
Mixed Amphiphilic Polymeric Nanoparticles of Chitosan, Poly(vinyl alcohol) and Poly(methyl methacrylate) for Intranasal Drug Delivery: A Preliminary In Vivo Study



Abstract
1. Introduction
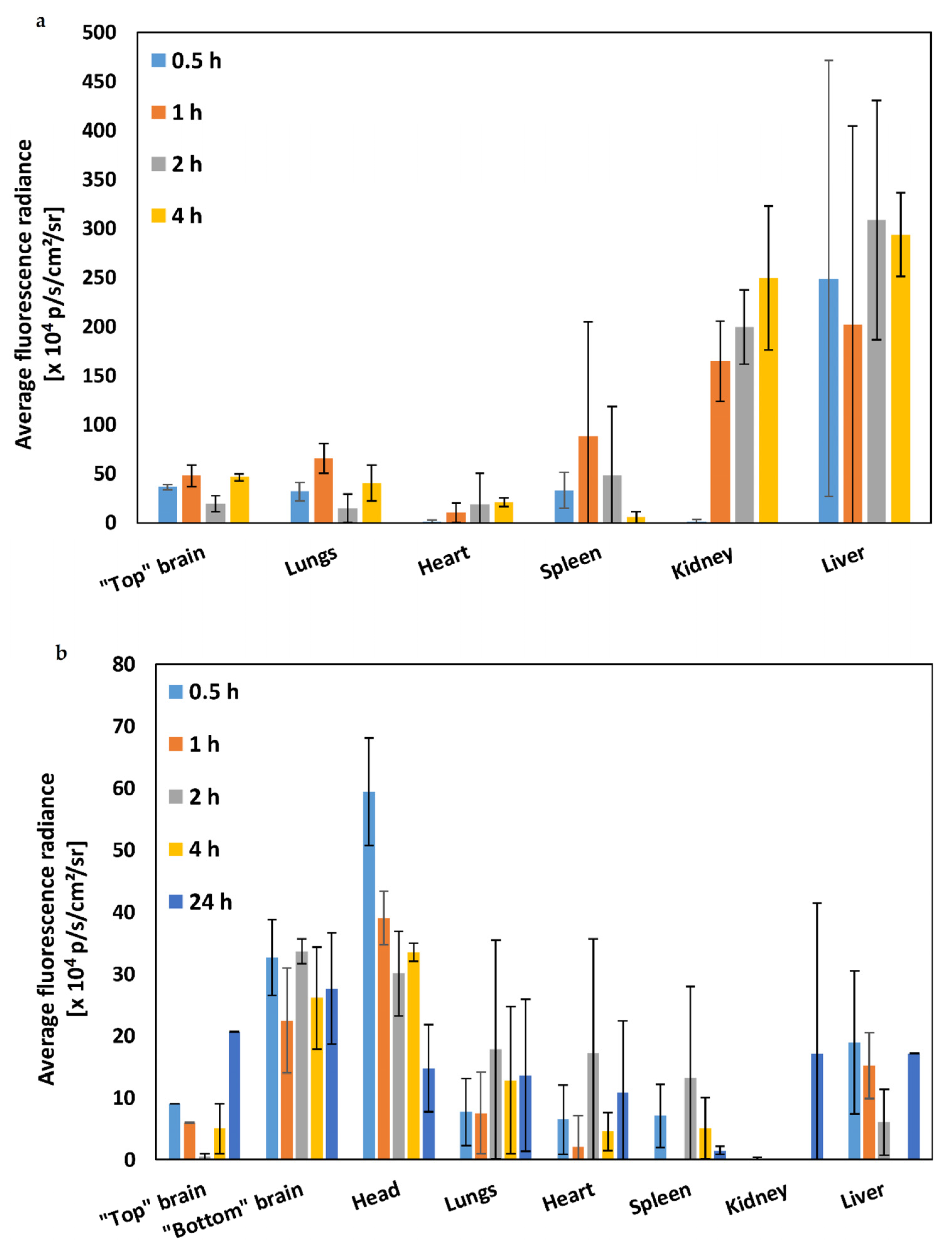
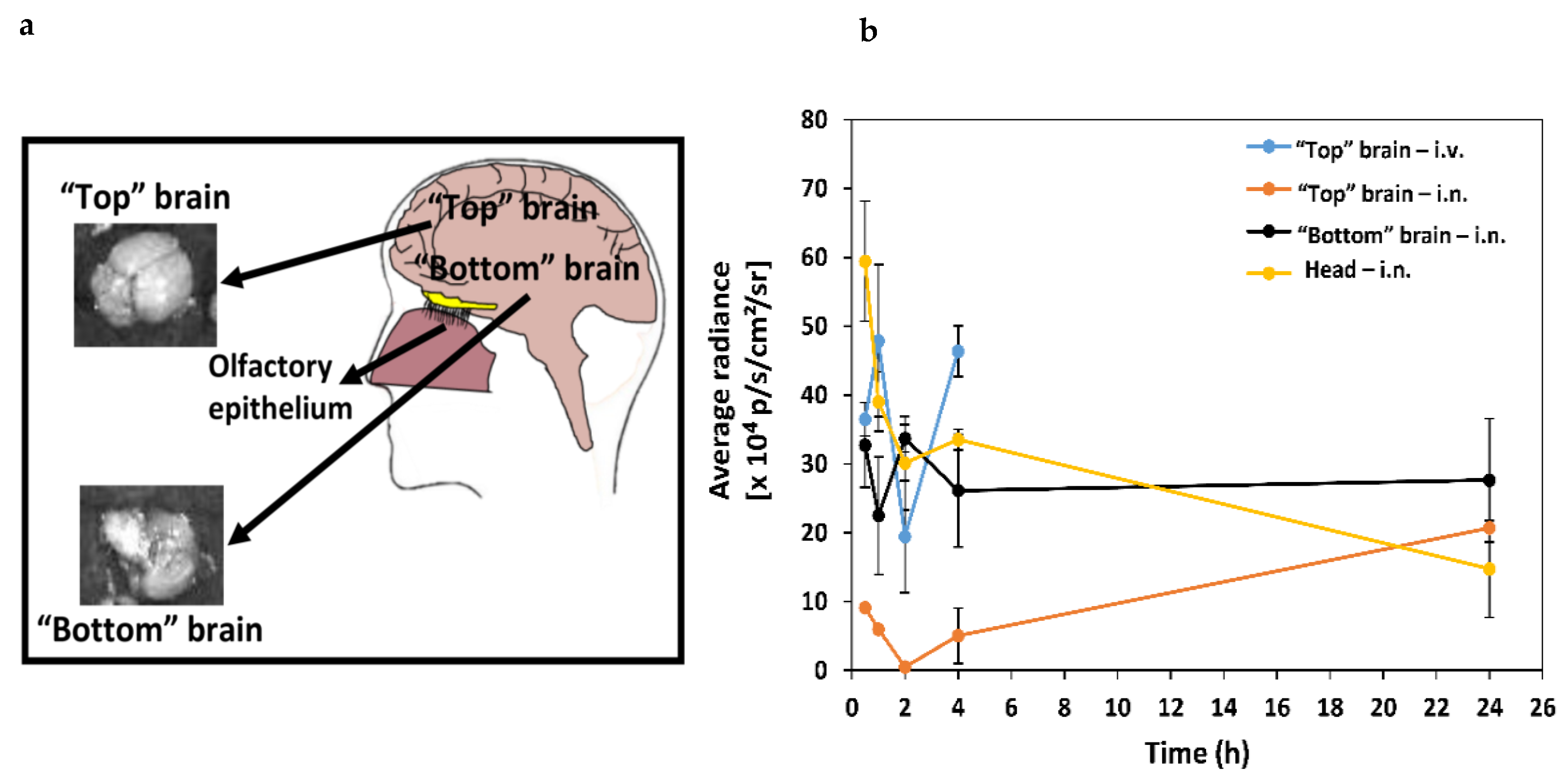
2. Results and Discussion
3. Methods
3.1. Synthesis of the Chitosan-g-Poly(methyl methacrylate) and Poly(vinyl alcohol)-g-Poly(methyl methacrylate) Copolymers
3.2. Preparation and Characterization of Mixed Chitosan-g-Poly(methyl methacrylate):Poly(vinyl alcohol)-g-Poly(methyl methacrylate) Nanoparticles
3.3. Biodistribution of Mixed Chitosan-g-Poly(methyl methacrylate):poly(vinyl alcohol)-g-Poly(methyl methacrylate) Nanoparticles
3.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blanco-Prieto, M. Drug Delivery to the Central Nervous System: A Review. J. Pharm. Sci. 2003, 6, 252–273. [Google Scholar]
- Pardridge, W. Non-Invasive Drug Delivery to the Human Brain Using Endogenous Blood-Brain Barrier Transport Systems. Pharm. Sci. Technol. Today 1999, 2, 49–59. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. Blood-Brain Barrier Shuttle Peptides: An Emerging Paradigm for Brain Delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Blood-Brain Barrier Active Efflux Transporters: ATP-Binding Cassette Gene Family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug Resistance-Associated Proteins: Expression and Function in the Central Nervous System. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef]
- Zeiadeh, I.; Najjar, A.; Karaman, R. Strategies for Enhancing the Permeation of CNS-Active Drugs through the Blood-Brain Barrier: A Review. Molecules 2018, 23, 1289. [Google Scholar] [CrossRef]
- Rapoport, S.I. Osmotic Opening of the Blood–Brain Barrier: Principles, Mechanism, and Therapeutic Applications. Cell Mol. Neurobiol. 2000, 20, 217–230. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Terasaki, T. Contribution of Carrier-Mediated Transport Systems to the Blood–Brain Barrier as a Supporting and Protecting Interface for the Brain; Importance for CNS Drug Discovery and Development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef]
- Tsuji, A. Small Molecular Drug Transfer Across the Blood-Brain Barrier via Carrier-Mediated Transport Systems. NeuroRx 2005, 2, 54–62. [Google Scholar] [CrossRef]
- Gomes, M.J.; Mendes, B.; Martins, S.; Sarmento, B. Nanoparticle Functionalization for Brain Targeting Drug Delivery and Diagnostic. In Handbook of Nanoparticles; Aliofkhazraei, M., Ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Gabathuler, R. Approaches to Transport Therapeutic Drugs Across the Blood-Brain Barrier to Treat Brain Diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef]
- Fang, F.; Zou, D.; Wang, W.; Yin, Y.; Yin, T.; Hao, S.; Wang, B.; Wang, G.; Wang, Y. Non-Invasive Approaches for Drug Delivery to the Brain Based on the Receptor Mediated Transport. Mater. Sci. Eng. C 2017, 76, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.H.; Sosa, C.; Liu, R.; Yao, N.; Priestley, R.D. Nanomedicine as a Non-Invasive Strategy for Drug Delivery Across the Blood Brain Barrier. Int. J. Pharm. 2016, 515, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Visser, C.C.; Appeldoorn, C.C.M.; Rip, J. Enhanced Brain Drug Delivery: Safely Crossing the Blood-Brain Barrier. Drug Discov. Today Technol. 2012, 9, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Wu, X.Y.; Bendayan, R. Nanotechnological Advances for the Delivery of CNS Therapeutics. Adv. Drug Deliv. Rev. 2012, 64, 686–700. [Google Scholar] [CrossRef]
- Saraiva, C.; Praca, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood-Brain Barrier to Treat Neurodegenerative Diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood Brain Barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef]
- Lucchini, R.G.; Dorman, D.C.; Elder, A.; Veronesi, B. Neurological Impacts from Inhalation of Pollutants and the Nose-Brain Connection. Neurotoxicology 2012, 33, 838–841. [Google Scholar] [CrossRef]
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Kreyling, W.; Cox, C. Translocation of Inhaled Ultrafine Particles to the Brain. Inhal. Toxicol. 2004, 16, 437–445. [Google Scholar] [CrossRef]
- Elder, A.; Gelein, R.; Silva, V.; Feikert, T.; Opanashuk, L.; Carter, J.; Potter, R.; Maynard, A.; Ito, Y.; Finkelstein, J.; et al. Translocation of Inhaled Ultrafine Manganese Oxide Particles to the Central Nervous System. Environ. Health Perspect. 2006, 114, 1172–1178. [Google Scholar] [CrossRef]
- Babadjouni, R.; Patel, A.; Liu, Q.; Shkirkova, K.; Lamorie-Foote, K.; Connor, M.; Hodis, D.M.; Cheng, H.; Sioutas, C.; Morgan, T.E.; et al. Nanoparticulate Matter Exposure Results in Neuroinflammatory Changes in the Corpus Callosum. PLoS ONE 2018, 13, e0206934. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in Nose-To-Brain Drug Delivery: A Review of Non-Clinical Brain Targeting Studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, H.; Li, F.; Lu, Y.; Qi, J.; Wu, W. An Update on the Role of Nanovehicles in Nose-to-Brain Drug Delivery. Drug Discov. Today 2018, 23, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H. Intranasal Delivery Bypasses the Blood-Brain Barrier to Target Therapeutic Agents to the Central Nervous System and Treat Neurodegenerative Disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.P.; Mundiña-Weilenmann, C.; Romero, E.L.; Morilla, M.J. Increased Brain Radioactivity by Intranasal P-labeled siRNA Dendriplexes Within in Situ-Forming Mucoadhesive Gels. Int. J. Nanomed. 2012, 7, 1373–1385. [Google Scholar]
- Kumar, A.; Pandey, A.N.; Jain, S.K. Nasal-Nanotechnology: Revolution for Efficient Therapeutics Delivery. Drug Deliv. 2016, 23, 671–683. [Google Scholar] [CrossRef]
- Chiappetta, D.A.; Hocht, C.; Opezzo, J.A.W.; Sosnik, A. Intranasal Administration of Antiretroviral-Loaded Micelles for Anatomical Targeting to the Brain in HIV. Nanomedicine 2013, 8, 223–237. [Google Scholar] [CrossRef]
- Kanazawa, T.; Taki, H.; Tanaka, K.; Takashima, Y.; Okada, H. Cell-Penetrating Peptide-Modified Block Copolymer Micelles Promote Direct Brain Delivery via Intranasal Administration. Pharm. Res. 2011, 28, 2130–2139. [Google Scholar] [CrossRef]
- Raskin Menaker, M.; Schlachet, I.; Sosnik, A. Mucoadhesive Nanogels by Ionotropic Crosslinking of Chitosan-g-Oligo(NiPAam) Polymeric Micelles as Novel Drug Nanocarriers. Nanomedicine 2016, 11, 217–233. [Google Scholar] [CrossRef]
- Moshe, H.; Davizon, Y.; Menaker Raskin, M.; Sosnik, A. Novel Poly(Vinyl Alcohol)-Based Amphiphilic Nanogels by Boric Acid Non-Covalent Crosslinking of Polymeric Micelles. Biomater. Sci. 2017, 5, 2295–2309. [Google Scholar] [CrossRef]
- Noi, I.; Schlachet, I.; Kumarasamy, M.; Sosnik, A. Permeability of Chitosan-g-Poly(Methyl Methacrylate) Amphiphilic Nanoparticles in a Model of Small Intestine In Vitro. Polymers 2018, 10, 478. [Google Scholar] [CrossRef]
- Schlachet, I.; Trousil, J.; Rak, D.; Knudsen, K.D.; Pavlova, E.; Nyström, B.; Sosnik, A. Chitosan-graft-Poly(Methyl Methacrylate) Amphiphilic Nanoparticles: Self-Association and Physicochemical Characterization. Carbohydr. Polym. 2019, 212, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Moshe Halamish, H.; Trousil, J.; Rak, D.; Knudsen, K.D.; Pavlova, E.; Nyström, B.; Štěpánek, P.; Sosnik, A. Self-Assembly and Nanostructure of Poly(Vinyl Alcohol)-graft-Poly(Methyl Methacrylate) Amphiphilic Nanoparticles. J. Colloid Interface Sci. 2019, 553, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, M.; Sosnik, A. The Nose-to-Brain Transport of Polymeric Nanoparticles is Mediated by Immune Sentinels and Not by Olfactory Sensory Neurons. Adv. Biosyst. 2019, 3, 1900123. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef]
- Ways, M.; Lau, W.M.; Khutoryanskiy, V.V. Chitosan and Its Derivatives for Application in Mucoadhesive Drug Delivery Systems. Polymers 2018, 10, 267. [Google Scholar] [CrossRef]
- Alam, S.; Khan, Z.I.; Mustafa, G.; Kumar, M.; Islam, F.; Bhatnagar, A.; Ahmad, F.J. Development and Evaluation of Thymoquinone-Encapsulated Chitosan Nanoparticles for Nose-to-Brain Targeting: A Pharmacoscintigraphic Study. Int. J. Nanomed. 2012, 7, 5705–5718. [Google Scholar] [CrossRef]
- Casettari, L.; Illum, L. Chitosan in Nasal Delivery Systems for Therapeutic Drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef]
- Rassu, G.; Soddu, E.; Cossu, M.; Gavini, E.; Giunchedi, P.; Dalpiaz, A. Particulate Formulations Based on Chitosan for Nose-to-Brain Delivery of Drugs. A Review. J. Drug Deliv. Sci. Technol. 2016, 32, 77–87. [Google Scholar] [CrossRef]
- Marques, C.; Som, C.; Schmutz, M.; Borges, O.; Borchard, G. How the Lack of Chitosan Characterization Precludes Implementation of the Safe-by-Design Concept. Front. Bioeng. Biotechnol. 2020, 8, 165. [Google Scholar] [CrossRef]
- Hu, Y.-L.; Qi, W.; Han, F.; Shao, J.-Z.; Gao, J.-Q. Toxicity Evaluation of Biodegradable Chitosan Nanoparticles Using a Zebrafish Embryo Model. Int. J. Nanomed. 2011, 6, 3351–3359. [Google Scholar]
- Schlachet, I.; Sosnik, A. Mixed Mucoadhesive Amphiphilic Polymeric Nanoparticles Cross a Model of Nasal Septum Epithelium In Vitro. ACS Appl. Mater. Interfaces 2019, 11, 21360–21371. [Google Scholar] [CrossRef] [PubMed]
- Schlachet, I. Innovative Nano-Biomaterials for the Improved Delivery of Antitumorals to the Central Nervous System in the Therapy of Pediatric Brain Tumors. Ph.D. Thesis, Technion-Israel Institute of Technology, Haifa, Israel, 2019. [Google Scholar]
- Gaucher, G.; Dufresne, M.-H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block Copolymer Micelles: Preparation, Characterization and Application in Drug Delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, Bioapplications, and Toxicity Evaluation of Chitosan-Based Nanoparticles. Int. J. Mol. Sci. 2019, 20, 5776. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors Controlling the Pharmacokinetics, Biodistribution and Intratumoral Penetration of Nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of Particle Size and Surface Charge on Cellular Uptake and Biodistribution of Polymeric Nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Bukchin, A.; Sanchez-Navarro, M.; Carrera, A.; Teixidó, M.; Carcaboso, A.M.; Giralt, E.; Sosnik, A. Amphiphilic Polymeric Nanoparticles Modified with a Retro-Enantio Peptide Shuttle Target the Brain of Mice. Chem. Mater. 2020, 32, 7679–7693. [Google Scholar] [CrossRef]
- Williams, R.M.; Shah, J.; Ng, B.D.; Minton, D.R.; Gudas, L.J.; Park, C.Y.; Heller, D.A. Mesoscale Nanoparticles Selectively Target the Renal Proximal Tubule Epithelium. Nano Lett. 2015, 15, 2358–2364. [Google Scholar] [CrossRef]
- Yadav, S.; Gattacceca, F.; Panicucci, R.; Amiji, M.M. Comparative Biodistribution and Pharmacokinetic Analysis of Cyclosporine-A in the Brain upon Intranasal or Intravenous Administration in an Oil-in-Water Nanoemulsion Formulation. Mol. Pharm. 2015, 12, 1523–1533. [Google Scholar] [CrossRef]
- Gao, X.; Chen, J.; Chen, J.; Wu, B.; Chen, H.; Jiang, X. Quantum Dots Bearing Lectin-Functionalized Nanoparticles as a Platform for In Vivo Brain Imaging. Bioconjug. Chem. 2008, 19, 2189–2195. [Google Scholar] [CrossRef]
- Sekerdag, E.; Lüle, S.; Bozdağ Pehlivan, S.; Öztürk, N.; Kara, A.; Kaffashi, A.; Vural, I.; Işıkay, I.; Yavuz, B.; Oguz, K.K.; et al. A Potential Non-Invasive Glioblastoma Treatment: Nose-to-Brain Delivery of Farnesylthiosalicylic Acid Incorporated Hybrid Nanoparticles. J. Control. Release 2017, 261, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.A.; Mazibuko, N.; Zelaya, F.; Vasilakopoulou, S.; Loveridge, J.; Oates, A.; Maltezos, S.; Mehta, M.; Wastling, S.; Howard, M.; et al. Effects of Route of Administration on Oxytocin-Induced Changes in Regional Cerebral Blood Flow in Humans. Nat. Commun. 2020, 11, 1160. [Google Scholar] [CrossRef] [PubMed]
- Shiga, H.; Taki, J.; Yamada, M.; Washiyama, K.; Amano, R.; Matsuura, Y.; Matsui, O.; Tatsutomi, S.; Yagi, S.; Tsuchida, A.; et al. Evaluation of the Olfactory Nerve Transport Function by SPECT-MRI Fusion Image with Nasal Thallium-201 Administration. Mol. Imaging Biol. 2011, 13, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.W.D.; Shao, Q.; Vig, P.J.S.; Bidwell, G.L. Intranasal Administration of Elastin-like Polypeptide for Therapeutic Delivery to the Central Nervous System. Drug Des. Devel. Ther. 2016, 10, 2803–2813. [Google Scholar] [CrossRef]
- Xia, H.; Gao, X.; Gu, G.; Liu, Z.; Zeng, N.; Hu, Q.; Song, Q.; Yao, L.; Pang, Z.; Jiang, X.; et al. Low molecular weight protamine-functionalized naoparticles for drug delivery to the brain after intranasal administration. Biomaterials 2011, 32, 9888–9898. [Google Scholar] [CrossRef]
- Md, S.; Khan, R.A.; Mustafa, G.; Chuttani, K.; Baboota, S.; Sahni, J.K.; Ali, J. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delivery: Pharmacodynamic, pharmacokinetic and scintigraphy study in mice model. Eur. J. Pharm. Sci. 2013, 48, 393–405. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-Modified MRNA Vaccines Induce Potent T Follicular Helper and Germinal Center B Cell Responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Brain Region | AUC0–4 h (× 104 p/s/cm2/sr) ± S.D. | AUC0–24 h (× 104 p/s/cm2/sr) ± S.D. |
|---|---|---|
| “Top” brain—i.v. | 130 ± 20 * | N.D. |
| “Top” brain—i.n. | 15 ± 4 | 41 ± 3 ***** |
| “Bottom” brain—i.n. | 110 ± 18 ** | 164 ± 7 **,****,***** |
| Head—i.n. | 138 ± 17 *** | 186 ± 6 ***** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlachet, I.; Moshe Halamish, H.; Sosnik, A. Mixed Amphiphilic Polymeric Nanoparticles of Chitosan, Poly(vinyl alcohol) and Poly(methyl methacrylate) for Intranasal Drug Delivery: A Preliminary In Vivo Study. Molecules 2020, 25, 4496. https://doi.org/10.3390/molecules25194496
Schlachet I, Moshe Halamish H, Sosnik A. Mixed Amphiphilic Polymeric Nanoparticles of Chitosan, Poly(vinyl alcohol) and Poly(methyl methacrylate) for Intranasal Drug Delivery: A Preliminary In Vivo Study. Molecules. 2020; 25(19):4496. https://doi.org/10.3390/molecules25194496
Chicago/Turabian StyleSchlachet, Inbar, Hen Moshe Halamish, and Alejandro Sosnik. 2020. "Mixed Amphiphilic Polymeric Nanoparticles of Chitosan, Poly(vinyl alcohol) and Poly(methyl methacrylate) for Intranasal Drug Delivery: A Preliminary In Vivo Study" Molecules 25, no. 19: 4496. https://doi.org/10.3390/molecules25194496
APA StyleSchlachet, I., Moshe Halamish, H., & Sosnik, A. (2020). Mixed Amphiphilic Polymeric Nanoparticles of Chitosan, Poly(vinyl alcohol) and Poly(methyl methacrylate) for Intranasal Drug Delivery: A Preliminary In Vivo Study. Molecules, 25(19), 4496. https://doi.org/10.3390/molecules25194496