Acrylamide Decreases Cell Viability, and Provides Oxidative Stress, DNA Damage, and Apoptosis in Human Colon Adenocarcinoma Cell Line Caco-2

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
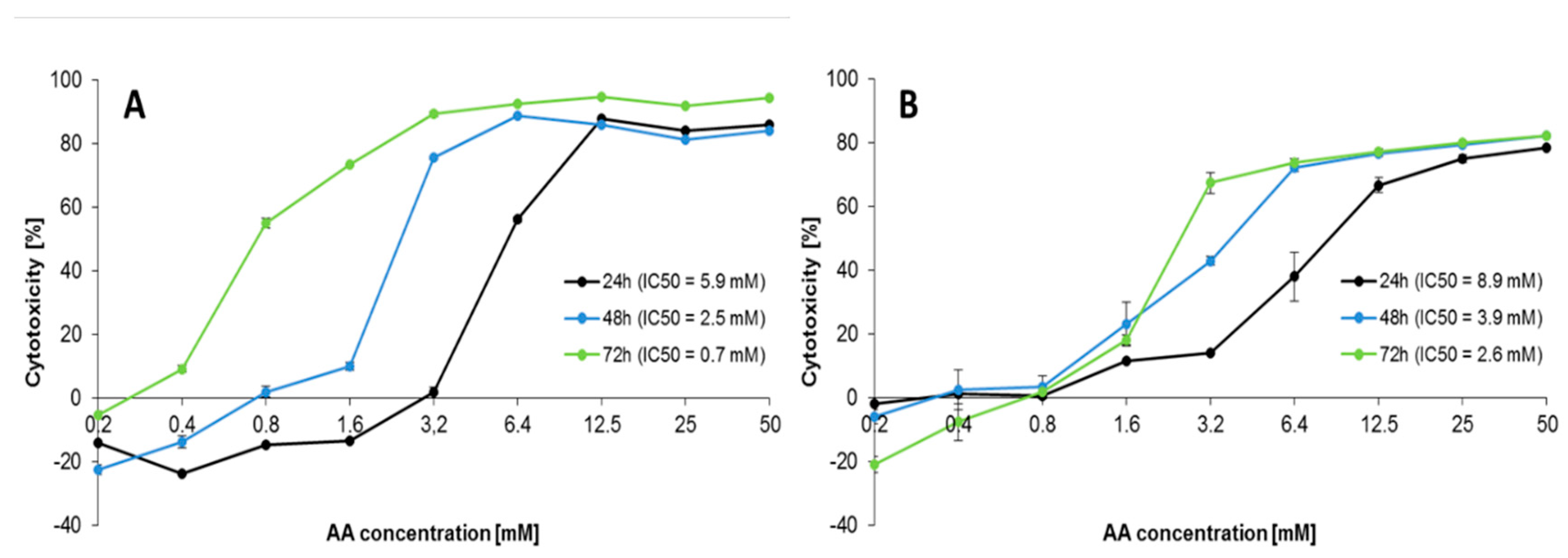
2.1. Effect of AA Treatment on Cell Proliferation
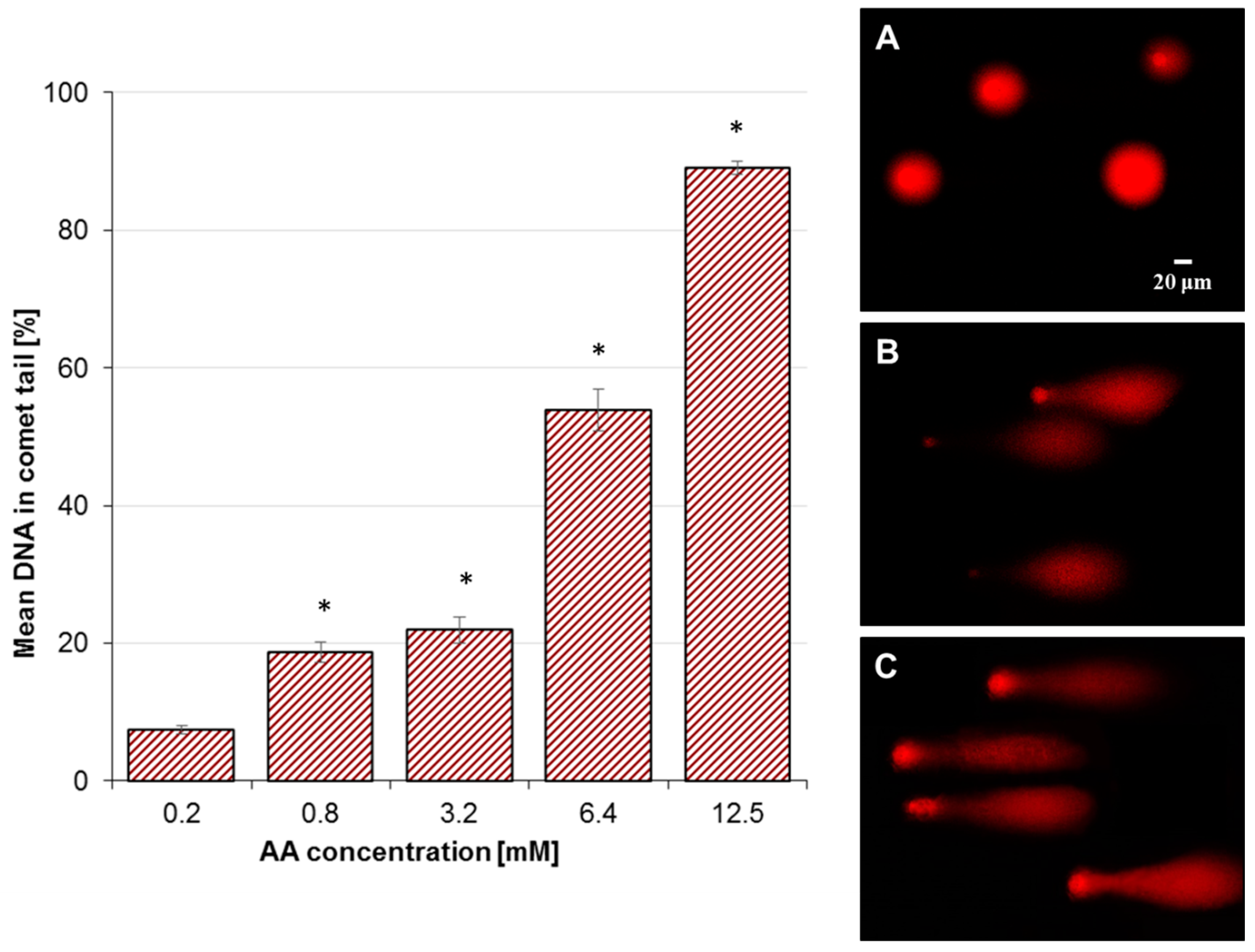
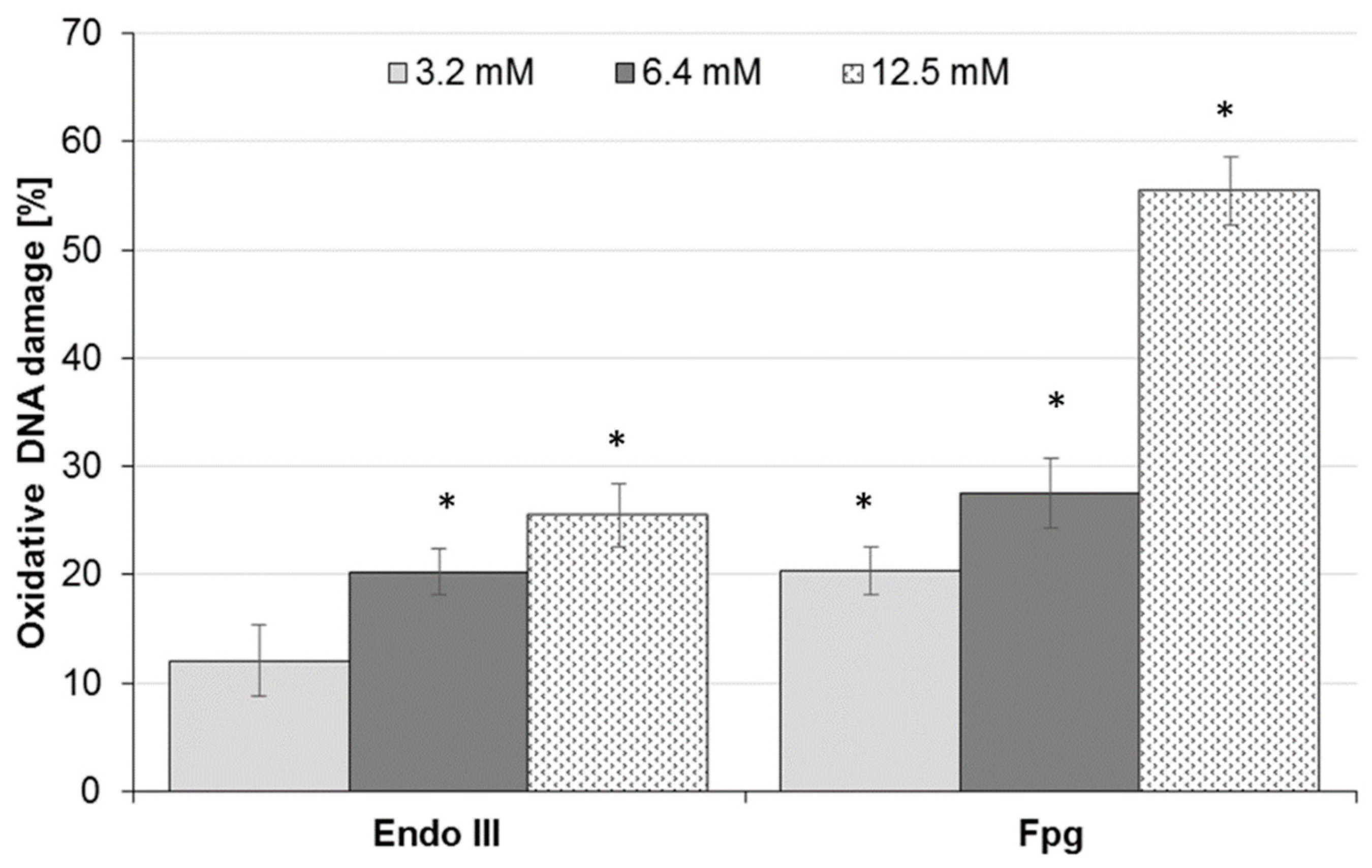
2.2. Effect of AA Treatment on Basal and Oxidative DNA Damage
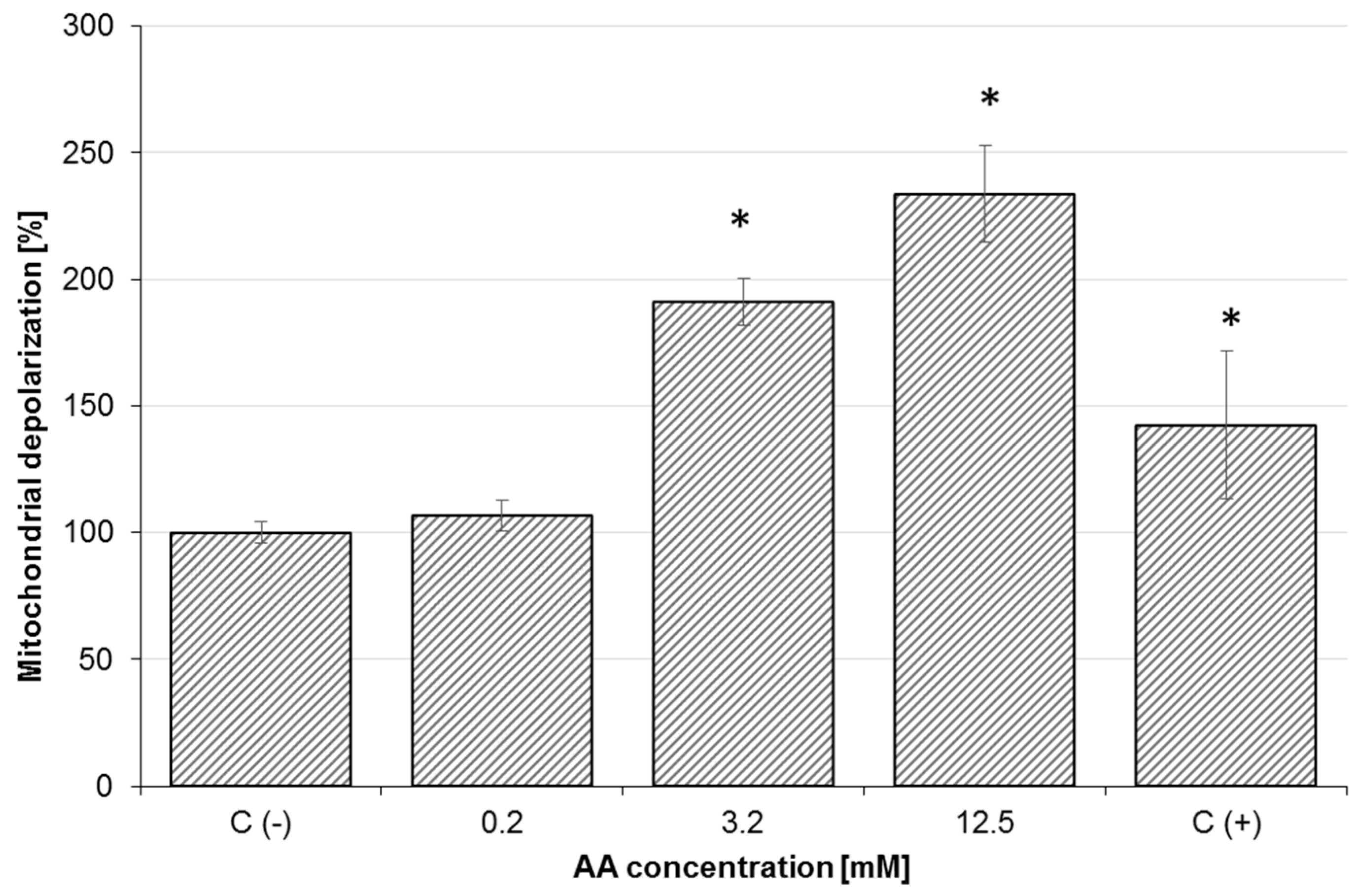
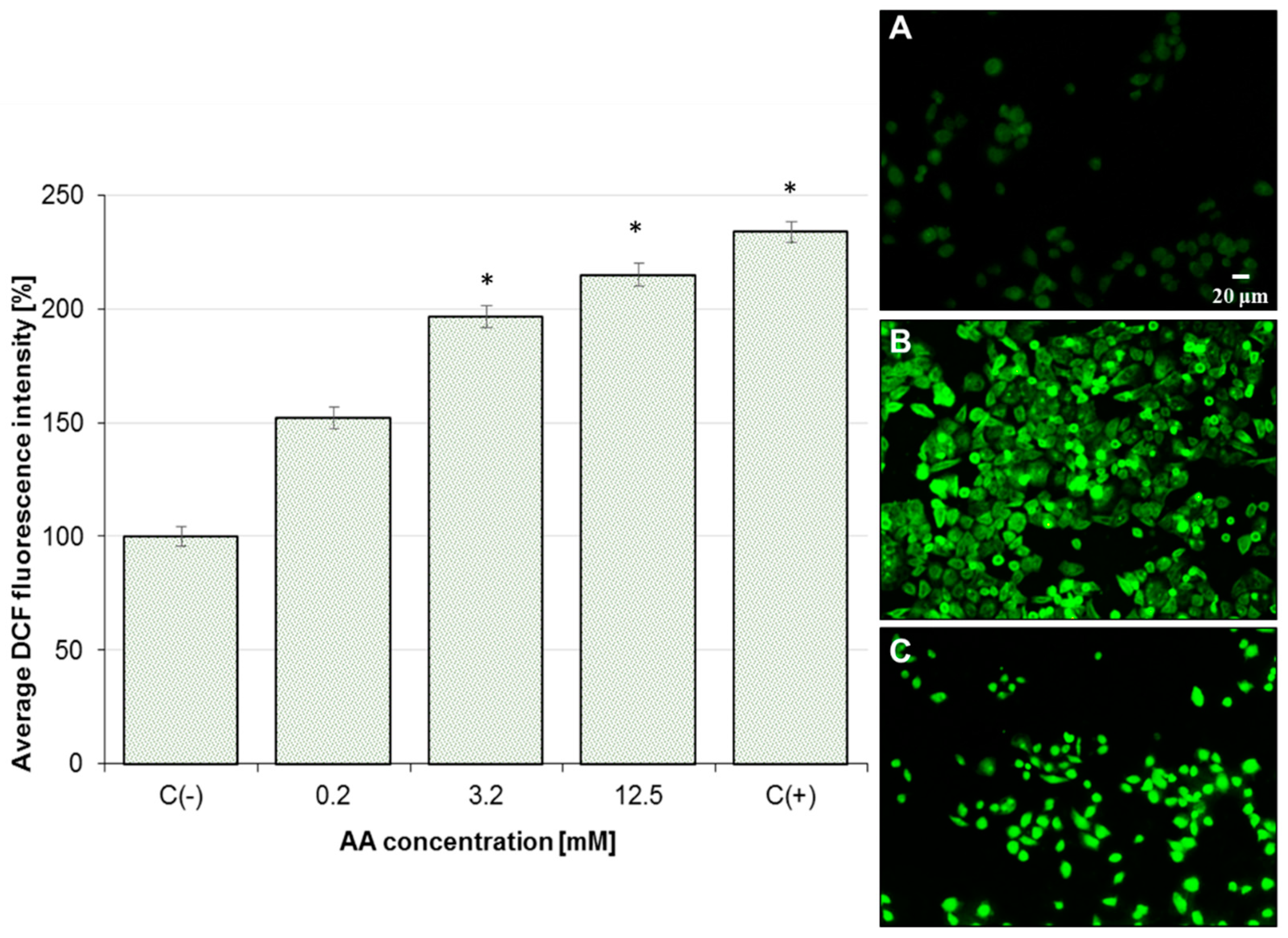
2.3. Effect of AA on Mitochondrial Membrane Potential (MMP) and Oxidative Stress
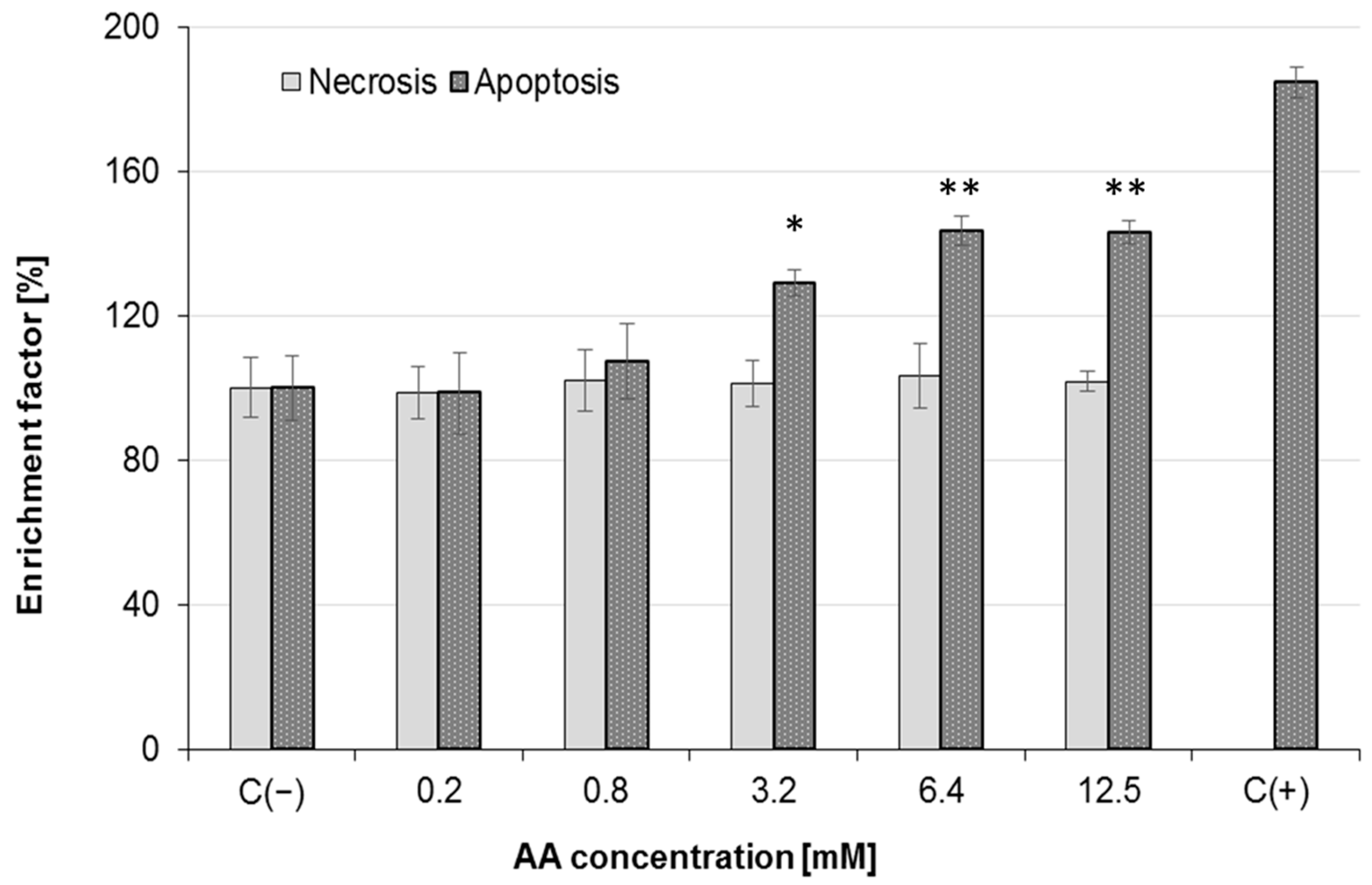
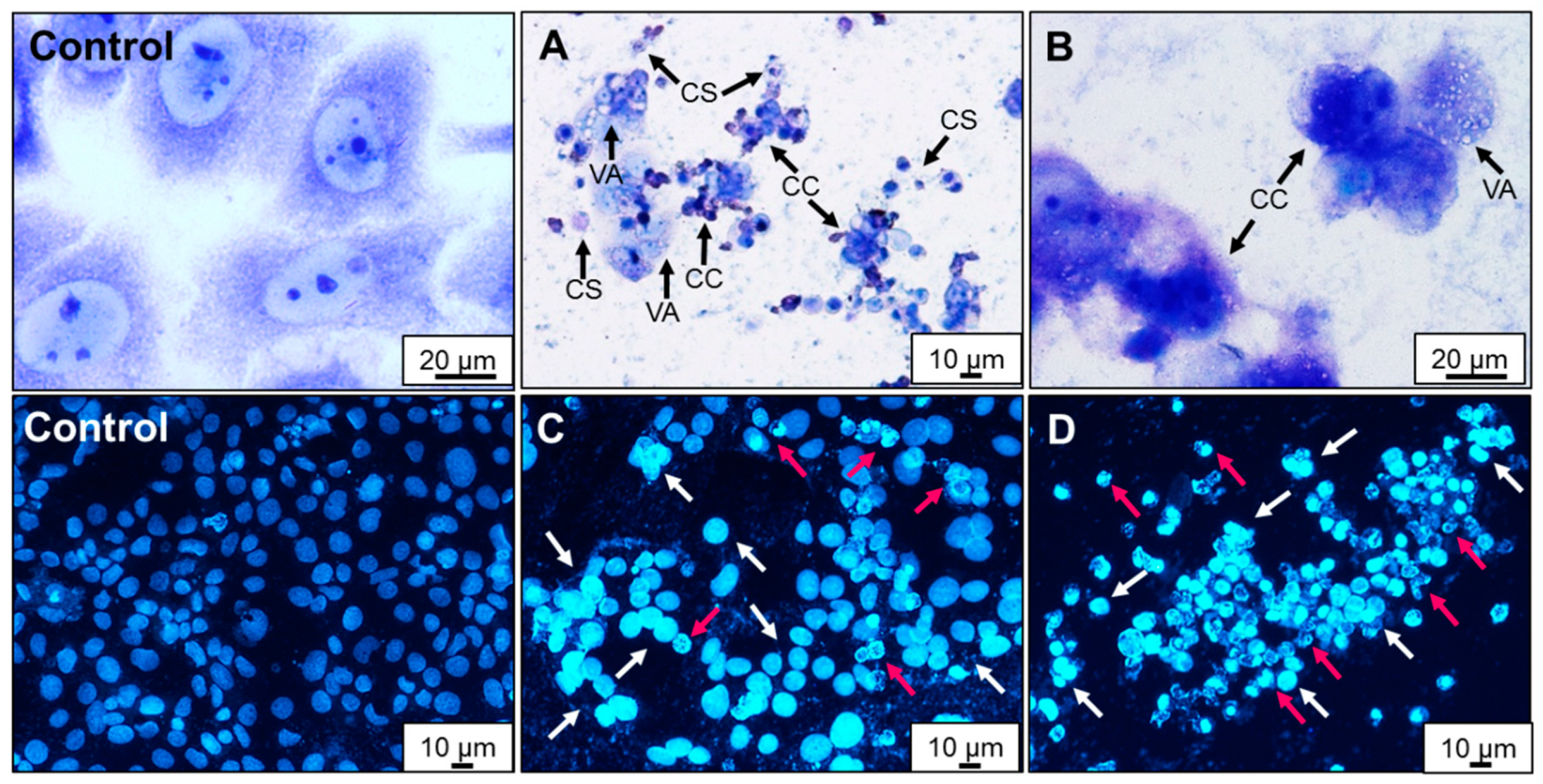
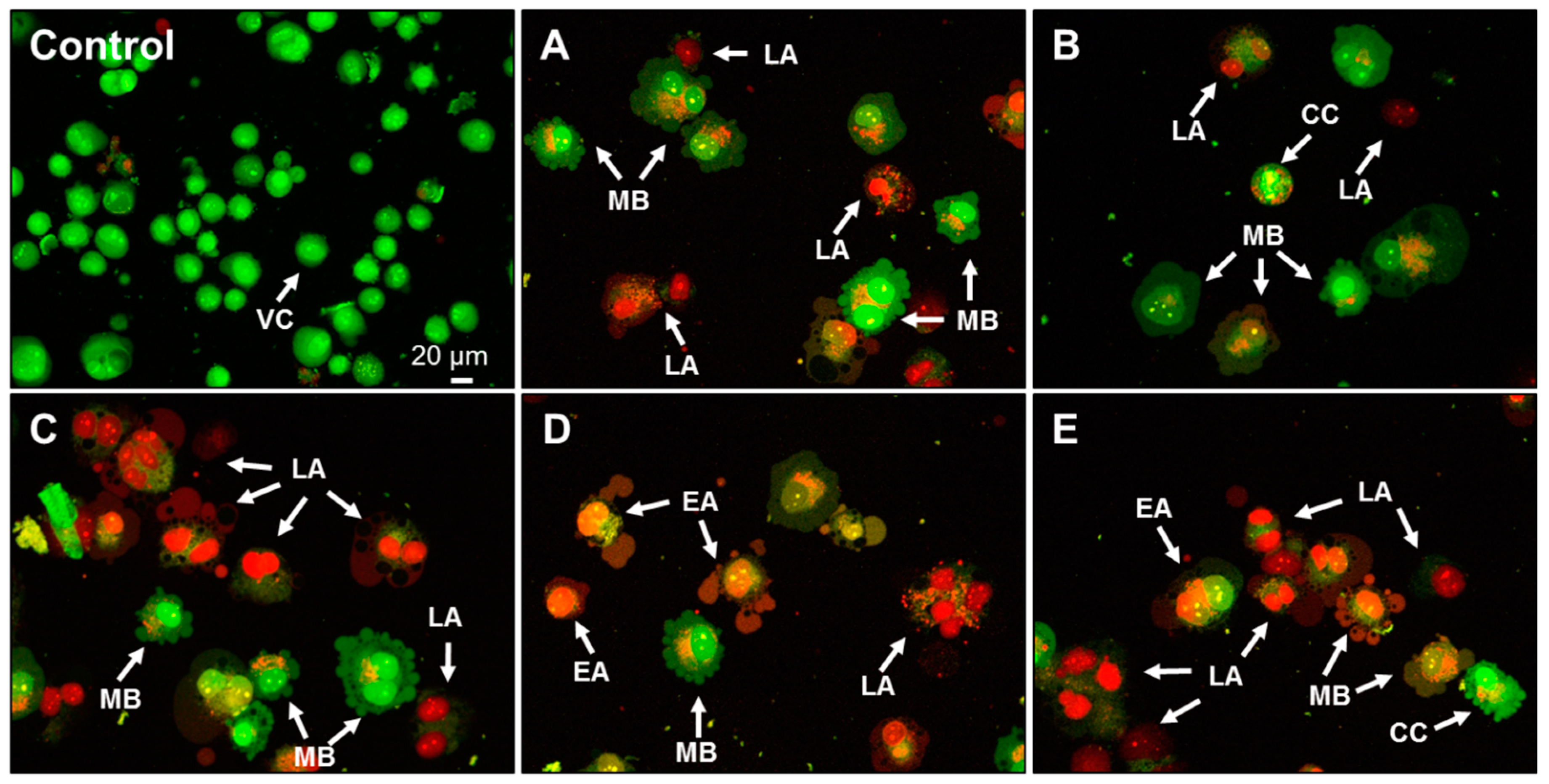
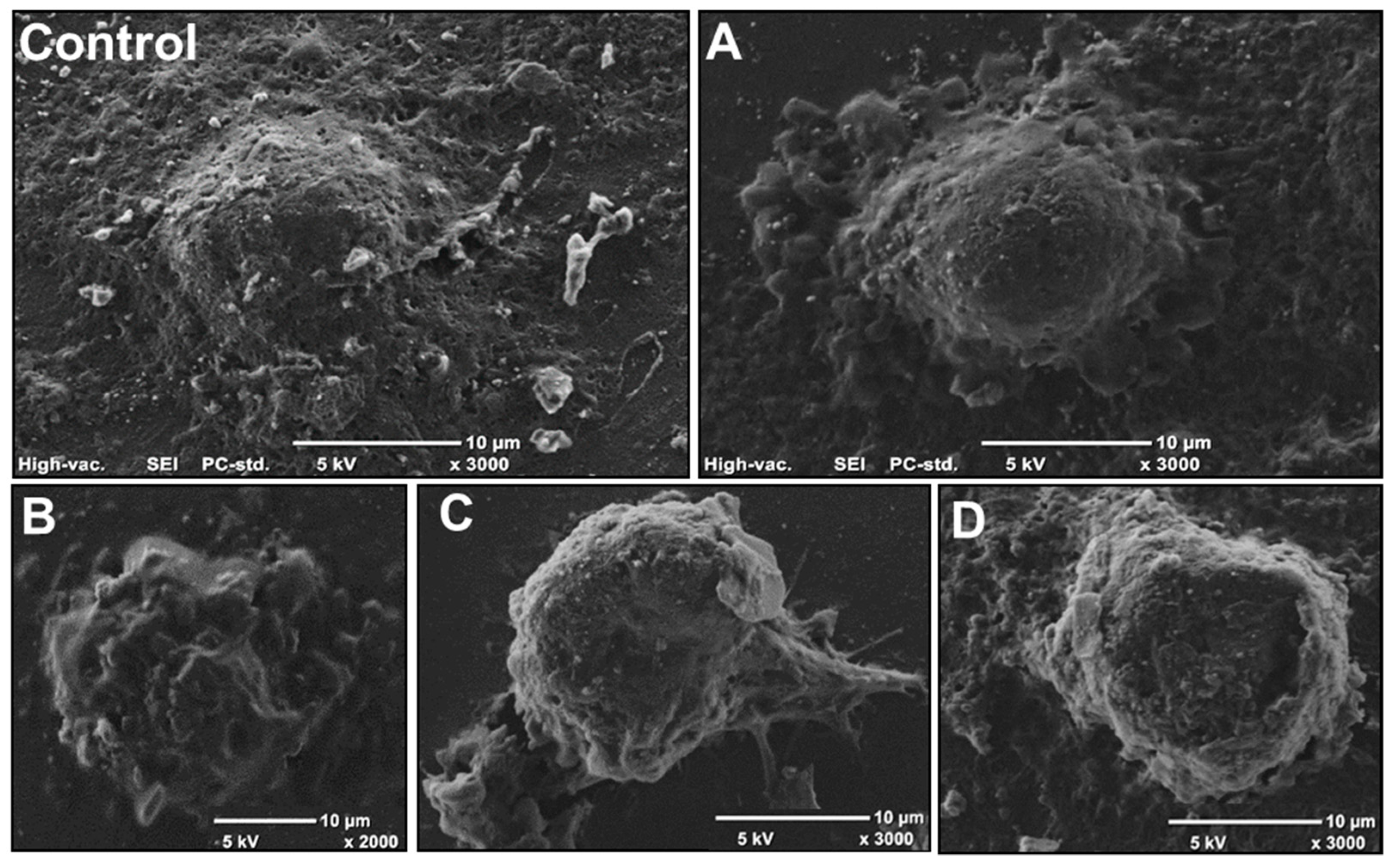
2.4. Effect of AA Treatment on Cell Death and Morphology
3. Materials and Methods
3.1. Chemicals and Culture Vessels
3.2. Caco-2 Cell Culture
3.3. MTT and PrestoBlue Assays
3.4. Cytotoxicity and Half Maximal Inhibitory Concentration (IC50)
3.5. Genotoxicity Testing (Comet Assay)
3.6. Oxidative DNA Damage
3.7. MMP Assay
3.8. ROS Generation
3.9. Apoptosis and Necrosis Detection
3.10. Giemsa/May–Grünwald Staining
3.11. DAPI Staining
3.12. AO/PI Double Staining
3.13. SEM
3.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartkiene, E.; Jakobsone, I.; Juodeikiene, G.; Vidmantiene, D.; Pugajeva, I.; Bartkevics, V. Study on the reduction of acrylamide in mixed rye bread by fermentation with bacteriocin-like inhibitory substances producing lactic acid bacteria in combination with Aspergillus niger glucoamylase. Food Control 2013, 30, 35–40. [Google Scholar] [CrossRef]
- Żyżelewicz, D.; Oracz, J.; Krysiak, W.; Budryn, G.; Nebesny, E. Effects of various roasting conditions on acrylamide, acrolein and polycyclic aromatic hydrocarbons content in cocoa bean and the derived chocolates. Dry. Technol. 2017, 35, 363–374. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; World Health Organization: Geneva, Switzerland, 1994; p. 560. [Google Scholar]
- Hogervorst, J.G.; Schouten, L.J.; Konings, E.J.; Goldbohm, R.A.; van den Brandt, P.A. A prospective study of dietary acrylamide intake and the risk of endometrial, ovarian, and breast cancer. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 2304–2313. [Google Scholar] [CrossRef] [PubMed]
- Hogervorst, J.G.; Schouten, L.J.; Konings, E.J.; Goldbohm, R.A.; van den Brandt, P.A. Dietary acrylamide intake and the risk of renal cell, bladder, and prostate cancer. Am. J. Clin. Nutr. 2008, 87, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Żyżelewicz, D.; Nebesny, E.; Oracz, J. Acrylamide—Formation, physicochemical and biological properties. Bromat. Chem. Toksykol. 2010, 3, 415–427. (In Polish) [Google Scholar]
- Hogervorst, J.G.; de Bruijn-Geraets, D.; Schouten, L.J.; van Engeland, M.; de Kok, T.M.; Goldbohm, R.A.; van den Brandt, P.A.; Weijenberg, M.P. Dietary acrylamide intake and the risk of colorectal cancer with specific mutations in KRAS and APC. Carcinogenesis 2014, 35, 1032–1038. [Google Scholar] [CrossRef]
- Duda-Chodak, A.; Wajda, Ł.; Tarko, T.; Sroka, P.; Satora, P. A review of the interactions between acrylamide, microorganisms and food components. Food Funct. 2016, 7, 1282–1295. [Google Scholar] [CrossRef]
- Puppel, N.; Tjaden, Z.; Fueller, F.; Marko, D. DNA strand breaking capacity of acrylamide and glycidamide in mammalian cells. Mutat. Res. 2005, 580, 71–80. [Google Scholar] [CrossRef]
- CAST–Council for Agricultural Science and Technology. Acrylamide in Food. 2006. Available online: http://www.cast-science.org/download.cfm?PublicationID=2914&File=f030b1a336fa49052cb068677cf12694a5a4 (accessed on 19 November 2019).
- Naruszewicz, M.; Zapolska-Downar, D.; Kośmider, A.; Nowicka, G.; Kozłowska-Wojciechowska, M.; Vikstrom, A.S.; Tornqvist, M. Chronic intake of potato chips in humans increases the production of reactive oxygen radicals by leucocytes and increases plasma C-reactive protein: A pilot study. Am. J. Clin. Nutr. 2009, 89, 773–777. [Google Scholar] [CrossRef]
- Huang, M.; Jiao, J.; Wang, J.; Xia, Z.; Zhang, Y. Characterization of acrylamide-induced oxidative stress and cardiovascular toxicity in zebrafish embryos. J. Hazard. Mater. 2018, 347, 451–460. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). Update on acrylamide levels in food from monitoring years 2007 to 2010. Eur. Food Safety Authority J. 2012, 10, 2938. [Google Scholar]
- FAO/WHO Expert Committee on Food Additives. Evaluations of the Joint FAO/WHO Expert Committee on Food Additives. 2011. Available online: http://apps.who.int/food-additives-contaminants-jecfa-database/chemical.aspx?chemID=5198 (accessed on 10 November 2019).
- Koszucka, A.; Nowak, A.; Nowak, I.; Motyl, I. Acrylamide in human diet, its metabolism, toxicity, inactivation and the associated European Union legal regulations in food industry. Crit. Rev. Food Sci. Nutr. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- NIH—U.S. National Library of Medicine. Acrylamide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/6579#section=UN-Classification (accessed on 15 January 2020).
- Jagerstad, M.; Skog, K. Genotoxicity of heat-processed foods. Mutat. Res. 2005, 574, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, L.; Shen, Y.; Su, H.; Li, Y.; Zhuang, J.; Zhang, L.; Zheng, X. Myricitrin inhibits acrylamide-mediated cytotoxicity in human Caco-2 cells by preventing oxidative stress. Biomed Res. Int. 2013, 2013, 724183. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ramiro, I.; Martín, M.Á.; Ramos, S.; Bravo, L.; Goya, L. Olive oil hydroxytyrosol reduces toxicity evoked by acrylamide in human Caco-2 cells by preventing oxidative stress. Toxicology 2011, 288, 43–48. [Google Scholar] [CrossRef]
- Qu, D.; Liu, C.; Jiang, M.; Feng, L.; Chen, Y.; Han, J. After in vitro digestion, jackfruit flake affords protection against acrylamide-induced oxidative damage. Molecules 2019, 24, 3322. [Google Scholar] [CrossRef]
- Xu, M.; McCanna, D.J.; Sivak, J.G. Use of the viability reagent PrestoBlue in comparison with alamarBlue and MTT to assess the viability of human corneal epithelial cells. J. Pharmacol. Toxicol. Methods 2015, 71, 1–7. [Google Scholar] [CrossRef]
- Sahinturk, V.; Kacar, S.; Vejselova, D.; Kutlu, H.M. Acrylamide exerts its cytotoxicity in NIH/3T3 fibroblast cells by apoptosis. Toxicol. Ind. Health. 2018, 34, 481–489. [Google Scholar] [CrossRef]
- Kacar, S.; Vejselova, D.; Kutlu, H.M.; Sahinturk, V. Acrylamide-derived cytotoxic, anti-proliferative, and apoptotic effects on A549 cells. Hum. Exp. Toxicol. 2018, 37, 468–474. [Google Scholar] [CrossRef]
- Kacar, S.; Sahinturk, V.; Kutlu, H.M. Effect of acrylamide on BEAS-2B normal human lung cells: Cytotoxic, oxidative, apoptotic and morphometric analysis. Acta Histochem. 2019, 121, 595–603. [Google Scholar] [CrossRef]
- Chen, J.-H.; Yang, C.-H.; Wang, Y.-S.; Lee, J.-G.; Chenga, C.-H.; Chou, C.-C. Acrylamide-induced mitochondria collapse and apoptosis in human astrocytoma cells. Food Chem. Toxicol. 2013, 51, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Attoff, K.; Kertika, D.; Lundqvist, J.; Oredsson, S.; Forsby, A. Acrylamide affects proliferation and differentiation of the neural progenitor cell line C17.2 and the neuroblastoma cell line SH-SY5Y. Toxicol. In Vitro 2016, 35, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, G.; Zou, C.; Liu, G.; Wu, W.; Yuan, T.; Liu, X. Acrylamide induces mitochondrial dysfunction and apoptosis in BV-2 microglial cells. Free Radic. Biol. Med. 2015, 84, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Mallepogu, V.; Jayasekhar Babu, P.; Doble, M.; Suman, B.; Nagalakshmamma, V.; Chalapathi, P.V.; Thyagaraju, K. Effects of acrylamide on cervical cancer (HeLa) cells proliferation and few marker enzymes. Austin J. Biotechnol. Bioeng. 2017, 4, 1087. [Google Scholar]
- Exon, J.H. A review of the toxicology of acrylamide. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 397–412. [Google Scholar] [CrossRef]
- Dearfield, K.L.; Douglas, G.R.; Ehling, U.H.; Moore, M.M.; Sega, G.A.; Brusick, D.J. Acrylamide: A review of its genotoxicity and an assessment of heritable genetic risk. Mutat. Res. 1995, 330, 71–99. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Jeffrey, D.; Shepard, K.; Chepelev, N.; Friedman, M.; Marroni, D.; Recio, L. Differential genotoxicity of acrylamide in the micronucleus and Pig-a gene mutation assays in F344 rats and B6C3F1 mice. Mutagenesis 2016, 31, 617–626. [Google Scholar] [CrossRef]
- Blasiak, J.; Gloc, E.; Wozniak, K.; Czechowska, A. Genotoxicity of acrylamide in human lymphocytes. Chem. Biol. Interact. 2004, 149, 137–149. [Google Scholar] [CrossRef]
- Jiang, L.; Cao, J.; An, Y.; Geng, C.; Qu, S.; Jiang, L.; Zhong, L. Genotoxicity of acrylamide in human hepatoma G2 (HepG2) cells. Toxicol. In Vitro 2007, 21, 1486–1492. [Google Scholar] [CrossRef]
- Recio, L.; Hobbs, C.; Caspary, W.; Witt, K.L. Dose-response assessment of four genotoxic chemicals in a combined mouse and rat micronucleus and comet assay protocol. J. Toxicol. Sci. 2010, 35, 149–162. [Google Scholar] [CrossRef]
- Pan, X.; Zhu, Z.; Lu, L.; Wang, D.; Lu, Q.; Yan, H. Melatonin attenuates oxidative damage induced by acrylamide in vitro and in vivo. Oxid. Med. Cell. Longevity 2015, 2015, 703709. [Google Scholar] [CrossRef]
- Shimamura, Y.; Iio, M.; Urahira, T.; Masuda, S. Inhibitory effects of Japanese horseradish (Wasabia japonica) on the formation and genotoxicity of a potent carcinogen, acrylamide. J. Sci. Food Agric. 2017, 97, 2419–2425. [Google Scholar] [CrossRef]
- El-Bohi, K.M.; Moustafa, G.G.; El-Sharkawi, N.I.; Sabik, L.M.E. Genotoxic effects of acrylamide in adult male albino rats liver. J. Am. Sci. 2011, 7, 1097–1108. [Google Scholar]
- Zamani, E.; Shaki, F.; Abedian Kenari, S.; Shokrzadeh, M. Acrylamide induces immunotoxicity through reactive oxygen species production and caspase-dependent apoptosis in mice splenocytes via the mitochondria-dependent signaling pathways. Biomed. Pharmacother. 2017, 94, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Seydi, E.; Rajabi, M.; Salimi, A.; Pourahmad, J. Involvement of mitochondrial-mediated caspase-3 activation and lysosomal labilization in acrylamide-induced liver toxicity. Toxicol. Environ. Chem. 2015, 97, 563–575. [Google Scholar] [CrossRef]
- Chen, W.; Su, H.; Xu, Y.; Jin, C. In vitro gastrointestinal digestion promotes the protective effect of blackberry extract against acrylamide-induced oxidative stress. Sci. Rep. 2017, 7, 40514. [Google Scholar] [CrossRef] [PubMed]
- Ghorbel, I.; Elwej, A.; Fendri, N.; Mnif, H.; Jamoussi, K.; Boudawara, T.; Grati Kamoun, Z.; Zeghal, N. Olive oil abrogates acrylamide induced nephrotoxicity by modulating biochemical and histological changes in rats. Ren. Fail. 2017, 39, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Erdemli, M.E.; Aksungur, Z.; Gul, M.; Yigitcan, B.; Bag, H.G.; Altinoz, E.; Turkoz, Y. The effects of acrylamide and vitamin E on kidneys in pregnancy: An experimental study. J. Matern. Fetal Neonatal. Med. 2019, 32, 3747–3756. [Google Scholar] [CrossRef]
- Koszucka, A.; Nowak, A. Thermal processing food-related toxicants: A Review. Crit. Rev. Food Sci. Nutr. 2019, 59, 3579–3596. [Google Scholar] [CrossRef]
- Pan, X.; Yan, D.; Wang, D.Y.; Wu, X.; Zhao, W.; Lu, Q.; Yan, H.W. Mitochondrion-mediated apoptosis induced by acrylamide is regulated by a balance between Nrf2 antioxidant and MAPK signaling pathways in PC12 cells. Mol. Neurobiol. 2016, 54, 4781–4794. [Google Scholar] [CrossRef]
- Pan, X.; Wu, X.; Yan, D.; Peng, C.; Rao, C.; Yan, H. Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: Involvement of the crosstalk between Nrf2 and NF-κB pathways regulated by MAPKs. Toxicol. Lett. 2018, 288, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, M.; Zou, F.; Bai, S.; Jiang, X.; Tian, L.; Ou, S.; Jiao, R.; Bai, W. Protection of cyanidin-3-O -glucoside against acrylamide- and glycidamide-induced reproductive toxicity in leydig cells. Food Chem. Toxicol. 2018, 119, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Baskić, D.; Popović, S.; Ristić, P.; Arsenijević, N.N. Analysis of cycloheximide-induced apoptosis in human leukocytes: Fluorescence microscopy using annexin V/propidium iodide versus acridin orange-ethidium bromide. Cell Biol. Int. 2006, 30, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, L.; Wang, H.; Chen, Q.; Wu, X.; Yan, X.; Chen, Y.; Xie, M. Protective effects of a Ganoderma atrum polysaccharide against acrylamide induced oxidative damage via a mitochondria mediated intrinsic apoptotic pathway in IEC-6 cells. Food Func. 2018, 9, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Sójka, M.; Klewicka, E.; Lipińska, L.; Klewicki, R.; Kołodziejczyk, K. Ellagitannins from Rubus idaeus L. Exert geno- and cytotoxic effects against human colon adenocarcinoma cell line Caco-2. J. Agric. Food Chem. 2017, 65, 2947–2955. [Google Scholar] [PubMed]
- Nowak, A.; Bakuła, T.; Matusiak, K.; Gałęcki, R.; Borowski, S.; Gutarowska, B. Odorous compounds from poultry manure induce DNA Damage, nuclear changes, and decrease cell membrane integrity in chicken liver hepatocellular carcinoma cells. Int. J. Environ. Res. Public Health 2017, 14, 933. [Google Scholar] [CrossRef]
- OECD Guidelines for the Testing of Chemicals, Section 4. Test No. 442D: In Vitro Skin Sensitisation ARE-Nrf2 Luciferase Test Method. Available online: https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-tg442d-508.pdf (accessed on 19 November 2019).
- Nowak, A.; Czyżowska, A.; Stańczyk, M. Protective activity of probiotic bacteria against 2-amino-3-methyl-3H-imidazo[4,5-f]quinoline (IQ) and 2-amino-1-methyl-6-phenyl-1H-imidazo[4,5-b]pyridine (PhIP)—An in vitro study. Food Addit. Contam. Part. A 2015, 32, 1927–1938. [Google Scholar] [CrossRef]
- Wu, D.; Yotnda, P. Production and detection of reactive oxygen species (ROS) in cancers. J. Vis. Exp. 2011, 57, e3357. [Google Scholar] [CrossRef]
- Osahor, A.; Deekonda, K.; Lee, C.W.; Sim, E.U.; Radu, A.; Narayanan, K. Rapid preparation of adherent mammalian cells for basic scanning electron microscopy (SEM) analysis. Anal. Biochem. 2017, 534, 46–48. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, A.; Zakłos-Szyda, M.; Żyżelewicz, D.; Koszucka, A.; Motyl, I. Acrylamide Decreases Cell Viability, and Provides Oxidative Stress, DNA Damage, and Apoptosis in Human Colon Adenocarcinoma Cell Line Caco-2. Molecules 2020, 25, 368. https://doi.org/10.3390/molecules25020368
Nowak A, Zakłos-Szyda M, Żyżelewicz D, Koszucka A, Motyl I. Acrylamide Decreases Cell Viability, and Provides Oxidative Stress, DNA Damage, and Apoptosis in Human Colon Adenocarcinoma Cell Line Caco-2. Molecules. 2020; 25(2):368. https://doi.org/10.3390/molecules25020368
Chicago/Turabian StyleNowak, Adriana, Małgorzata Zakłos-Szyda, Dorota Żyżelewicz, Agnieszka Koszucka, and Ilona Motyl. 2020. "Acrylamide Decreases Cell Viability, and Provides Oxidative Stress, DNA Damage, and Apoptosis in Human Colon Adenocarcinoma Cell Line Caco-2" Molecules 25, no. 2: 368. https://doi.org/10.3390/molecules25020368
APA StyleNowak, A., Zakłos-Szyda, M., Żyżelewicz, D., Koszucka, A., & Motyl, I. (2020). Acrylamide Decreases Cell Viability, and Provides Oxidative Stress, DNA Damage, and Apoptosis in Human Colon Adenocarcinoma Cell Line Caco-2. Molecules, 25(2), 368. https://doi.org/10.3390/molecules25020368