
Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib

Abstract
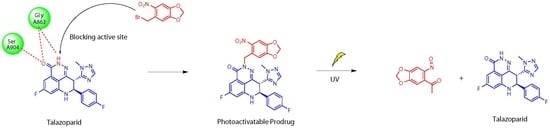
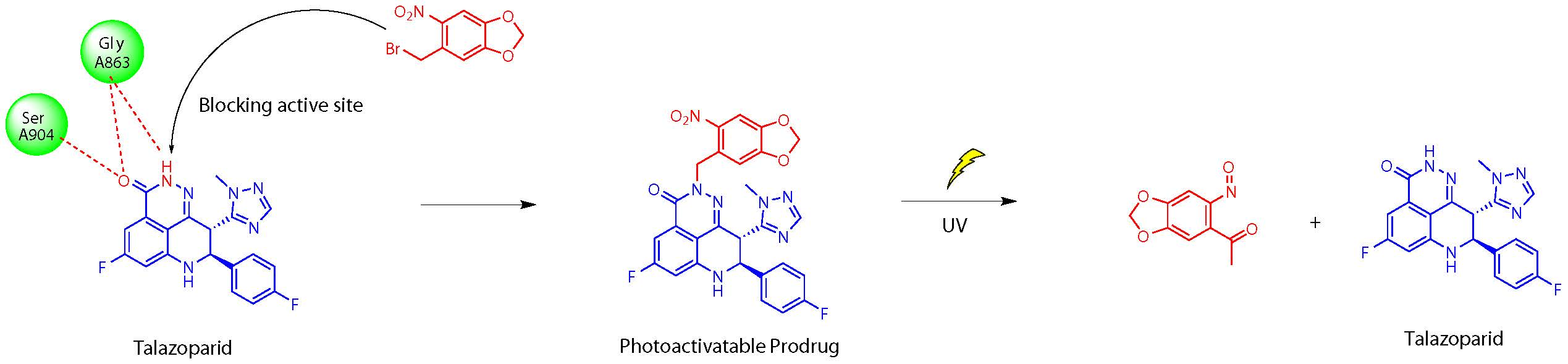
1. Introduction
2. Results and Discussion
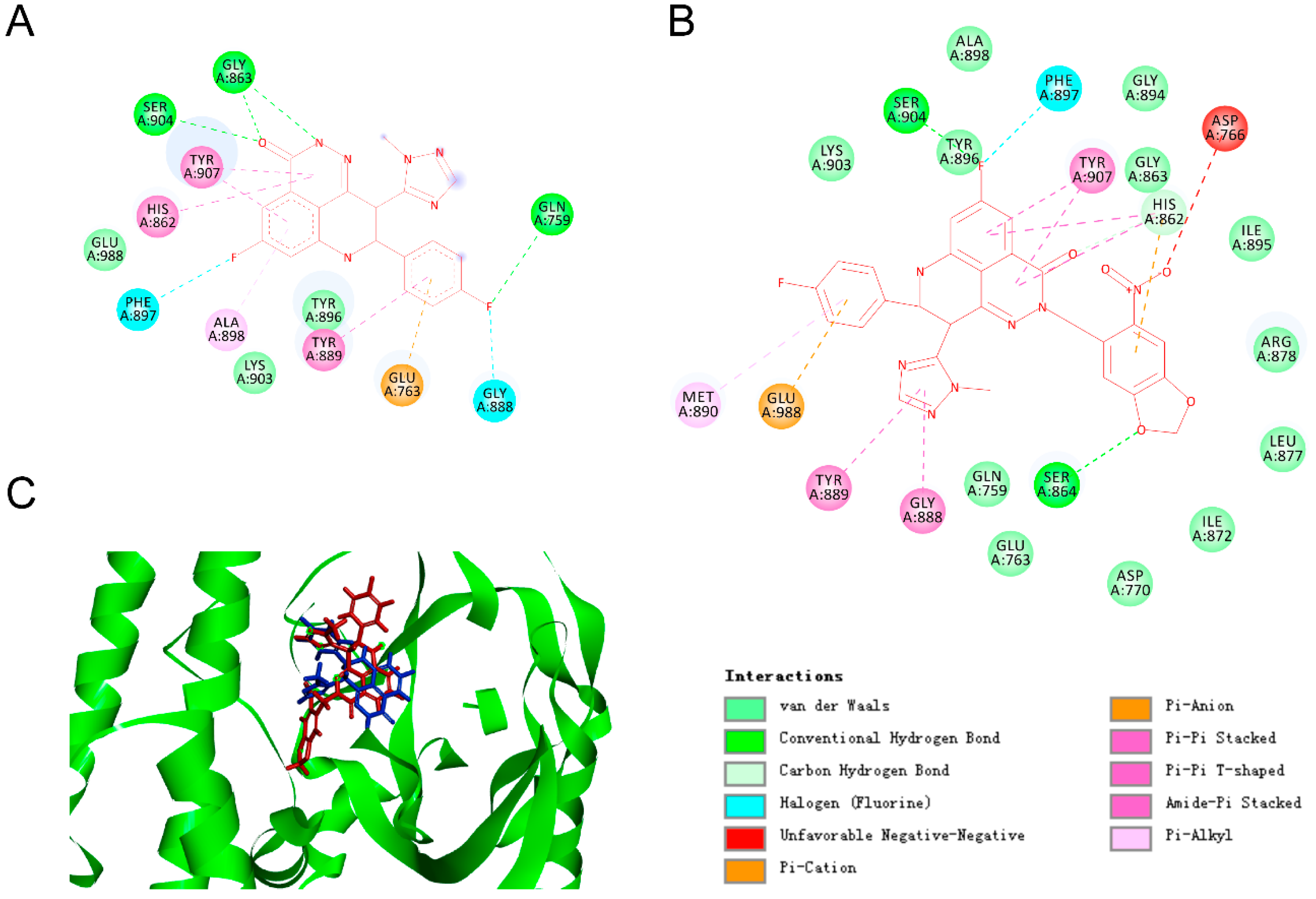
2.1. Molecular Modelling
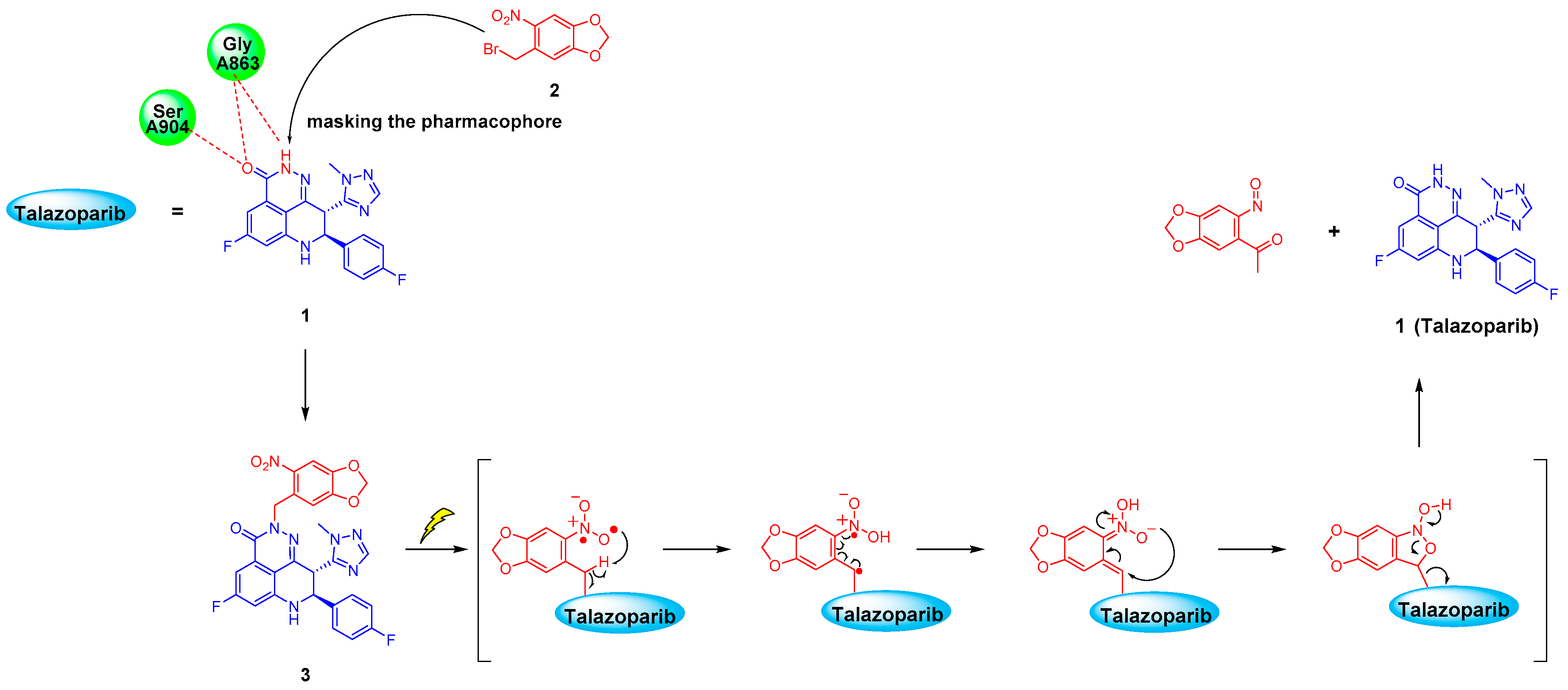
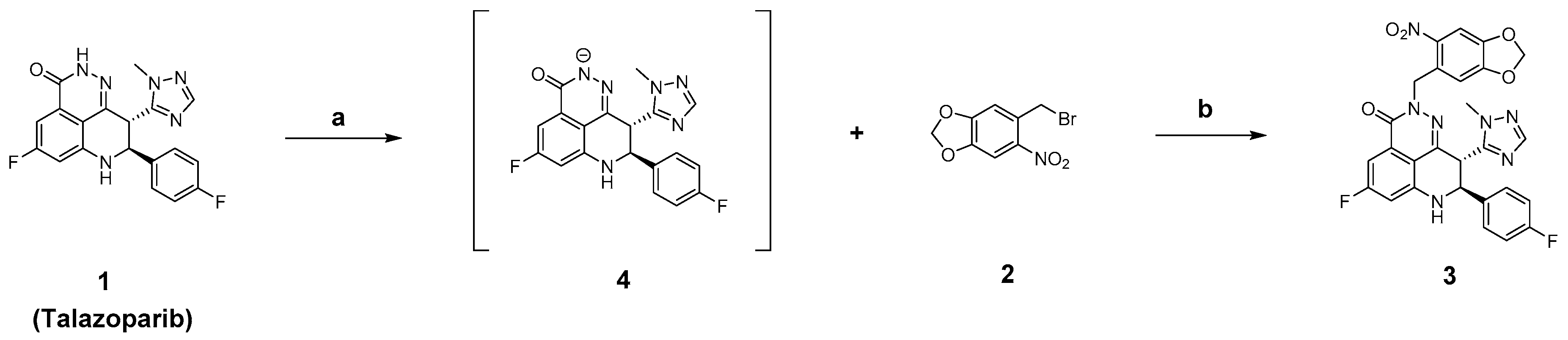
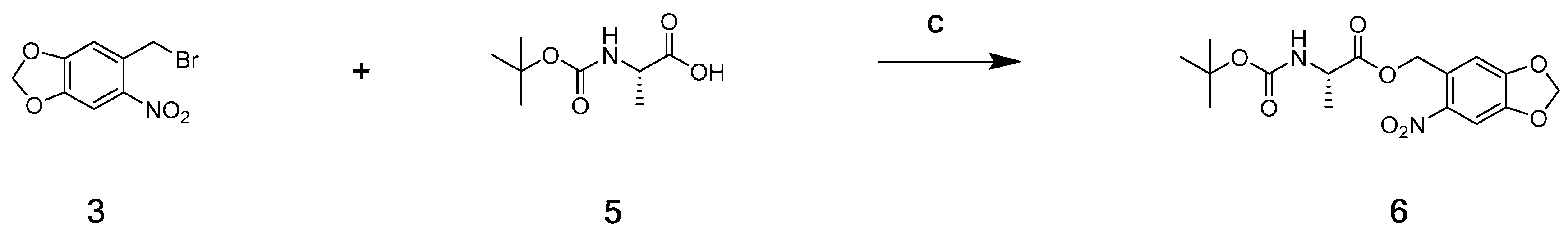
2.2. Chemistry
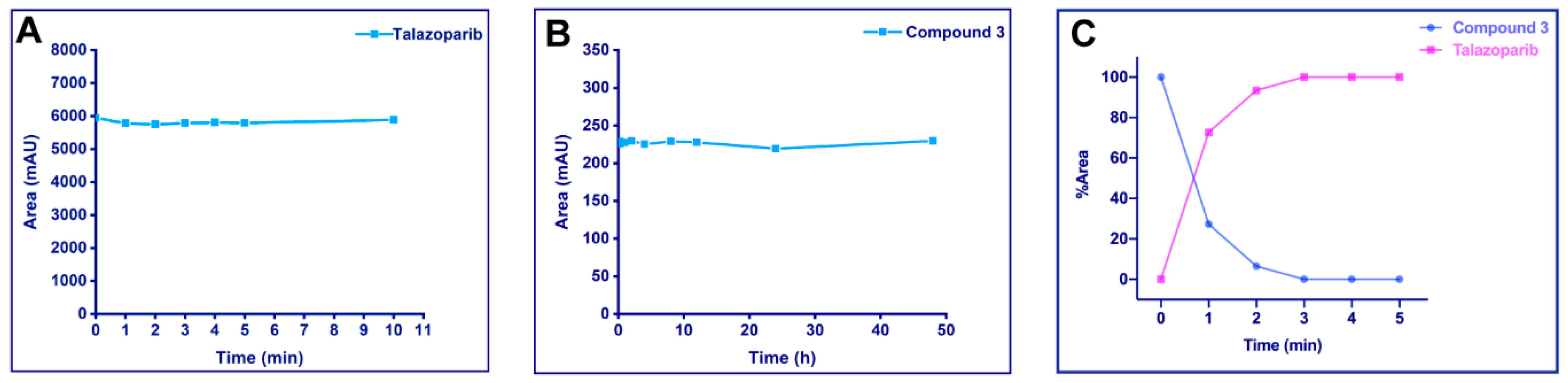
2.3. Stability Assays and UV Cleavage Test In Vitro
2.3.1. UV Stability of Talazoparib
2.3.2. Stability of the Photoactivatable Prodrug in Phosphate-Buffered Saline
2.3.3. UV Release of the Photoactivatable Prodrug
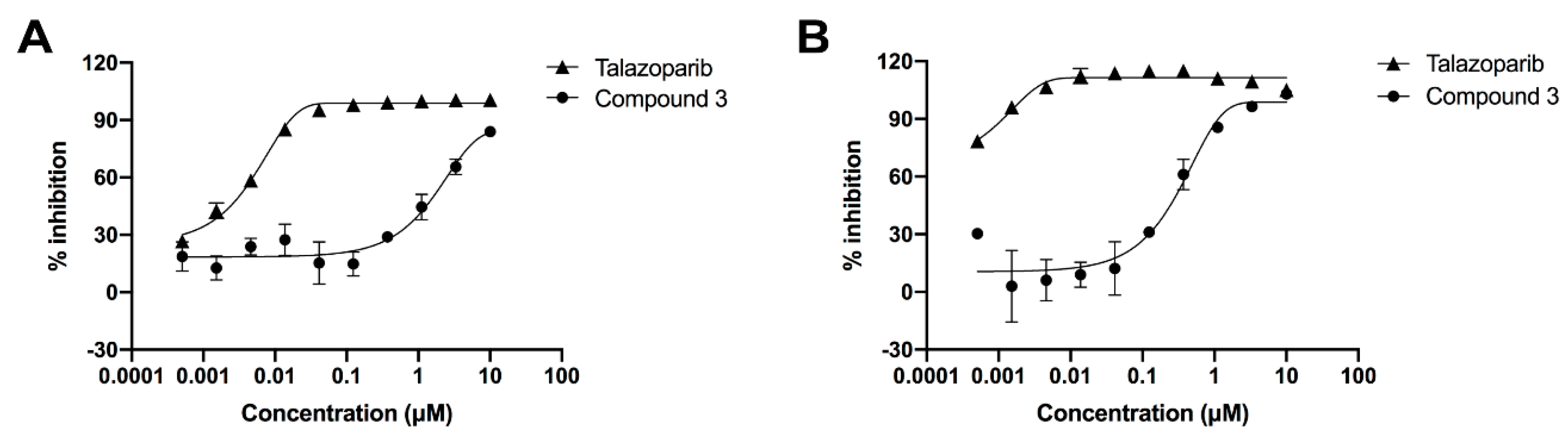
2.4. Enzymatic Experiments In Vitro
2.4.1. Inhibition of the PARP-1 Enzyme
2.4.2. PARylation Assay
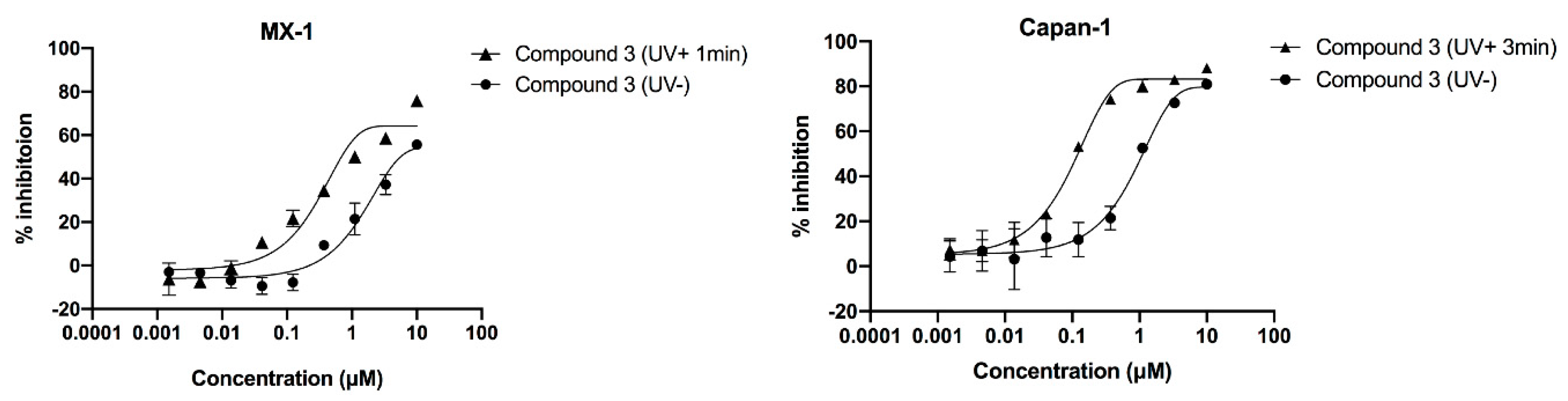
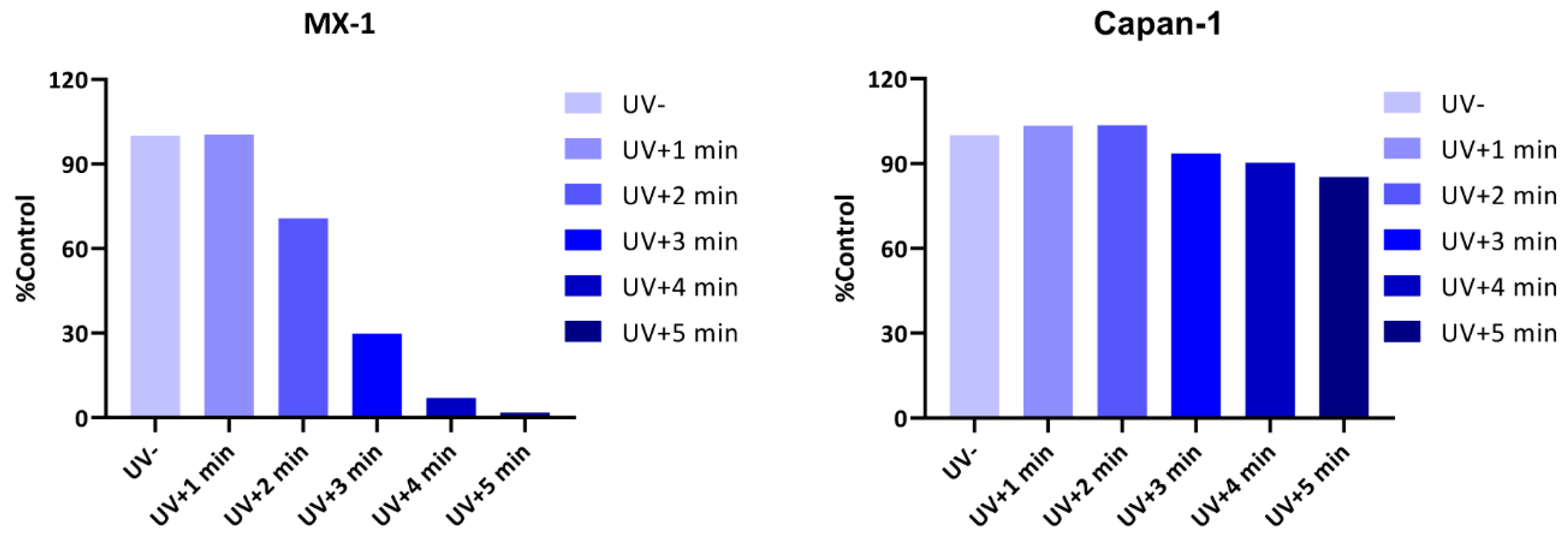
2.5. Compound’s Cytotoxicity Assay in the Absence and Presence of UV Irradiation
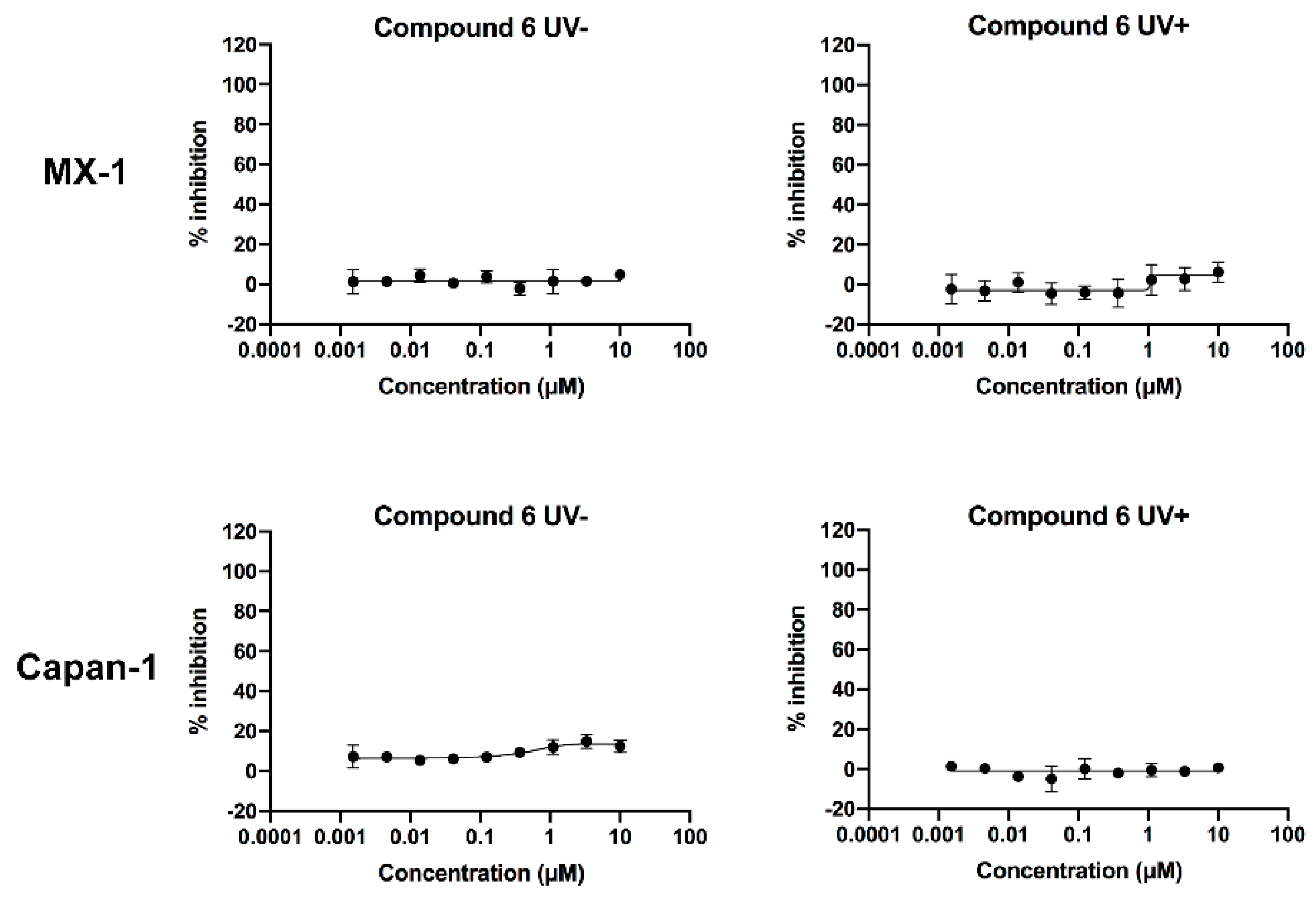
2.6. Verification of the Toxicity of Leaving Photoactivatable Protecting Groups
3. Materials and Methods
3.1. Molecular Docking
3.2. Chemistry
3.2.1. Synthesis of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2-((6-nitrobenzo[d][1,3]dioxol-5-yl)methyl)-2,7,8,9-tetrahydro-3H pyrido[4,3,2-de]phthalazin-3-one (3)
3.2.2. (6-Nitrobenzo[d][1,3]dioxol-5-yl)methyl (tert-butoxycarbonyl)-l-alaninate (6)
3.3. Talazoparib UV Stability Assays
3.4. Phosphate-Buffered Saline Stability of the Photoactivatable Prodrug
3.5. UV Cleavage Test for Photoactivatable Prodrug
3.6. PARP-1 Enzyme Inhibition Assay
3.7. PARylation Assay
3.8. Cell Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sulai, N.H.; Tan, A.R. Development of poly(ADP-ribose) polymerase inhibitors in the treatment of BRCA-mutated breast cancer. Clin. Adv. Hematol. Oncol. 2018, 16, 491–501. [Google Scholar]
- Althaus, F.R.; Richter, C. ADP-ribosylation of proteins. Enzymology and biological significance. Mol. Biol. Biochem. Biophys. 1987, 37, 1–237. [Google Scholar] [PubMed]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Rhee, H.K.; Lim, S.Y.; Jung, M.J.; Kwon, Y.; Kim, M.H.; Choo, H.Y. Synthesis of isoquinolinone-based tetracycles as poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors. Bioorg. Med. Chem. 2009, 17, 7537–7541. [Google Scholar] [CrossRef]
- Liu, J.F.; Tolaney, S.M.; Birrer, M.; Fleming, G.F.; Buss, M.K.; Dahlberg, S.E.; Lee, H.; Whalen, C.; Tyburski, K.; Winer, E.; et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur. J. Cancer 2013, 49, 2972–2978. [Google Scholar] [CrossRef]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Vélez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A Small-Molecule Inhibitor of WEE1, AZD1775, Synergizes with Olaparib by Impairing Homologous Recombination and Enhancing DNA Damage and Apoptosis in Acute Leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Inc. TALZENNATM (Talazoparib) Capsules, for Oral Use: US Prescribing Information. Available online: http://www.fda.gov/ (accessed on 18 October 2018).
- US FDA. FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer [Media Release]. Available online: http://www.fda.gov/ (accessed on 16 October 2018).
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Pelliccioli, A.P.; Wirz, J. Photoremovable protecting groups: Reaction mechanisms and applications. Photochem. Photobiol. Sci. 2002, 1, 441–458. [Google Scholar] [CrossRef]
- Mayer, G.; Heckel, A. Biologically active molecules with a “light switch”. Angew. Chem. Int. Ed. 2006, 45, 4900–4921. [Google Scholar] [CrossRef]
- Ellis-Davies, G.C. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–628. [Google Scholar] [CrossRef]
- Kaplan, J.H.; Forbush, B.; Hoffman, J.F. Rapid photolytic release of adenosine 5’-triphosphate from a protected analog: Utilization by the sodium:potassium pump of human red blood cell ghosts. Biochemistry 1978, 17, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Engels, J.; Schlaeger, E.J. Synthesis, structure, and reactivity of adenosine cyclic 3′,5′-phosphate benzyl triesters. J. Med. Chem. 1977, 20, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, B.; Horbert, R.; Döbber, A.; Kuhl, L.; Peifer, C. Photoactivatable Caged Prodrugs of VEGFR-2 Kinase Inhibitors. Molecules 2016, 21, 570. [Google Scholar] [CrossRef] [PubMed]
- Horbert, R.; Pinchuk, B.; Davies, P.; Alessi, D.; Peifer, C. Photoactivatable Prodrugs of Antimelanoma Agent Vemurafenib. ACS Chem. Biol. 2015, 10, 2099–2107. [Google Scholar] [CrossRef]
- Shin, W.S.; Han, J.; Kumar, R.; Lee, G.G.; Sessler, J.L.; Kim, J.-H.; Kim, J.S. Programmed activation of cancer cell apoptosis: A tumor-targeted phototherapeutic topoisomerase I inhibitor. Sci. Rep. 2016, 6, 29018. [Google Scholar] [CrossRef]
- Ibsen, S.; Zahavy, E.; Wrasdilo, W.; Berns, M.; Chan, M.; Esener, S. A Novel Doxorubicin Prodrug with Controllable Photolysis Activation for Cancer Chemotherapy. Pharm. Res. 2010, 27, 1848–1860. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ananthaswamy, H.N. Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol. 2004, 195, 298–308. [Google Scholar] [CrossRef]
- Bliman, D.; Nilsson, J.R.; Kettunen, P.; Andréasson, J.; Grøtli, M. A Caged Ret Kinase Inhibitor and its Effect on Motoneuron Development in Zebrafish Embryos. Sci. Rep. 2015, 5, 13109. [Google Scholar] [CrossRef]
- Aoyagi-Scharber, M.; Gardberg, A.S.; Yip, B.K.; Wang, B.; Shen, Y.; Fitzpatrick, P.A. Structural basis for the inhibition of poly(ADP-ribose) polymerases 1 and 2 by BMN 673, a potent inhibitor derived from dihydropyridophthalazinone. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1143–1149. [Google Scholar] [CrossRef]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-te trahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar]
- Jones, P.; Altamura, S.; Boueres, J.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compound | Inhibition of PARP-1 Enzyme |
|---|---|
| IC50 (μM) | |
| Talazoparib | 0.005 |
| 3 | 1.919 |
| Test Compound | Inhibit Polymerization of PAR |
|---|---|
| IC50 (μM) | |
| Talazoparib | <0.0005 |
| 3 | 0.329 |
| Test Compound | MX-1 Cell (UV + 1 min) IC50 (μM) | Capan-1 Cell (UV + 3 min IC50 (μM) |
|---|---|---|
| Talazoparib (UV-) | 0.015 | 0.003 |
| 3 (UV-) | 1.873 | 0.863 |
| 3 (UV+) | 0.577 | 0.092 |
| Test Compound | MX-1 Cell IC50 (μM) | Capan-1 Cell IC50 (μM) |
|---|---|---|
| 6 (UV-) | >10 | >10 |
| 6 (UV+) | >10 | >10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xiao, D.; Liu, L.; Xie, F.; Li, W.; Sun, W.; Yang, X.; Zhou, X. Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib. Molecules 2020, 25, 407. https://doi.org/10.3390/molecules25020407
Li J, Xiao D, Liu L, Xie F, Li W, Sun W, Yang X, Zhou X. Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib. Molecules. 2020; 25(2):407. https://doi.org/10.3390/molecules25020407
Chicago/Turabian StyleLi, Jiaguo, Dian Xiao, Lianqi Liu, Fei Xie, Wei Li, Wei Sun, Xiaohong Yang, and Xinbo Zhou. 2020. "Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib" Molecules 25, no. 2: 407. https://doi.org/10.3390/molecules25020407
APA StyleLi, J., Xiao, D., Liu, L., Xie, F., Li, W., Sun, W., Yang, X., & Zhou, X. (2020). Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib. Molecules, 25(2), 407. https://doi.org/10.3390/molecules25020407