
The Structural and Magnetic Properties of FeII and CoII Complexes with 2-(furan-2-yl)-5-pyridin-2-yl-1,3,4-oxadiazole


Abstract
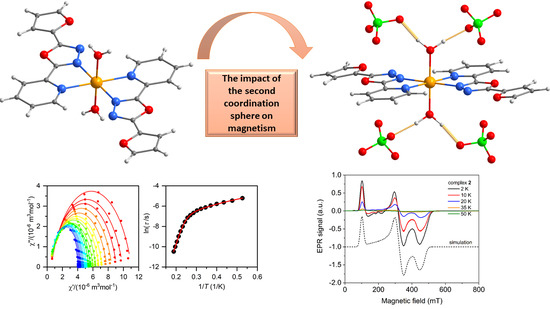
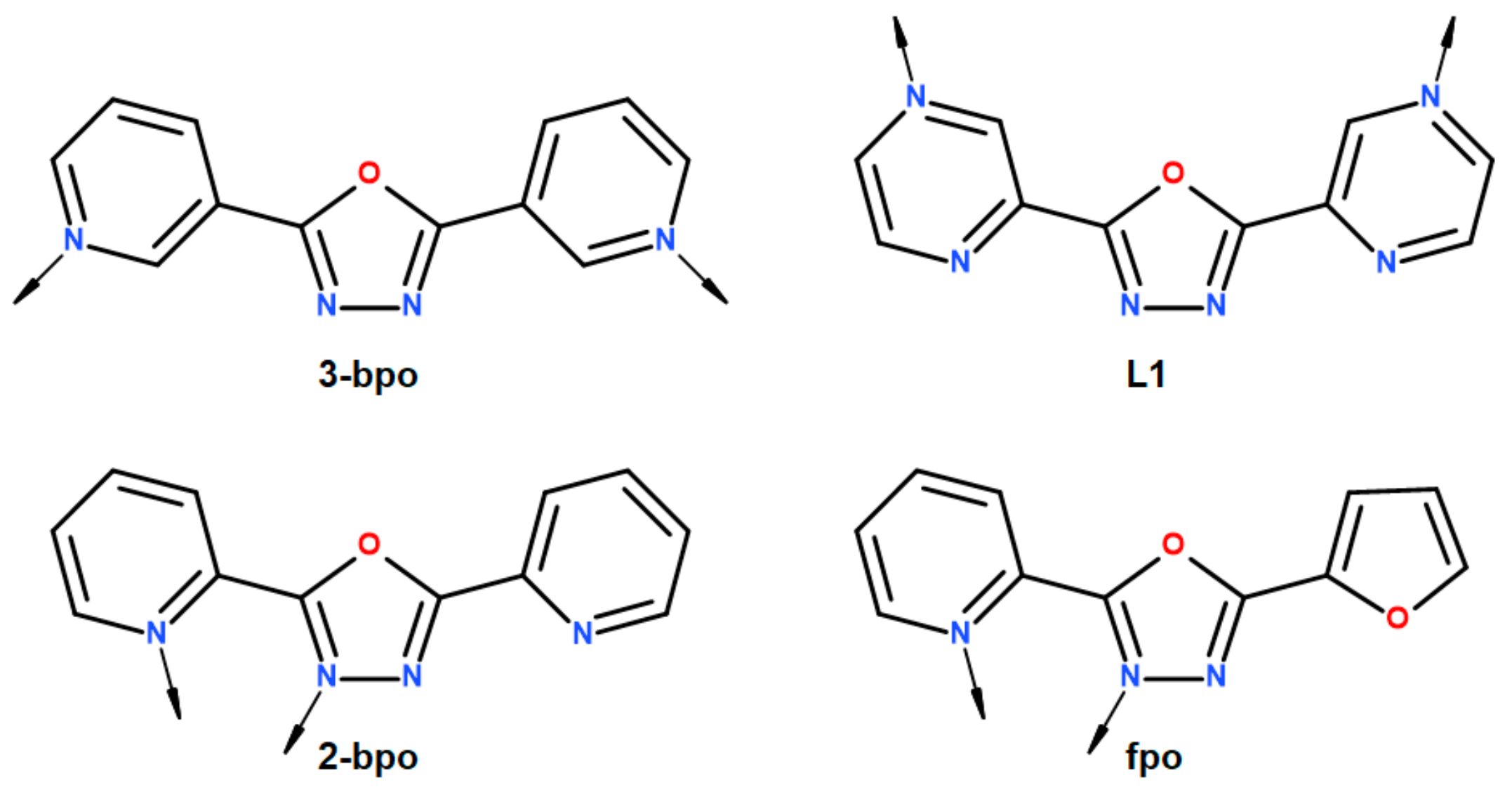
1. Introduction
2. Results and Discussion
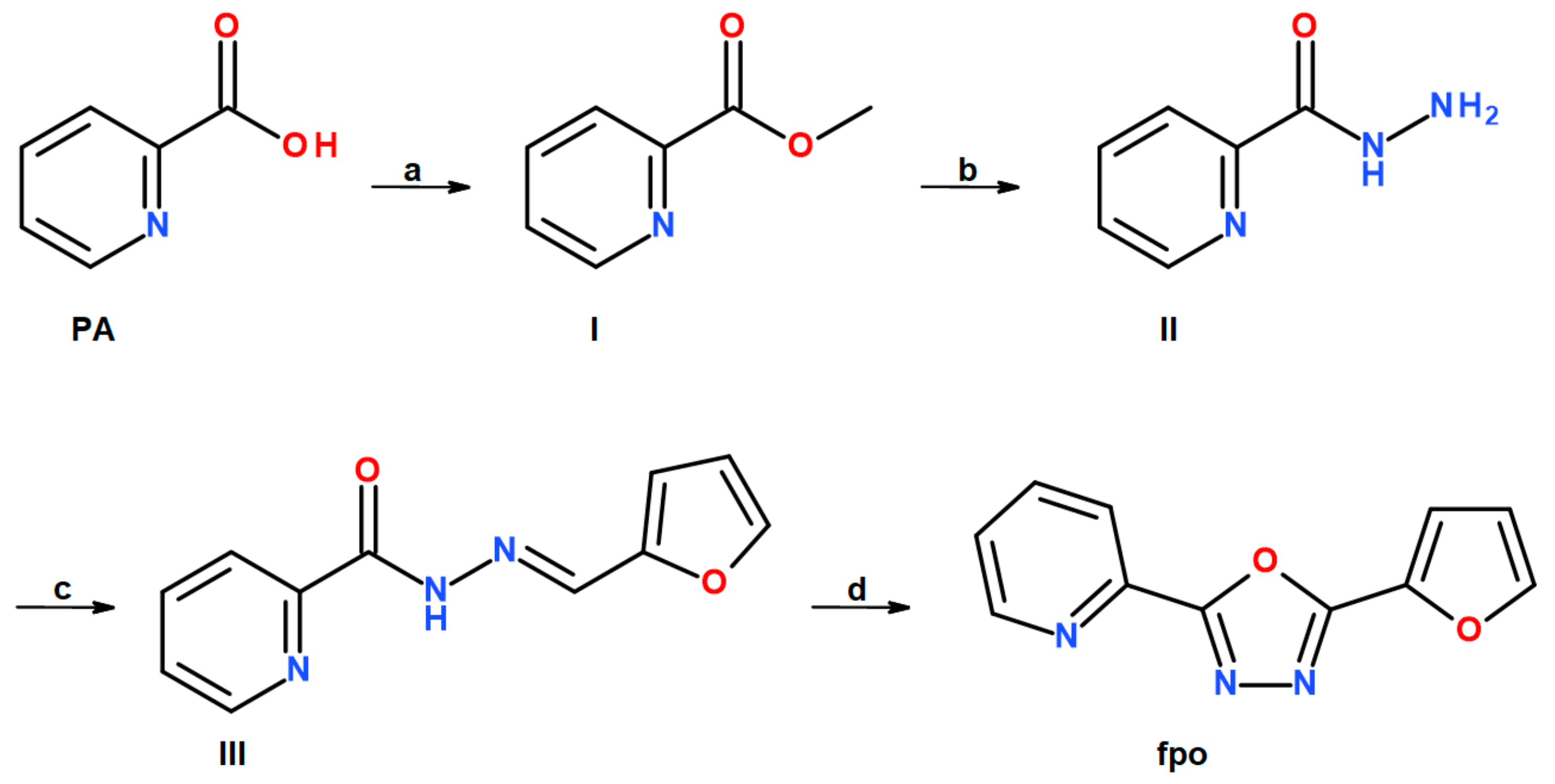
2.1. Synthesis
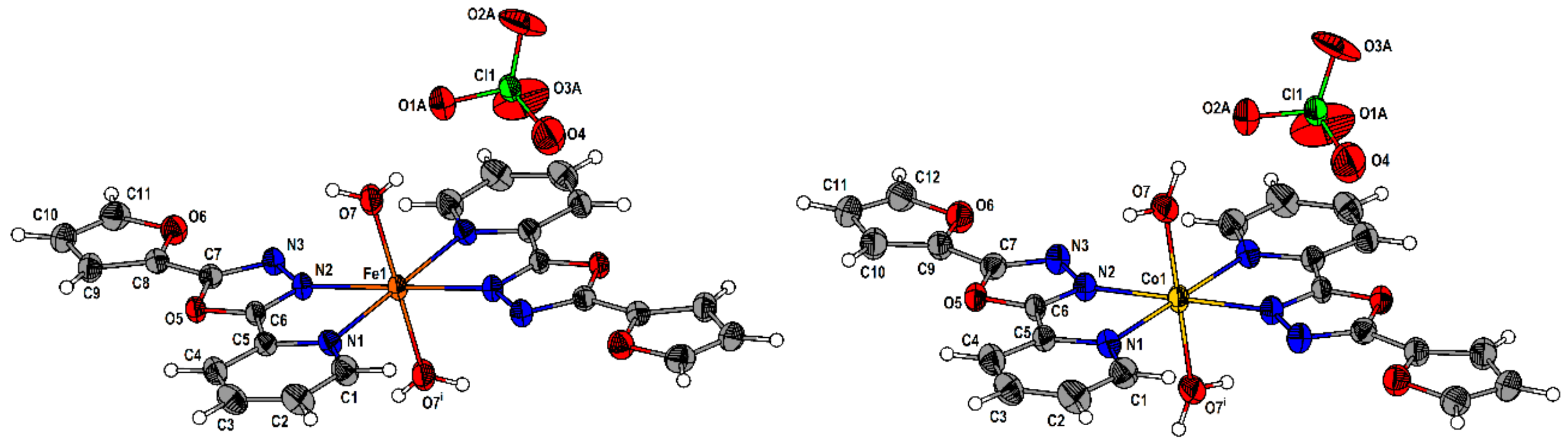
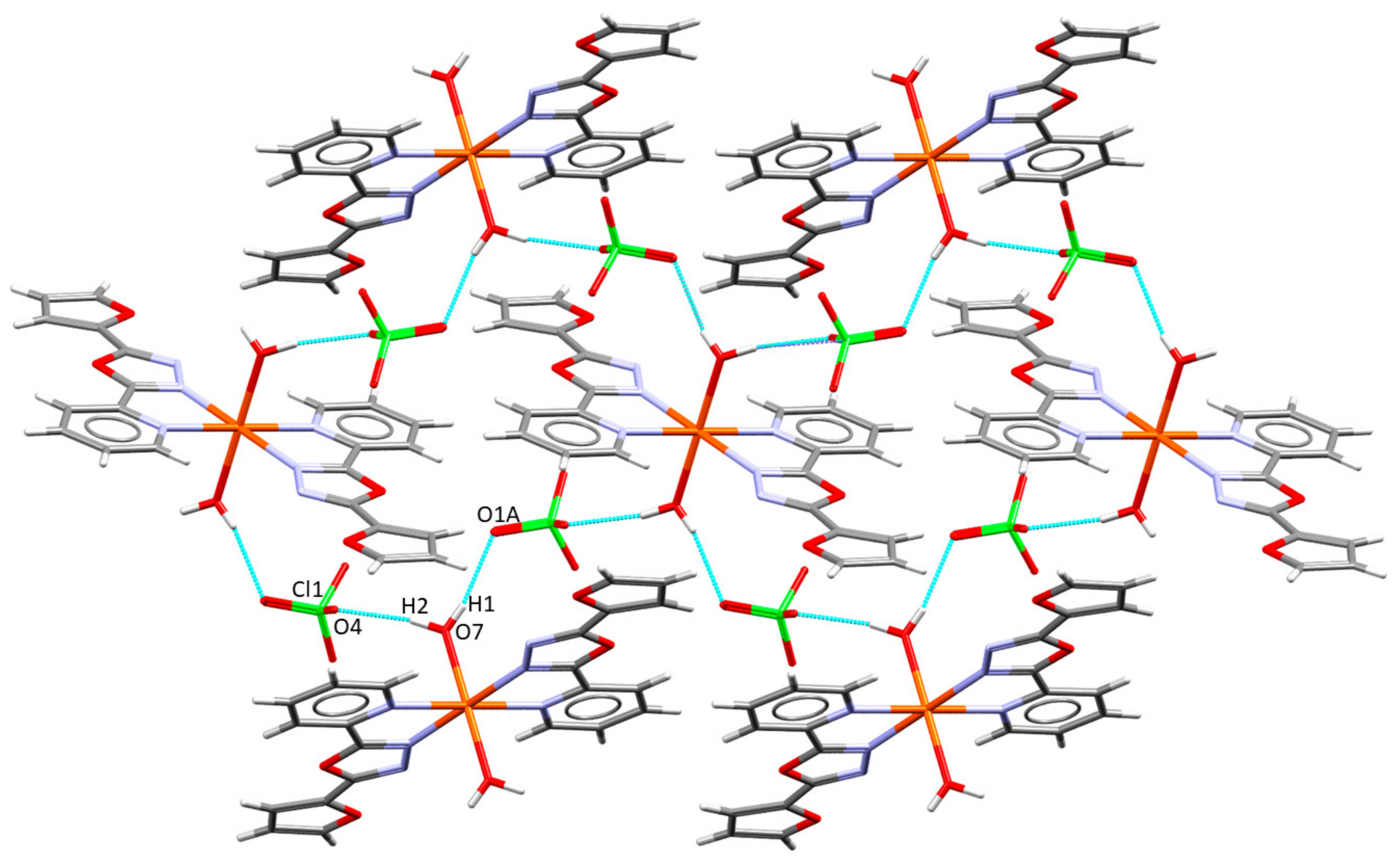
2.2. Description of the Molecular and Crystal Structure
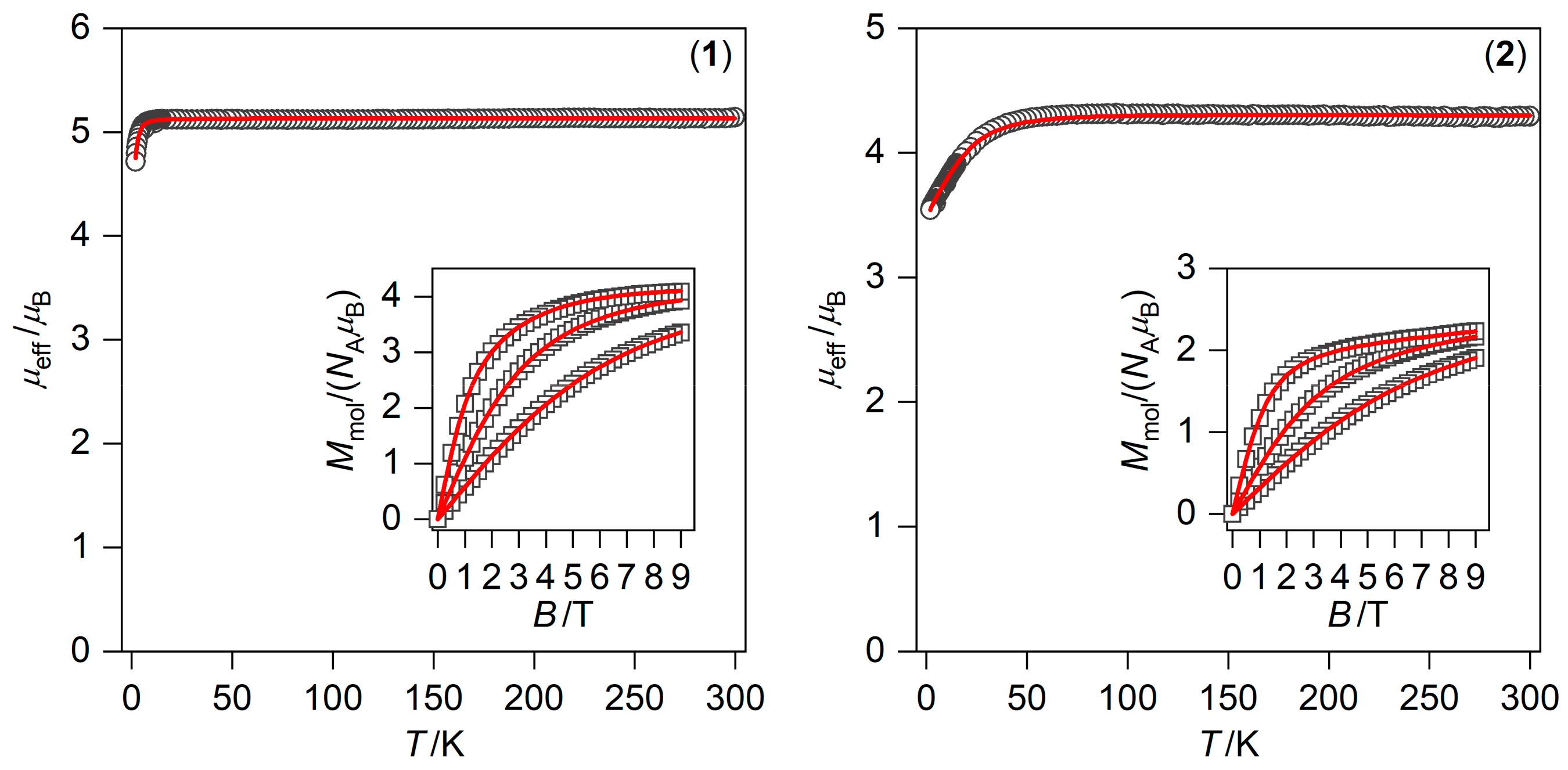
2.3. Static and Dynamic Magnetic Properties
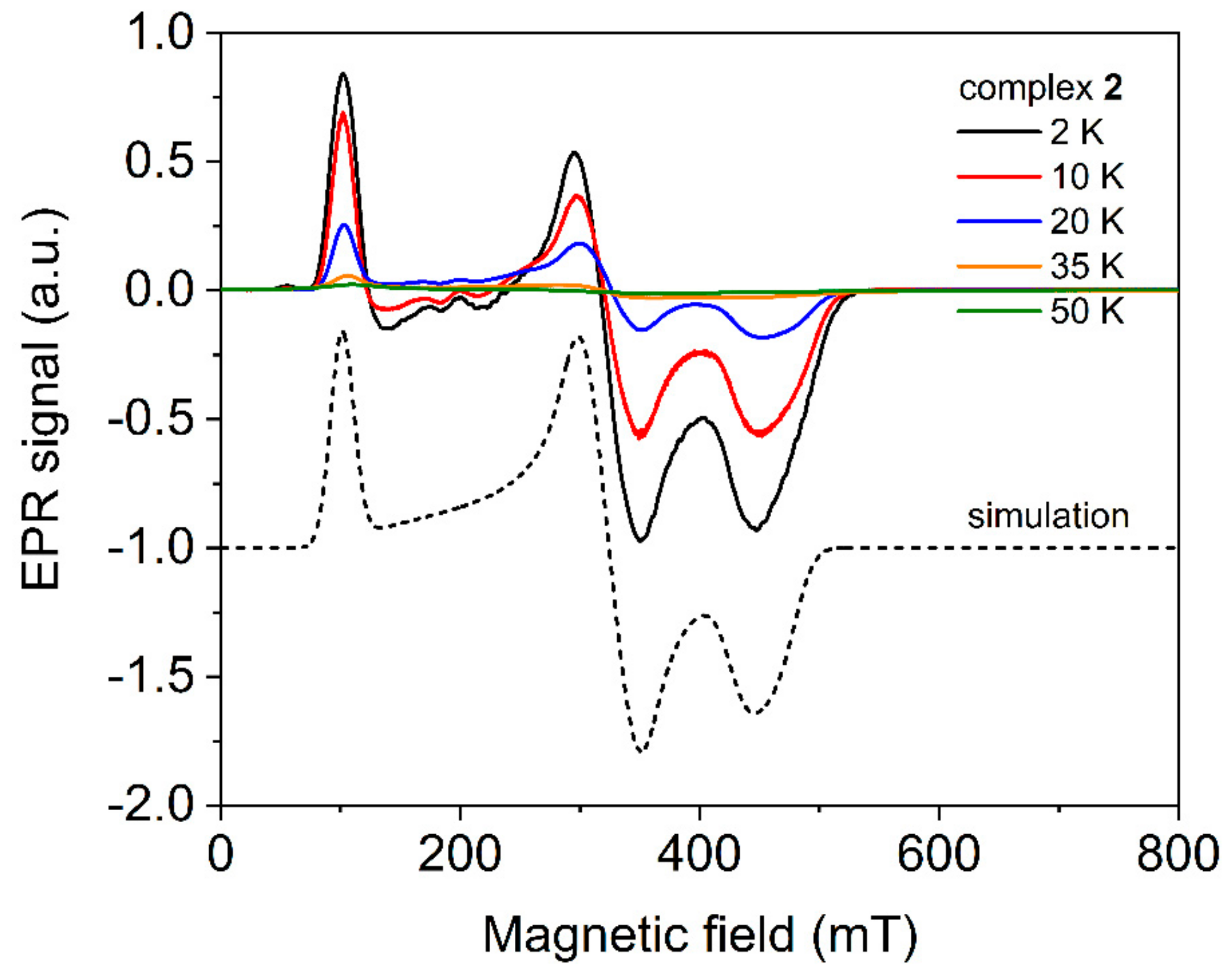
2.4. Electron Paramagnetic Spectroscopy
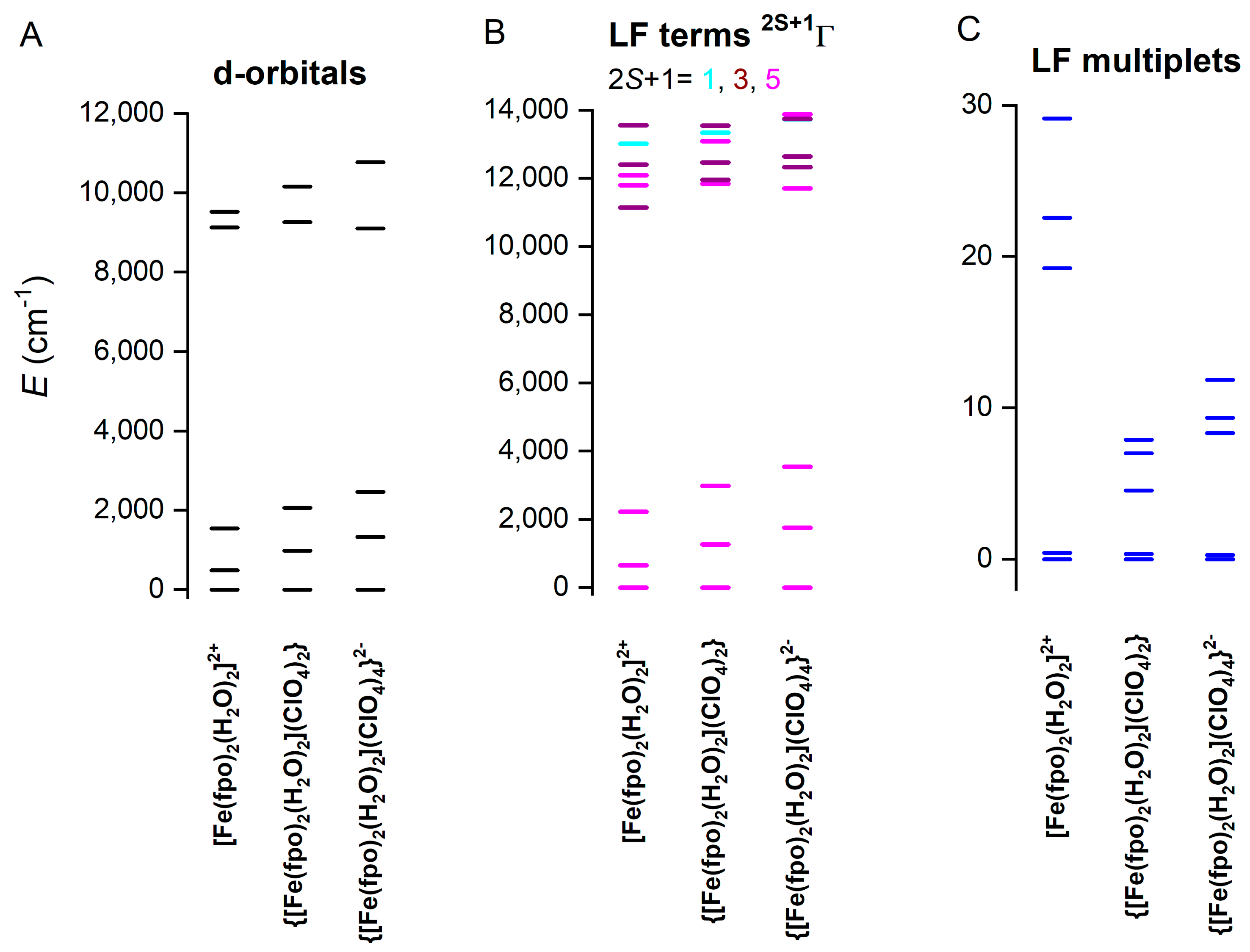
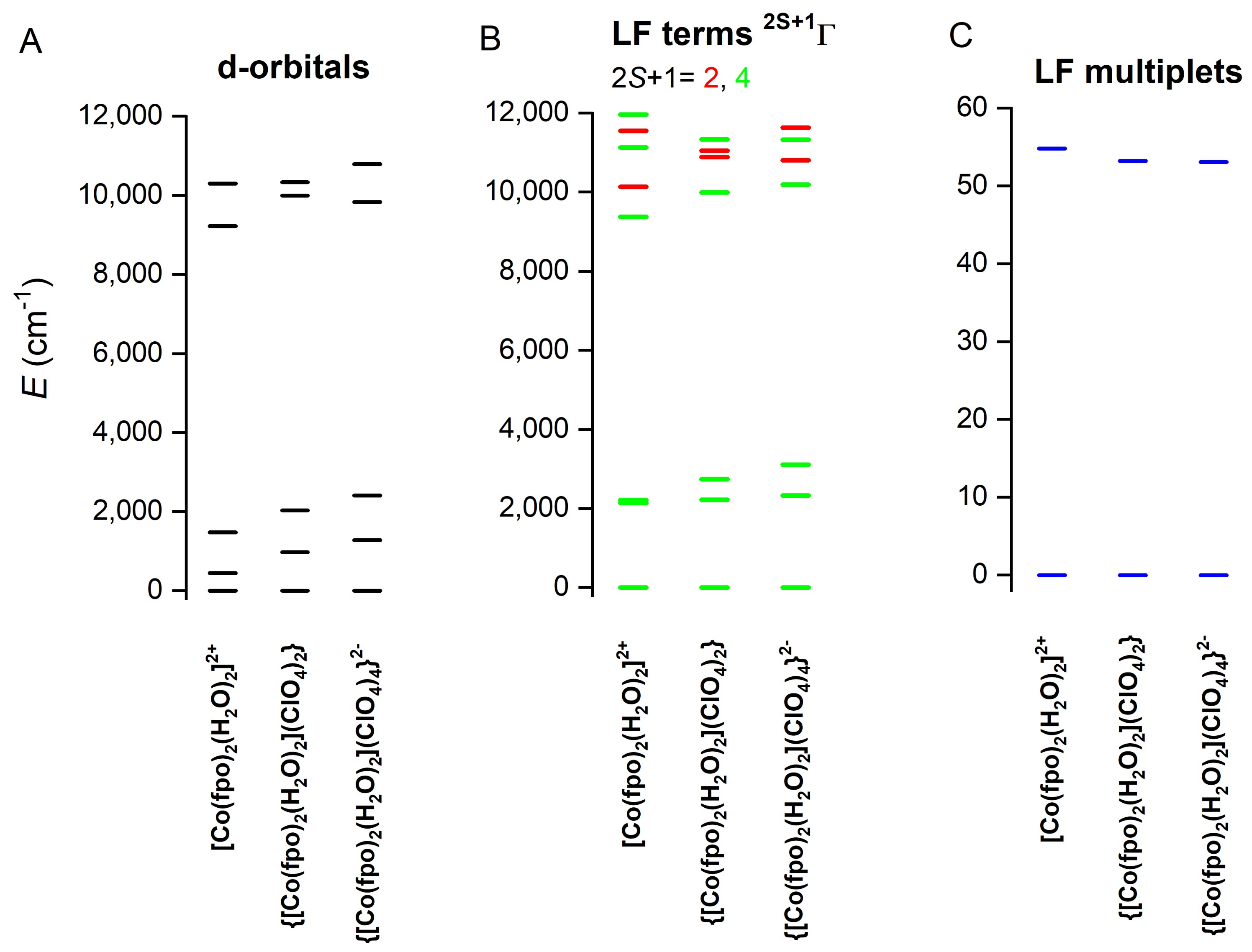
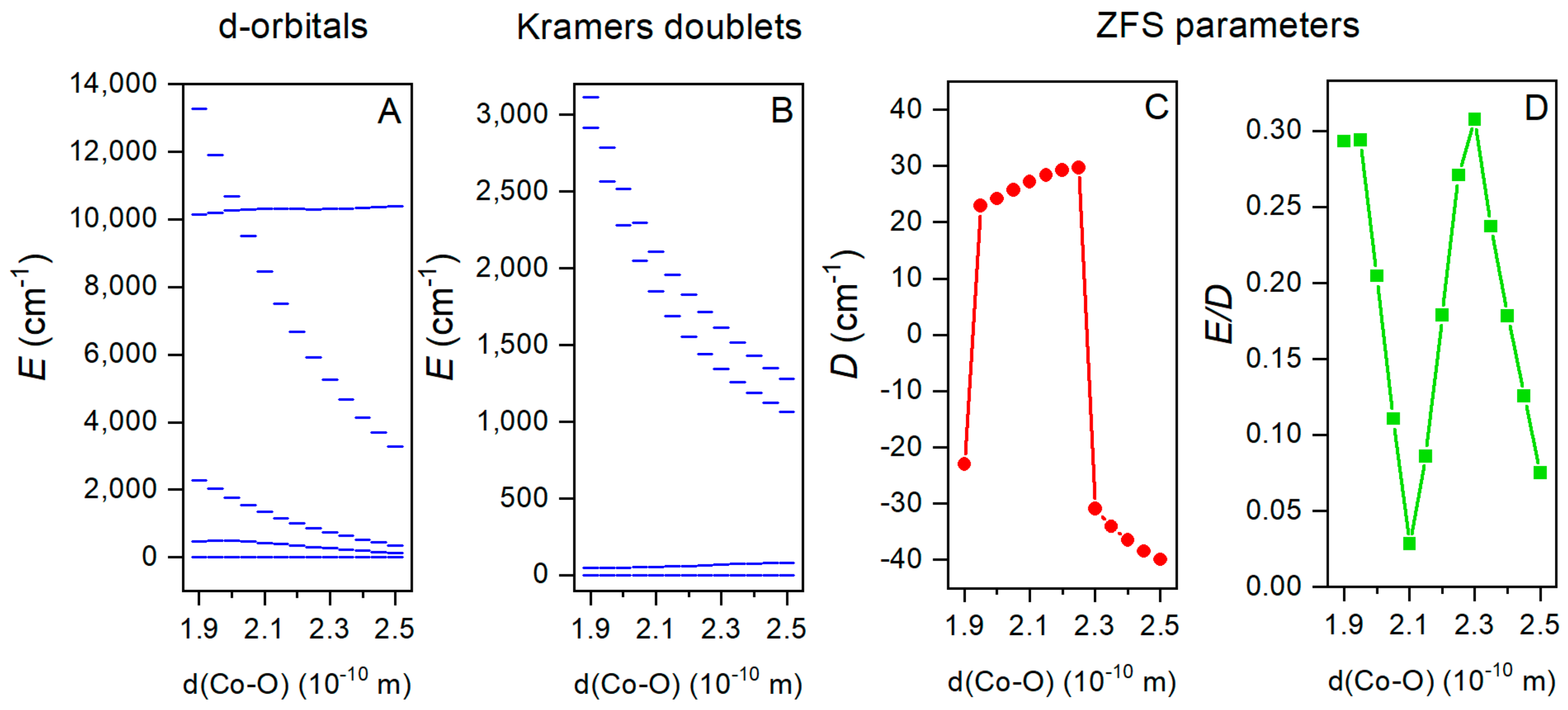
2.5. Theoretical Calculations
3. Materials and Methods
3.1. Preparation of Organic Ligands
3.1.1. Methyl Picolinate (I)
3.1.2. Picolinic Acid Hydrazide (II)
3.1.3. Picolinic Acid 2-(2-furanylmethylene)hydrazide (III)
3.1.4. 2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (fpo)
3.2. Preparation of the Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartolomé, J.; Luis, F.; Fernández, J.F. Molecular Magnets: Physics and Applications; Springer Verlag: Berlin, Germany, 2016. [Google Scholar]
- Ardavan, A.; Rival, O.; Morton, J.J.L.; Blundell, S.J.; Tyryshkin, A.M.; Timco, G.A.; Winpenny, R.E.P. Will spin-relaxation times in molecular magnets permit quantum information processing? Phys. Rev. Lett. 2007, 98, 057201. [Google Scholar] [CrossRef] [PubMed]
- Kahn, O. Spin-transition polymers: From molecular materials toward memory devices. Science 1998, 279, 44–48. [Google Scholar] [CrossRef]
- Bartual-Murgui, C.; Akou, A.; Thibault, C.; Molnár, G.; Vieu, C.; Salmon, L.; Bousseksou, A. Spin-crossover metal–organic frameworks: Promising materials for designing gas sensors. J. Mater. Chem. C 2015, 3, 1277–1285. [Google Scholar] [CrossRef]
- Linares, J.; Codjovi, E.; Garcia, Y. Pressure and temperature spin crossover sensors with optical detection. Sensors 2012, 12, 4479–4492. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Dey, A.; Shanmugam, M.; Narayanan, R.S.; Chandrasekhar, V. Cobalt(II) Complexes as Single-Ion Magnets. In Organometallic Magnets. Topics in Organometallic Chemistry; Chandrasekhar, V., Pointillart, F., Eds.; Springer: Cham, Germany, 2018; Volume 64, pp. 35–75. [Google Scholar]
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; Wiley: Chichester, UK, 2013. [Google Scholar]
- Yao, X.-N.; Du, J.-Z.; Zhang, Y.-Q.; Leng, X.-B.; Yang, M.-W.; Jiang, S.-D.; Wang, Z.-X.; Ouyang, Z.-W.; Deng, L.; Wang, B.-W.; et al. Two-coordinate Co(II) imido complexes as outstanding single-molecule magnets. J. Am. Chem. Soc. 2016, 139, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.; Ziegenbalg, S.; Aliabadi, A.; Schnegg, A.; Görls, H.; Plass, W. Magnetic relaxation in cobalt(ii)-based single-ion magnets influenced by distortion of the pseudotetrahedral [N2O2] coordination environment. Dalton Trans. 2018, 47, 10861–10873. [Google Scholar] [CrossRef]
- Chandrasekhar, V.; Dey, A.; Mota, A.J.; Colacio, E. Slow magnetic relaxation in Co(III)–Co(II) mixed-valence dinuclear complexes with a CoIIO5X (X = Cl, Br, NO3) distorted-octahedral coordination sphere. Inorg. Chem. 2013, 52, 4554–4561. [Google Scholar] [CrossRef]
- Kobayashi, F.; Ohtani, R.; Nakamura, M.; Lindoy, L.F.; Hayami, S. Slow magnetic relaxation triggered by a structural phase transition in long-chain-alkylated cobalt(II) single-ion magnets. Inorg. Chem. 2019, 58, 7409–7415. [Google Scholar] [CrossRef]
- Halcrow, M.A. Structure:function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef]
- Bar, A.K.; Pichon, C.; Sutter, J.-P. Magnetic anisotropy in two—to eight-coordinated transition–metal complexes: Recent developments in molecular magnetism. Coord. Chem. Rev. 2016, 308, 346–380. [Google Scholar] [CrossRef]
- Herchel, R.; Váhovská, L.; Potočňák, I.; Trávníček, Z. Slow magnetic relaxation in octahedral cobalt(II) field-induced single-ion magnet with positive axial and large rhombic anisotropy. Inorg. Chem. 2014, 53, 5896–5898. [Google Scholar] [CrossRef] [PubMed]
- Váhovská, L.; Vitushkina, S.; Potočňák, I.; Trávníček, Z.; Herchel, R. Effect of linear and non-linear pseudohalides on the structural and magnetic properties of Co(II) hexacoordinate single-molecule magnets. Dalton Trans. 2018, 47, 1498–1512. [Google Scholar] [CrossRef] [PubMed]
- Váhovská, L.; Bukrynov, O.; Potočňák, I.; Čižmár, E.; Kliuikov, A.; Vitushkina, S.; Dušek, M.; Herchel, R. New cobalt(II) field-induced single-molecule magnet and the first example of a cobalt(III) complex with tridentate binding of a deprotonated 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole ligand. Eur. J. Inorg. Chem. 2018, 2019, 250–261. [Google Scholar] [CrossRef]
- Du, M.; Wang, Q.; Li, C.-P.; Zhao, X.-J.; Ribas, J. Coordination assemblies of CoII/CuII/ZnII/CdIIwith 2,5-bipyridyl-1,3,4-oxadiazole and dicyanamide anion: Structural diversification and properties. Cryst. Growth Des. 2010, 10, 3285–3296. [Google Scholar] [CrossRef]
- Li, C.-P.; Zhao, X.-H.; Chen, X.-D.; Yu, Q.; Du, M. Metal-involved solvothermal interconversions of pyrazinyl substituted azole derivatives: Controllability and mechanism. Crys. Growth Des. 2010, 10, 5034–5042. [Google Scholar] [CrossRef]
- Gudasi, K.; Patil, M.; Vadavi, R.; Shenoy, R.; Patil, S. Transition metal complexes with a new tridentate ligand, 5-[6-(5-mercapto- 1,3,4-oxadiazol-2-yl)pyridin-2-yl]-1,3,4-oxadiazole-2-thiol. J. Serb. Chem. Soc. 2007, 72, 357–366. [Google Scholar] [CrossRef]
- Mathew, G.; Suseelan, M.S.; Krishnan, R. Synthesis and characterization of cobalt (II) complexes of chromen-2-one-3-carboxy hydrazide and 2-(chromen-2′-onyl)-5-(aryl) 1,3,4-oxadiazole derivatives. Indian J. Chem. A 2006, 45, 2040–2044. [Google Scholar]
- Wang, Y.-T.; Tang, G.-M.; Ma, W.-Y.; Wan, W.-Z. Synthesis and characterization of two new coordination supramolecular structures from a versatile unsymmetric 5-(4-pyridyl)-1,3,4-oxadiazole-2-thione (Hpot) ligand. Polyhedron 2007, 26, 782–790. [Google Scholar] [CrossRef]
- Incarvito, C.; Rheingold, A.L.; Qin, C.J.; Gavrilova, A.L.; Bosnich, B. Bimetallic reactivity. On the use of oxadiazoles as binucleating ligands. Inorg. Chem. 2001, 40, 1386–1390. [Google Scholar] [CrossRef]
- Incarvito, C.; Rheingold, A.L.; Gavrilova, A.L.; Qin, C.J.; Bosnich, B. Bimetallic reactivity. One-site addition Two-metal oxidation reactions using a Di-Co(II) complex of a binucleating ligand with 5-and 6-coordinate sites. Inorg. Chem. 2001, 40, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Subha, C.; Selvaraj, A. Synthesis and characterization of Co (II) And Ni (II) complexes of 2, 5-substituted 1, 3, 4-oxadiazole derivatives. Res. J. Chem. Sci. 2014, 4, 34–38. [Google Scholar]
- Goswami, S.; Jena, H.S.; Konar, S. Study of Heterogeneous Catalysis by Iron-squarate based 3D metal organic framework for the transformation of tetrazines to oxadiazole derivatives. Inorg. Chem. 2014, 53, 7071–7073. [Google Scholar] [CrossRef] [PubMed]
- Klingele, J.; Kaase, D.; Schmucker, M.; Lan, Y.; Chastanet, G.; Létard, J.-F. Thermal spin crossover and LIESST effect observed in complexes [Fe(LCh)2(NCX)2] [LCh = 2,5-Di(2-pyridyl)-1,3,4-Chalcadiazole; Ch = O, S, Se; X = S, Se, BH3]. Inorg. Chem. 2013, 52, 6000–6010. [Google Scholar] [CrossRef]
- Köhler, C.; Rentschler, E. The first 1,3,4-oxadiazole based dinuclear iron(II) complexes showing spin crossover behavior with hysteresis. Eur. J. Inorg. Chem. 2015, 2016, 1955–1960. [Google Scholar] [CrossRef]
- Phung, Q.M.; Domingo, A.; Pierloot, K. Dinuclear iron(II) spin-crossover compounds: A theoretical study. Chem. Eur. J. 2017, 24, 5183–5190. [Google Scholar] [CrossRef]
- Mani, G.S.; Donthiboina, K.; Shankaraiah, N.; Kamal, A. Iodine-promoted one-pot synthesis of 1,3,4-oxadiazole scaffolds via sp3 C–H functionalization of azaarenes. New J. Chem. 2019, 43, 15999–16006. [Google Scholar] [CrossRef]
- Lu, G.-P.; Lin, Y.-M. A base-induced ring-opening process of 2-substituted-1,3,4-oxadiazoles for the generation of nitriles at room temperature. J. Chem. Res. 2014, 38, 371–374. [Google Scholar] [CrossRef]
- Yu, W.; Huang, G.; Zhang, Y.; Liu, H.; Dong, L.; Yu, X.; Li, Y.; Chang, J. I2-mediated oxidative C–O bond formation for the synthesis of 1,3,4-oxadiazoles from aldehydes and hydrazides. J. Org. Chem. 2013, 78, 10337–10343. [Google Scholar] [CrossRef]
- Dijken, D.J.V.; Kovaříček, P.; Ihrig, S.P.; Hecht, S. Acylhydrazones as widely tunable photoswitches. J. Am. Chem. Soc. 2015, 137, 14982–14991. [Google Scholar] [CrossRef]
- Boča, R. Theoretical Foundations of Molecular Magnetism; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Zhang, Y.-Z.; Gómez-Coca, S.; Brown, A.J.; Saber, M.R.; Zhang, X.; Dunbar, K.R. Trigonal antiprismatic Co(ii) single molecule magnets with large uniaxial anisotropies: Importance of Raman and tunneling mechanisms. Chem. Sci. 2016, 7, 6519–6527. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Boča, R. Magnetic Parameters and Magnetic Functions in Mononuclear Complexes Beyond the Spin-Hamiltonian Formalism. In Structure and Bonding; Magnetic Functions Beyond the Spin-Hamiltonian; Mingos, D.M.P., Ed.; Springer: Heidelberg/Berlin, Germany, 2005; Volume 117, pp. 1–264. [Google Scholar]
- Palii, A.V.; Korchagin, D.V.; Yureva, E.A.; Akimov, A.V.; Misochko, E.Y.; Shilov, G.V.; Talantsev, A.D.; Morgunov, R.B.; Aldoshin, S.M.; Tsukerblat, B.S. Single-ion magnet Et4N[CoII(hfac)3] with nonuniaxial Anisotropy: Synthesis, experimental characterization, and theoretical modeling. Inorg. Chem. 2016, 55, 9696–9706. [Google Scholar] [CrossRef] [PubMed]
- Titiš, J.; Boča, R. Magnetostructuraldcorrelations in hexacoordinated cobalt(II) complexes. Inorg. Chem. 2011, 50, 11838–11845. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.P. N-electron valence state perturbation theory: A fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.P. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 117, 9138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 33, 8822. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 34, 7406. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Suturina, E.A.; Maganas, D.; Bill, E.; Atanasov, M.; Neese, F. Magneto-structural correlations in a series of pseudotetrahedral [CoII(XR)4]2–single molecule magnets: An ab initio ligand field study. Inorg. Chem. 2015, 54, 9948–9961. [Google Scholar] [CrossRef] [PubMed]
- Petit, S.; Pilet, G.; Luneau, D.; Chibotaru, L.F.; Ungur, L. A dinuclear cobalt(ii) complex of calix [8] arenes exibiting strong magnetic anisotropy. Dalton Trans. 2007, 40, 4582–4588. [Google Scholar] [CrossRef]
- Mondal, A.; Kharwar, A.K.; Konar, S. Sizeable effect of lattice solvent on field induced slow magnetic relaxation in seven coordinated CoII complexes. Inorg. Chem. 2019, 58, 10686–10693. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. BIOVIA Draw, Version 19.1; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Menges, F. Spectragryph—Optical Spectroscopy Software, Version 1.2.12. 2019. Available online: http://www.effemm2.de/spectragryph/ (accessed on 1 October 2019).
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; John Wiley & Sons LTD: Chichester, UK, 2015. [Google Scholar]
- iNMR. Available online: http://www.inmr.net (accessed on 3 December 2019).
- CrysAlisPro, Version 1.171.39.9g Rigaku Oxford Diffraction. 2015. Available online: https://www.rigaku.com/en/products/smc/crysalis. (accessed on 26 September 2016).
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 1999. [Google Scholar]
Sample Availability: Not available |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 |
|---|---|---|
| Chemical formula | C22H18Cl2FeN6O14 | C22H18Cl2CoN6O14 |
| Formula weight | 717.17 | 720.25 |
| Temperature [K] | 293(2) | 293(2) |
| λ (Mo, Kα) [Å] | 0.71073 | 0.71073 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/c | P21/c |
| a [Å] | 8.8543(18) | 8.8827(9) |
| b [Å] | 13.627(3) | 13.6502(12) |
| c [Å] | 11.840(2) | 11.7754(10) |
| α [°] | 90 | 90 |
| β [°] | 91.26(3) | 91.469(7) |
| γ [°] | 90 | 90 |
| Volume [Å3] | 1428.2(5) | 1427.3(2) |
| Z | 2 | 2 |
| D [g·cm−3] | 1.668 | 1.662 |
| μ [mm−1] | 0.80 | 0.86 |
| F (000) | 728 | 726 |
| Reflections collected | 5761 | 5605 |
| Independent reflections | 3198 | 3165 |
| Rint | 0.017 | 0.065 |
| R[F2 > 2σ(F2)] | 0.049 | 0.072 |
| wR(F2) | 0.133 | 0.215 |
| Σ | 1.02 | 1.02 |
| D | E/D | g1 | g2 | g3 | δ b | Δ1–5 c | Δ1–6 c | |
|---|---|---|---|---|---|---|---|---|
| [Fe(fpo)2(H2O)2]2+ | ||||||||
| CAS(6,5) | −7.05 | 0.077 | 2.045 | 2.076 | 2.234 | 654 | 29.1 | 679 |
| CAS(6,10) | −6.02 | 0.082 | 2.045 | 2.072 | 2.225 | 623 | 24.9 | 645 |
| {[Fe(fpo)2(H2O)2](ClO4)2} | ||||||||
| CAS(6,5) | −1.83 | 0.223 | 2.055 | 2.084 | 2.110 | 1267 | 7.90 | 1280 |
| CAS(6,10) | −2.04 | 0.158 | 2.054 | 2.079 | 2.104 | 1238 | 6.26 | 1196 |
| {[Fe(fpo)2(H2O)2](ClO4)4}2− | ||||||||
| CAS(6,5) | −2.91 | 0.060 | 2.058 | 2.073 | 2.081 | 1761 | 11.9 | 1773 |
| CAS(6,10) | −3.06 | 0.061 | 2.057 | 2.069 | 2.076 | 1727 | 12.5 | 1739 |
| Experimentally Determined Parameters | ||||||||
| +1.24 | 0.32 | 2.094 | 2.094 | 2.094 | ||||
| −1.23 | 0.31 | 2.094 | 2.094 | 2.094 | ||||
| D | E/D | g1 | g2 | g3 | δ b | Δ1–4 c | Δ1–5 c | |
|---|---|---|---|---|---|---|---|---|
| [Co(fpo)2(H2O)2]2+ | ||||||||
| CAS(7,5) | 27.2 | 0.076 | 2.090 | 2.352 | 2.374 | 2138 | 54.8 | 1964 |
| CAS(7,10) | 27.1 | 0.084 | 2.091 | 2.356 | 2.383 | 1967 | 54.7 | 1825 |
| {[Co(fpo)2(H2O)2](ClO4)2} | ||||||||
| CAS(7,5) | 23.3 | 0.318 | 2.092 | 2.283 | 2.385 | 2223 | 53.2 | 2187 |
| CAS(7,10) | 23.3 | 0.307 | 2.093 | 2.291 | 2.392 | 2068 | 52.8 | 2031 |
| {[Co(fpo)2(H2O)2](ClO4)4}2− | ||||||||
| CAS(7,5) | −24.9 | 0.212 | 2.096 | 2.242 | 2.390 | 2331 | 53.1 | 2331 |
| CAS(7,10) | −24.4 | 0.222 | 2.098 | 2.249 | 2.396 | 2160 | 52.3 | 2160 |
| Experimentally determined parameters | ||||||||
| +18.9 | 0.33 | 2.31 | 2.31 | 2.02 | ||||
| −21.2 | 0.32 | 2.13 | 2.13 | 2.37 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoufalý, P.; Čižmár, E.; Kuchár, J.; Herchel, R. The Structural and Magnetic Properties of FeII and CoII Complexes with 2-(furan-2-yl)-5-pyridin-2-yl-1,3,4-oxadiazole. Molecules 2020, 25, 277. https://doi.org/10.3390/molecules25020277
Zoufalý P, Čižmár E, Kuchár J, Herchel R. The Structural and Magnetic Properties of FeII and CoII Complexes with 2-(furan-2-yl)-5-pyridin-2-yl-1,3,4-oxadiazole. Molecules. 2020; 25(2):277. https://doi.org/10.3390/molecules25020277
Chicago/Turabian StyleZoufalý, Pavel, Erik Čižmár, Juraj Kuchár, and Radovan Herchel. 2020. "The Structural and Magnetic Properties of FeII and CoII Complexes with 2-(furan-2-yl)-5-pyridin-2-yl-1,3,4-oxadiazole" Molecules 25, no. 2: 277. https://doi.org/10.3390/molecules25020277
APA StyleZoufalý, P., Čižmár, E., Kuchár, J., & Herchel, R. (2020). The Structural and Magnetic Properties of FeII and CoII Complexes with 2-(furan-2-yl)-5-pyridin-2-yl-1,3,4-oxadiazole. Molecules, 25(2), 277. https://doi.org/10.3390/molecules25020277