
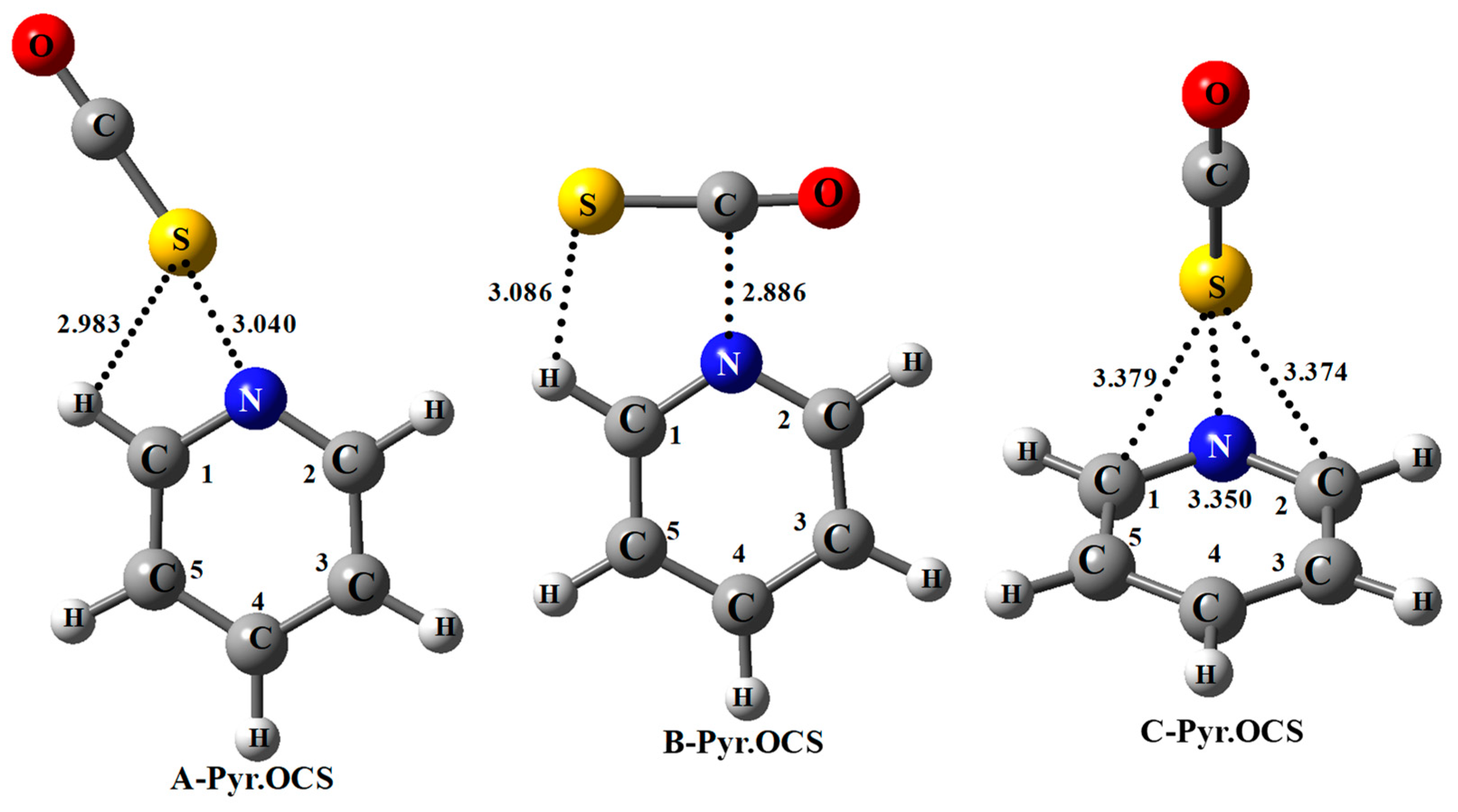
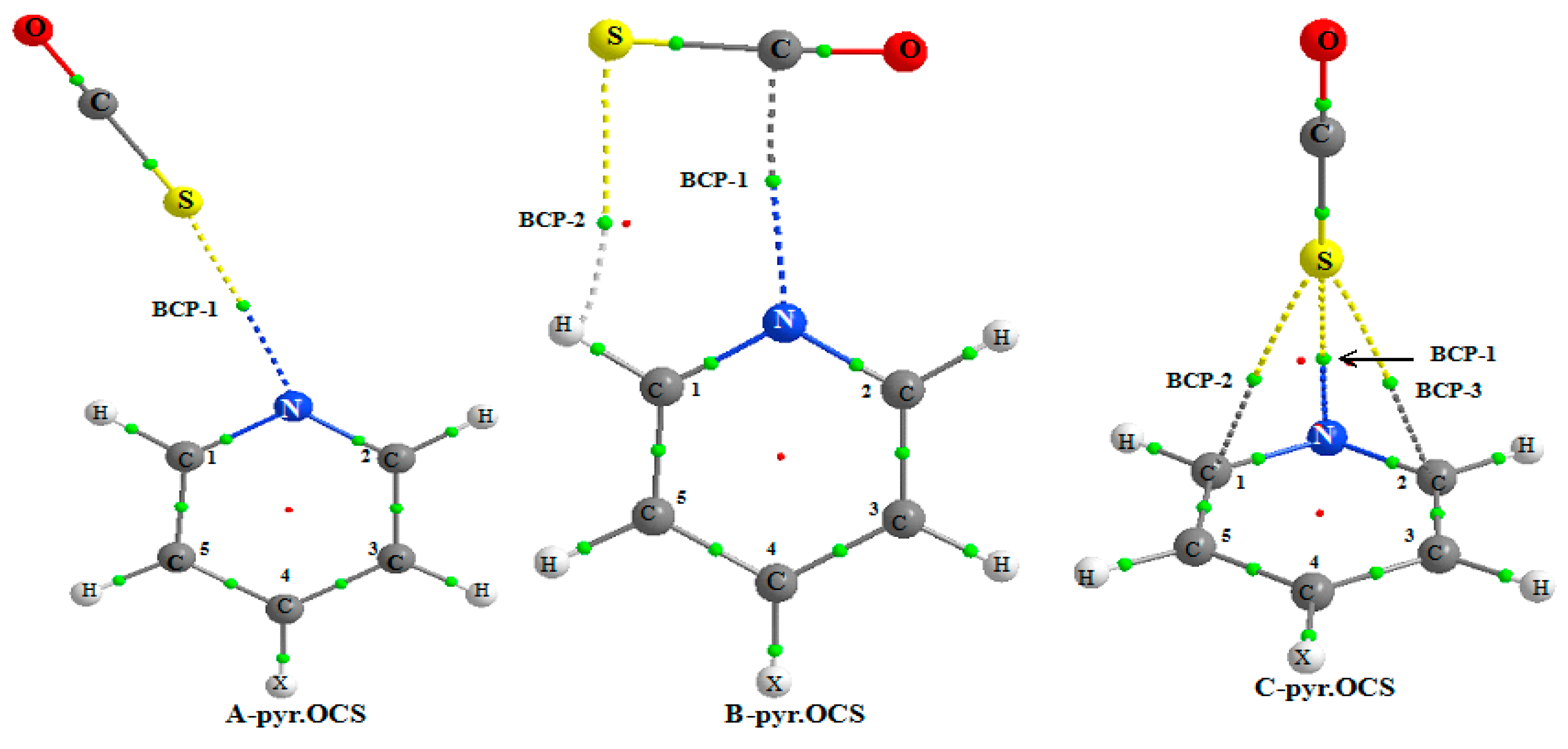
Figure 3 shows the AIM picture for the complex of non-substituted pyridine and the existence of BCPs for the three
A,
B, and
C systems.
For all the A complexes, a BCP-1 is obtained between the N and S atoms. A second BCP-2 is predicted between the S and H1 atom, only for the NO2-pyr.OCS complex. This indicates that the S…H1 interaction is stronger in this system, as expected from the higher acidity of the ortho C–H bond due to the presence of a strong electron withdrawing group at pyridine. The S…HC1 hydrogen bond is far from linear, the S…HC1 angle increasing slightly from 106.4° to 108.5° from the NH2-pyr.OCS to the NO2-pyr.OCS system. This also suggests that the S…HC1 hydrogen bond is slightly stronger in this last system.
The values of electron density and its Laplacian fulfill the criteria expected for closed–shell interaction [
53,
54]. The ρ(r
c) values for BCP-1 are comprised between 0.0130 and 0.0117 a.u., whereas BCP-2 could be detected only for the NO
2-pyr.OCS interaction. The ρ(r
c) values for BCP-1 in pyridine.OCS complexes are somewhat greater than those observed for the pyridine.CS
2 complexes, thus indicating that the N…S bond is stronger in the former system. The positive values of H(r
c) and G(r
c)/ρ(r
c) ratio further indicate further that the N…S interaction is primarily electrostatic in nature. The binding energy (in kJ/mol) of the pyridine.OCS complexes is strongly correlated with the ρ(r
c) values (in me) for BCP-1 as given below.
The NBO analysis provides interesting data on the nature of the interaction in the
A complexes.
Table 4 reports the NBO charges on the S, C, and O atoms of OCS, the charge transfer from pyridines to OCS along with the variation of the C=O and C=S distances.
Let us observe that the charge transfer (CT) in pyr.OCS is significantly larger than the CT in pyridines.CS2 systems where the values are comprised between 7.4 and 3.4 me. This is obviously due to the presence of stronger σ-hole at the S atom of OCS compared to CS2.
The charge transfer (me) is related to the V
s,min values of the electrostatic potential (a.u) of pyridines by the equation:
Table 5 reports the most relevant hyperconjugation energies in the
A systems.
The NBO analysis confirms the existence of both N…S and C1H…S interactions, the N…S interaction being the stronger one. This is in agreement with the correlation between the binding energies and the hyperconjugation energies from the LP(N) to σ*(C=S):
Comparison of the data of
Table 4 and
Table 5 shows that in agreement with the LP(N)→σ*(C=S) charge transfer, the charges on the S atom are decreasing. The calculations also reveal a charge transfer from the LP(S) to σ*(C1H); this charge transfer increases from NH
2-pyridine to NO
2-pyridine. This increase is in agreement with the decrease of the intermolecular S…HC1 distance and is most probably due to the increase in acidity of the C1H bond.
It is surprising that despite the fact that the LP(N)→σ*(C=S) charge transfer is sensitive to the substitution, the elongation of the C=S bond remains more or less constant. A possible explanation is that the increase of σ*(C=S) occupation is nearly compensated by a decrease in π*(C=S) occupation [
23]. Another explanation is an electrostatic attractive interaction between the C and S atoms [
55].
A SAPT analysis was performed to gain further insight into the nature of the interaction. The results are reported in
S.I.2 of Supplementary Information. The interaction energies are slightly higher than the binding energies calculated at the MP2 level because of distortion energy and higher order electron correlation at the SAPT level. This analysis shows that the electrostatic interaction decreases from NH
2-pyridine (−21.11 kJ/mol) to NO
2-pyridine (−16.58 kJ/mol) as expected from the V
s,min values. However, the contribution of the electrostatic energy to the total energy decreases slightly from NH
2-pyridine (46%) to NO
2-pyridine (44%) while the contribution of dispersion energy increases, from 39% to 44%.
2.4. B-Complexes of Pyridines.OCS
As shown in
Figure 2, the
B complex is formed due to the interaction of the positive electrostatic potential of the C atom of OCS with the N atom of pyridine, resulting in a weak N…C tetrel bond. The binding energies of pyridine.OCS
B complexes range between −10.78 and −11.81 kJ/mol and are related to the PA and IP of the pyridines (correlation equations are given in
S.I.3 of Supplementary Information). In the
B structure (
Figure 2), the N…C distances are shorter than the sum of the van der Waals radii (3.25 Å) and range between 2.869 and 2.907 Å. These distances indicate the formation of N…C tetrel bonds. The binding energies decrease with increasing N…C intermolecular distances. The binding energies of the
B complexes of pyr.OCS are almost 2 kJ/mol greater than that for the corresponding pyr.CS
2 complexes, because of stronger π-hole at the C-atom of OCS compared to CS
2. In fact, for pyridine, F-pyridine, NC-pyridine, and NO
2-pyridine, the
B complex is slightly more stable than the corresponding
A complex; which is in complete contrast with the
B complexes of pyridines.CS
2 where the
B complex was found to be the weakest [
46]. Although the S…HC1 distances are relatively long (between 3.066 and 3.127 Å), our further analysis suggests the formation of S…HC1 hydrogen bonds. This is in agreement with the small elongation of the C1H bond comprised between 0.33 and 0.47 Å.
The basic structure of the
B complex of pyridines.CS
2 and pyridines.OCS systems is the same. Both involve the formation of N…C tetrel bonds. The fundamental difference is the direction of the charge transfer which in the pyridines.CS
2 systems is directed from CS
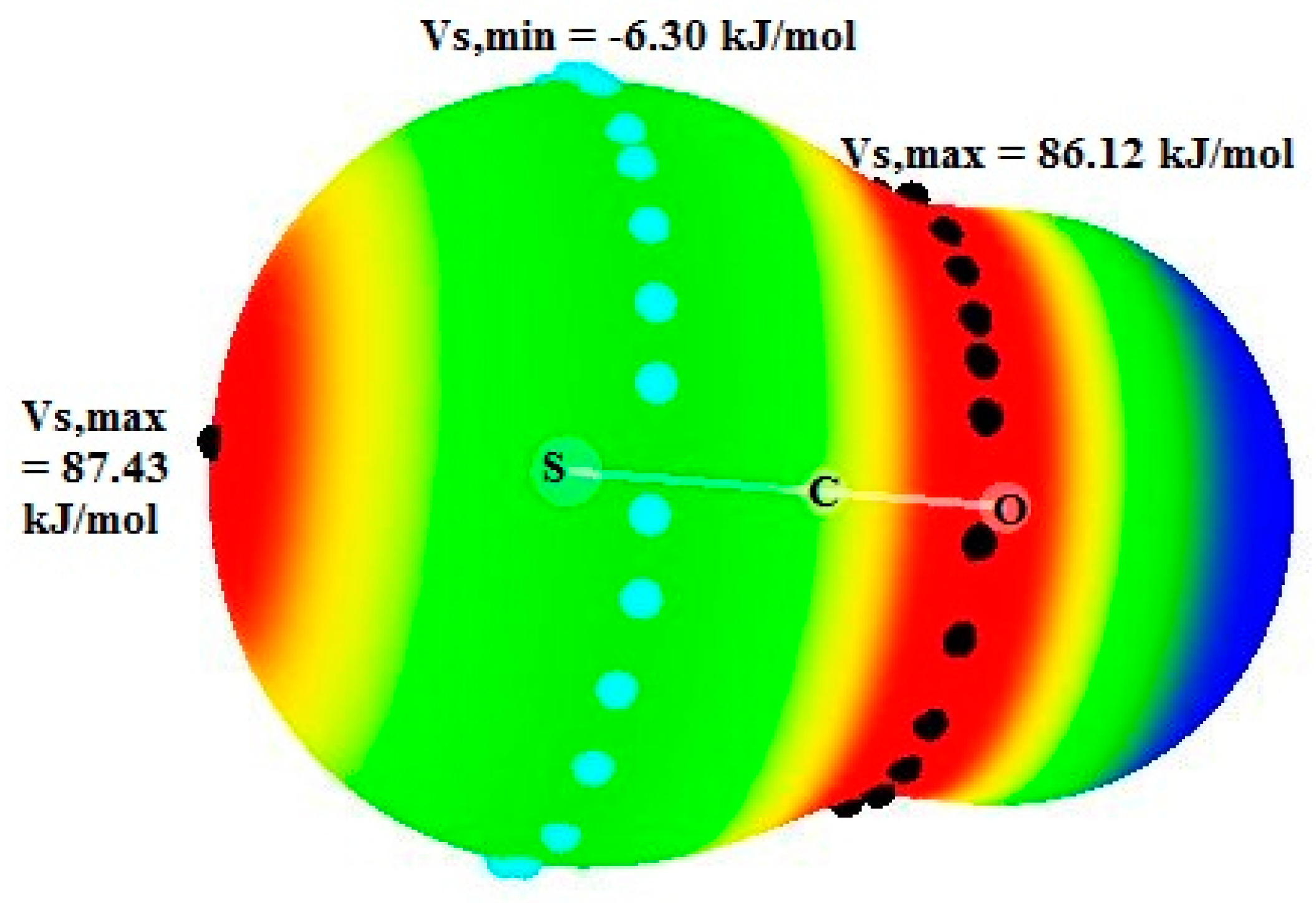
2 to pyridines and in the pyridines.OCS systems from pyridines to OCS. This may be due to the fact that the electrostatic potential around the C atom is equal to 86.12 kJ/mol in OCS, which is much higher than this potential in CS
2 (23.10 kJ/mol) [
46]. This agrees with the charges on the C atom, respectively equal to 0.468 e in OCS and −0.342 e in isolated CS
2.
Table 6 reports the results obtained from the AIM analysis. Two BCPs (as illustrated in
Figure 3) are predicted, the first one between the N and S atoms (BCP-1) and the second one (BCP-2) between the S and H1 atoms (at the exception of CH
3-pyridine). The values of the electron density and its Laplacian are larger for BCP-1 than for BCP-2. Furthermore, the binding energy is found to be linearly correlated with the ρ(r
c) value of BCP-1. These two factors indicate that the N...C tetrel bond is the primary interaction in
B complex. The ρ(r
c) values of BCP-1 of pyr.OCS and pyr.CS
2 complexes range between 13.7 to 12.7 and 10.2 to 9.8 me respectively, indicating that the tetrel bond is stronger for the former complex.
Table 7 reports the charges on the S, C, and O atoms, the charge transfer from pyridine to OCS along with the variations of the C=S and C=O distances for the
B complex of pyridines.OCS systems.
These results indicate a moderate charge transfer, from 4.2 to 8.5 me. Surprisingly, the positive charges on the C atom are increasing in complex formation, whereas the O and S atoms of OCS are gaining electron. The elongation of the C=S bond and the contraction of the C=O bond remains approximately the same for all the complexes.
Table 8 reports the most important hyperconjugation energies in the
B complexes. There is in all the systems a non-negligible hyperconjugation to the Rydberg orbitals of the N atom from σ (C–O) (4 kJ/mol) and from σ(C–S) bonds (2.5 kJ/mol). The values of the binding energies are related to the hyperconjugation energies to the σ* (CO) orbitals by the relation:
This indicates that the N atom is the main interaction site. Despite the relatively long S…HC1 distances, the NBO analysis reveals a non-negligible charge transfer to the σ*(C1H) orbital in agreement with the elongation of the C1H bond of c.a 0.35 mÅ for all the complexes. This charge transfer is about the same as in the A complexes.
No CH…C hydrogen bonds between the CH bond of pyridines and the C atom of OCS could be detected in the present system. This bond was predicted in the complexes between diazines (pyrazine, pyrimidine, and pyridazine) and CS
2 [
28]. These interactions are weak (−ΔE between 3.1 and 5.3 kJ/mol). The formation of CH…C hydrogen bonds in diazines. CS
2 systems can be explained by the presence of two N atoms in the heterocyclic ring which increases the acidity of the CH bond by delocalization of the two N LPs of diazines to the σ*(CH) orbital.
We also want to mention the work recently published [
34] on the interaction between substituted azine molecules HN(CH)SX (X = F, NC, Cl, CN, CCH, H) and OCS. Calculations performed at the MP2 level with the aug-cc-pVTZ basis set indicate that the complex is cyclic. A tetrel bond N…C is formed between the N of the azine molecule and the C atom of OCS and a chalcogen bond O…S is formed between the O atom of OCS and the S atom of the azine molecule. In contrast with the present results, the S atom of OCS is not involved in the interaction. A charge transfer is calculated from the LP of the N atom to the π*(C=S) and π*(C=O) orbitals.
The results of SAPT calculations are reported in
S.I.4 of the Supplementary Information. They are very similar to the results obtained for the
A complexes. The contribution of the electrostatic energy remains almost constant (44%–45%) of the total energy and the contribution of the dispersion energy slightly increases from NH
2-pyridine (43%) to NO
2-pyridine (46%).
2.5. C-Complexes of Pyridines.OCS
In this case of pyridine.OCS interaction, the OCS molecule is nearly perpendicular to the pyridine ring, the NSCO dihedral angle being equal to 179°. The binding energies are comprised between −13.33 kJ/mol and −10.76 kJ/mol.
For the complex between pyridine and OCS, the AIM analysis indicates the existence of a BCP between the N and S atoms and between the S and C1 and C2 atom. For other systems, the AIM picture indicates the existence of a BCP between the C4 or C5 atoms and the S atom. The electron density fluctuates between 0.0068 and 0.0081 a.u. and its Laplacian between 0.0232 and 0.0282 a.u. This π interaction induces a weak charge transfer from pyridine to OCS, ranging from 6.84 to 4.35 me.
The correlations between binding energies and PA or IP of the pyridines are characterized by worse correlation coefficients (given in
S.I.5 of Supplementary Information). This is also the case for the correlation between the binding energies and V
s,min values:
This suggests that the contribution of the N…S interaction to the total binding energy is smaller in these complexes.
NBO results of the
C complexes are listed in
Table 9. The CT for these complexes is quite moderate and range between 4.4 and 6.8 me. On complex formation, the electron density on the C1 and C2 atoms of pyridine ring increases slightly whereas the charge on N atom of pyridine almost remains unchanged. The positive charge on the S atom of the OCS molecule increases for the –NH
2, −CH
3, −H, and –F substituted complexes whereas it decreases for the –CN and –NO
2 substituted complexes. The negative charge on the O atom of the OCS molecule increases to some extent (between 0.02 and 15 me) on complex formation with the exception of –NO
2 complex where a decrease in electron density of the O atom has been observed. Moreover, complex formation results in a very negligible increase in the C=S bond length except for the –NH
2 (0.33 mÅ) and –H (0.08 mÅ) substituted complexes where a slight contraction of the C=S bond has been predicted. The C=O bond distances also gets elongated in all the complexes within a margin ranging between 0.09 and 2.37 mÅ. As mentioned in
Table 9, the major source of stabilization for these complexes comes from the π(C2-C3)→σ*(C-S), π(C4-C5)→σ*(C-S), and π(C1-N)→σ*(C-S) hyperconjugation energies but the small contribution from π(C2-C3)→π*(C-O) orbital interaction cannot be overlooked. It should be noted that the occupation of the σ*(C-S) orbital in isolated OCS is 0.0102 e. This occupation increases slightly in all the complexes as mentioned in
Table 9, resulting in a small increase in the C=S bond length.
The results of a SAPT analysis are reported in
S.I.6 of the Supplementary Information. In the case of
C complexes, we observe a great difference in the nature of interaction with
A and
B complexes. The contribution of the electrostatic energy to the total binding energy fluctuates between 30% (NH
2-pyr) and 25% (NO
2-pyr); whereas the contribution of the dispersion energy ranges between 58% and 65% and is much larger than that for the
A and
B complexes. This was to be expected for an interaction involving mainly π-electrons.
2.6. Vibrational Data
Some relevant vibrational data will be discussed here.
Table 10 reports the frequency shifts of the ν(C=O) and ν(C=S) vibrations of the
A,
B, and
C complexes.
For the much stronger OCS…Cl
− interaction, Δν(C=S) and Δν (C=O) values of 31.4 and 63 cm
−1 were reported [
32].
From these results and from comparing
Table 4 and
Table 7, it appears that the red shift in ν(C=S) in
A and
B complexes of pyridine.OCS is due to the elongation of the C=S bond owing to the increase in the σ*(C-S) population. The Δν(C=S) is negligible for the
C complexes. For the
A and
C complexes, the Δν(C=O) are negative and corresponds to an elongation of the C=O bond. On the other hand, the positive Δν(C=O) values for the
B complexes correspond to a contraction of the C=O bond upon complex formation.
The deformation vibration δ(OCS) is degenerate in the isolated OCS molecule and its frequency is predicted at 513.3 cm−1. In complexes A and C, this vibration remains degenerate and is blueshifted between 17.7 and 14.5 cm−1 in A complexes and between 8.5 and 4.1 cm−1 in C complexes. In the B complexes, the degeneracy is removed and the vibration is split into two components. The first component is redshifted between 32.3 cm−1 and 24.3 cm−1; the second component is blueshifted by small amounts, between 3.7 and 2.7 cm−1.
The pyridine vibrations are shifted by small amounts, between 1 and 5 cm
−1. Interestingly, the vibration predicted at 3197 cm
−1 which is predominantly the v(CH) vibration is calculated at 3202 cm
−1 in the
A-pyr.OCS complex. The red shift of 5 cm
−1 can be explained by the formation of a weak S…H1C hydrogen bond and the charge transfer to the σ*(C1H) orbital which elongates the C1H bond. In contrast, in the pyridine.Cl
− complex, the ν(C1H) vibration is blueshifted by 25 cm
−1 [
32]; this blue shift can be explained by a decrease of the anomeric effect because the LP of the N atom is partially involved in the formation of the intermolecular N…Cl
− bond.







{kind=link}
{kind=link}
{kind=link}
{kind=link}