
Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes


Abstract
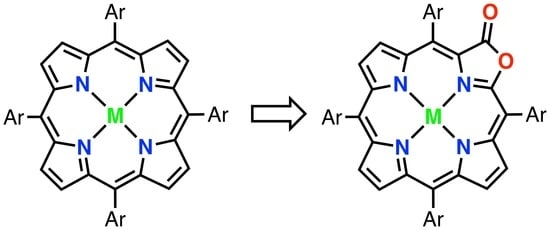
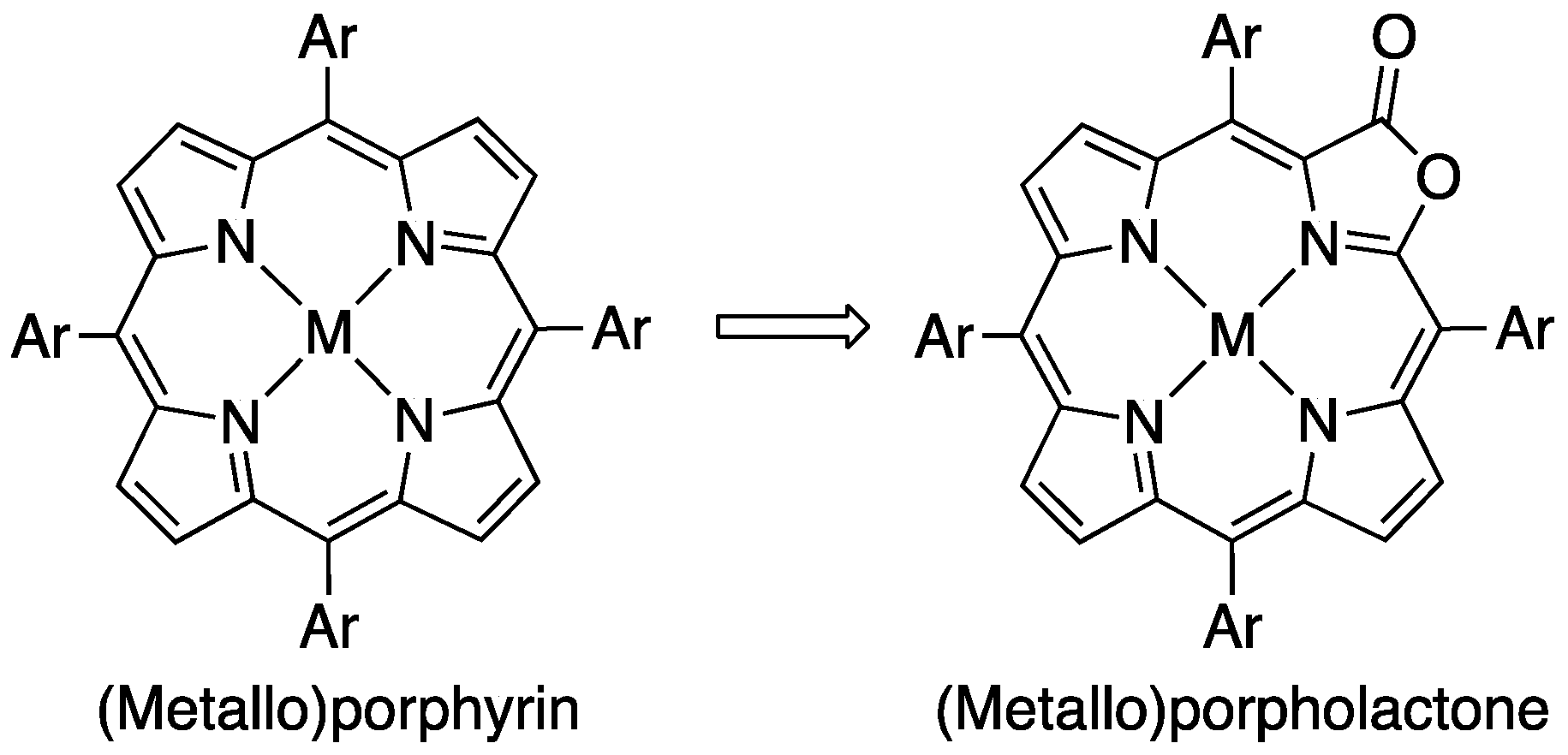
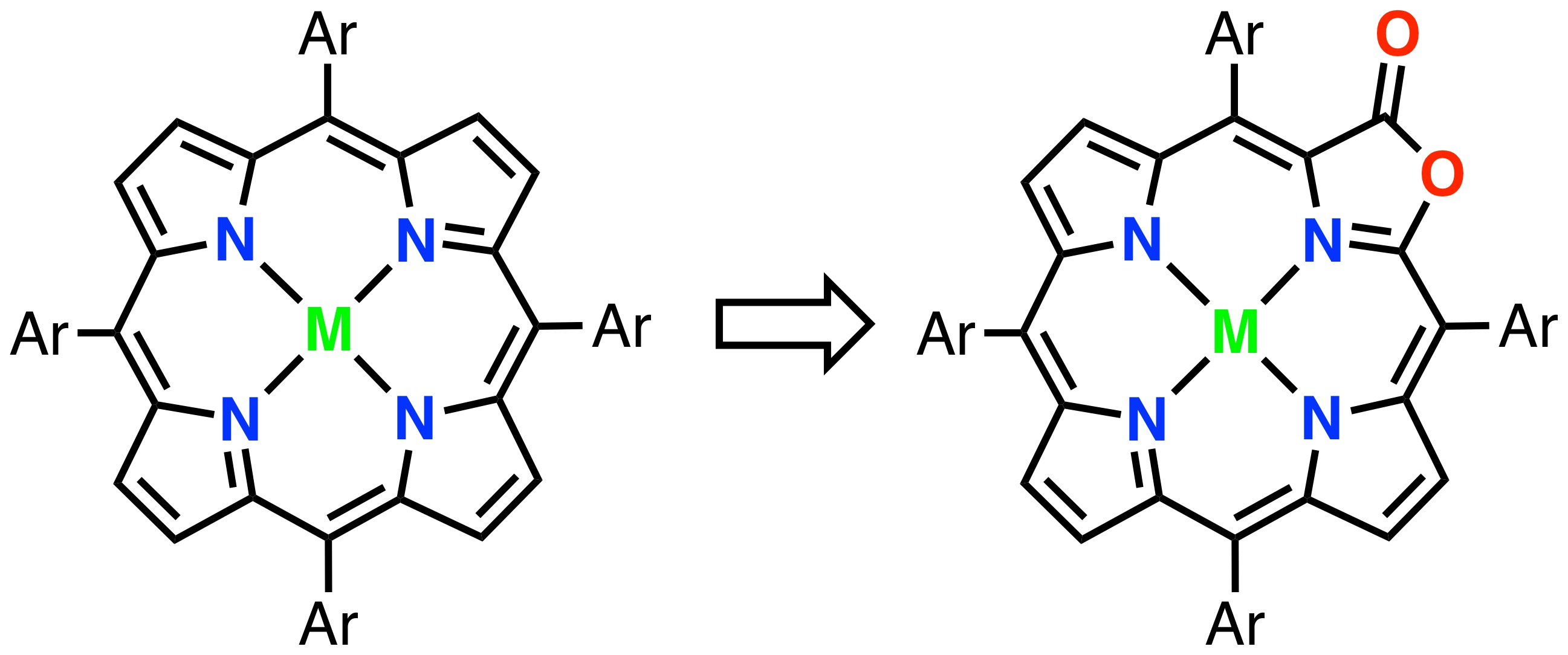{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
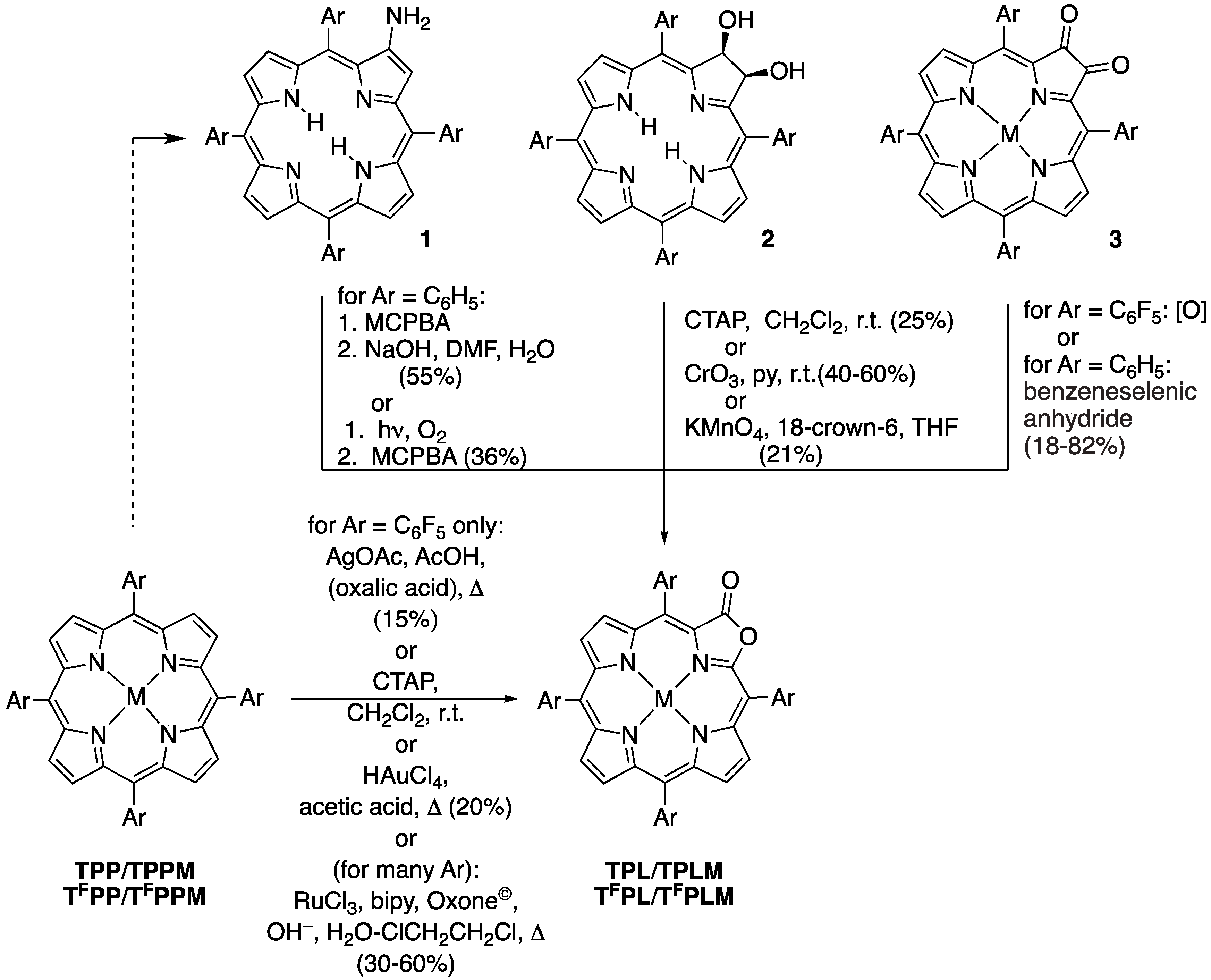
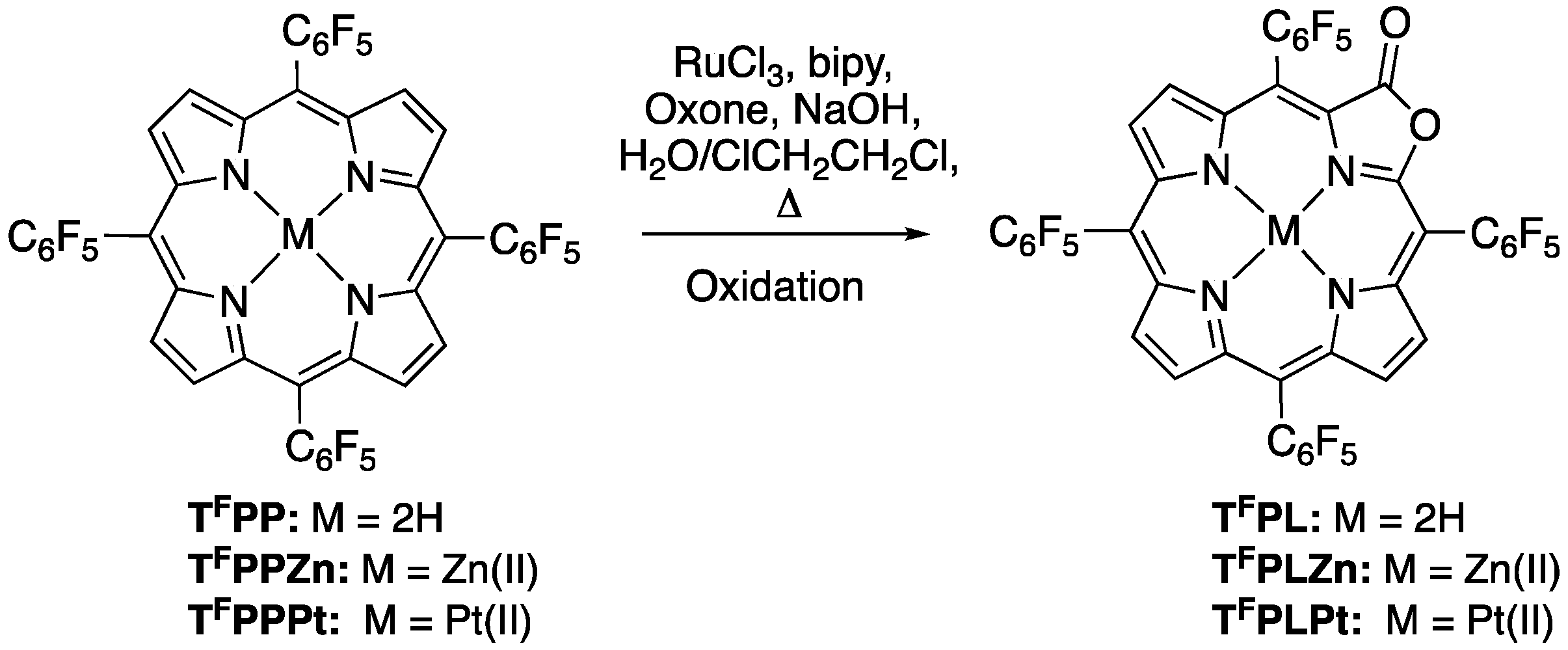
2.1. The Most Efficient Pathway toward TFPL/TFPLZn/TFPLPt
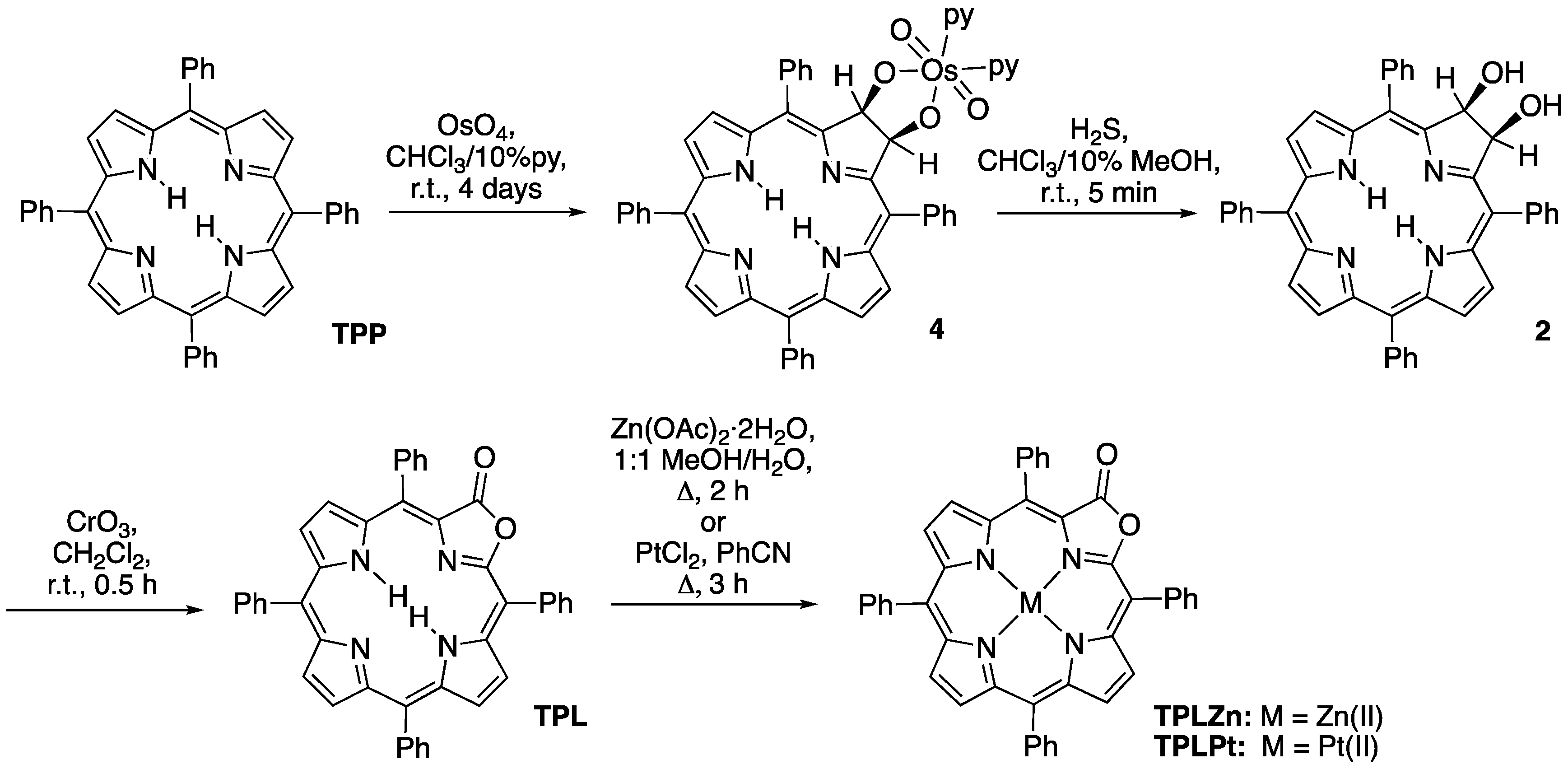
2.2. The Most Efficient Pathway toward TPL/TPLZn/TPLPt
3. Materials and Methods
3.1. Materials
3.2. Two-Step Procedure for Synthesis of meso-Tetraphenyl-2-Oxa-3-Oxoporphyrin (TPL) by OsO4-Mediated Dihydroxylation, Followed by CrO3 Oxidation
3.2.1. Dihydroxylation of TPP; Synthesis of meso-Tetraphenyl-2,3-cis-dihydroxychlorin 2
3.2.2. Synthesis of meso-Tetraphenyl-2-oxa-3-oxoporphyrin (TPL) via CrO3 Oxidation of Diol 2
3.3. Preparation of Metalloporpholactone TPLZn via Zinc Insertion into Free Base TPL
3.4. Preparation of Metalloporpholactone TPLPt via Platinum Insertion into Free Base TPL
3.5. One-Step Synthesis of meso-Tetrakis(pentafluorophenyl)-2-oxa-3-oxoporphyrin TFPL by RuCl3/Oxone®-Mediated Oxidation. General Procedure
3.6. Synthesis of [meso-Tetrakis(pentafluorophenyl)-2-oxa-3-oxoporphyrinato]zinc(II) (TFPLZn)
3.7. Synthesis of [meso-Tetrakis(pentafluorophenyl)-2-oxa-3-oxoporphyrinato]platinum(II) (TFPLPt)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AcOH | acetic acid |
| AgOAc | silver acetate |
| bipy | 2,2′-bipyridine |
| 18-C-6 | 18-crown-6 crown ether |
| CTAP | cetyltrimethylammonium permanganate |
| DMF | dimethyl formamide |
| MCPBA | m-chloroperoxybenzoic acid |
| PhCN | benzonitrile |
| py | pyridine |
References
- Brückner, C.; Akhigbe, J.; Samankumara, L. Syntheses and Structures of Porphyrin Analogues Containing Non-pyrrolic Heterocycles. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NY, USA, 2014; pp. 1–276. [Google Scholar]
- Crossley, M.J.; King, L.G. Novel Heterocyclic Systems from Selective Oxidation at the β-Pyrrolic Position of Porphyrins. J. Chem. Soc. Chem. Commun. 1984, 920–922. [Google Scholar] [CrossRef]
- Jayaraj, K.; Gold, A.; Austin, R.N.; Ball, L.M.; Terner, J.; Mandon, D.; Weiss, R.; Fischer, J.; DeCian, A.; Bill, E.; et al. Compound I and Compound II Analogues from Porpholactones. Inorg. Chem. 1997, 36, 4555–4566. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C.; Ogikubo, J.; McCarthy, J.R.; Akhigbe, J.; Hyland, M.A.; Daddario, P.; Worlinsky, J.L.; Zeller, M.; Engle, J.T.; Ziegler, C.J.; et al. meso-Arylporpholactones and their Reduction Products. J. Org. Chem. 2012, 77, 6480–6494. [Google Scholar] [CrossRef] [PubMed]
- Gouterman, M.; Hall, R.J.; Khalil, G.E.; Martin, P.C.; Shankland, E.G.; Cerny, R.L. Tetrakis(pentafluorophenyl)porpholactone. J. Am. Chem. Soc. 1989, 111, 3702–3707. [Google Scholar] [CrossRef]
- Ning, Y.; Jin, G.-Q.; Zhang, J.-L. Porpholactone Chemistry: An Emerging Approach to Bioinspired Photosensitizers with Tunable Near-Infrared Photophysical Properties. Acc. Chem. Res. 2019, 52, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Worlinsky, J.L.; Halepas, S.; Ghandehari, M.; Khalil, G.; Brückner, C. High pH Sensing with Water-soluble Porpholactone Derivatives and their Incorporation into a Nafion® Optode Membrane. Analyst 2015, 140, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.; Brückner, C. Modifications of Porphyrins and Hydroporphyrins for their Solubilization in Aqueous Media. Molecules 2017, 22, 980. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grazynski, L. O-Confused Oxaporphyrins. An Intermediate in Formation of 3-Substituted 2-Oxa-21-Carbaporphyrins. J. Org. Chem. 2005, 70, 9123–9130. [Google Scholar] [CrossRef]
- Lara, K.K.; Rinaldo, C.R.; Brückner, C. meso-Tetraaryl-7,8-dihydroxydithiachlorins: First Examples of Heterochlorins. Tetrahedron Lett. 2003, 44, 7793–7797. [Google Scholar] [CrossRef]
- Lara, K.K.; Rinaldo, C.K.; Brückner, C. meso-Tetraaryl-7,8-diol-dithiachlorins and their Pyrrole-modified Derivatives: Synthesis and Spectroscopic Comparison to their Aza-analogues. Tetrahedron 2005, 61, 2529–2539. [Google Scholar] [CrossRef]
- Sharma, M.; Meehan, E.; Mercado, B.Q.; Brückner, C. β-Alkyloxazolochlorins: Revisiting the Ozonation of Octaalkylporphyrins, and Beyond. Chem. Eur. J. 2016, 22, 11706–11718. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Osuka, A. Subporpholactone, Subporpholactam, Imidazolosubporphyrin, and Iridium Complexes of Imidazolosubporphyrin: Formation of Iridium Carbene Complexes. Angew. Chem. Int. Ed. 2018, 57, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.-S.; Chang, Y.; Chen, J.-Z.; Tian, J.; Mack, J.; Cheng, X.; Shen, Z.; Zhang, J.-L. Porphodilactones as Synthetic Chlorophylls: Relative Orientation of β-Substituents on a Pyrrolic Ring Tunes NIR Absorption. J. Am. Chem. Soc. 2014, 136, 9598–9607. [Google Scholar] [CrossRef] [PubMed]
- Guberman-Pfeffer, M.J.; Lalisse, R.F.; Hewage, N.; Brückner, C.; Gascón, J.A. Origins of the Electronic Modulations of Bacterio- and Isobacteriodilactone Regioisomers. J. Phys. Chem. A 2019, 123, 7470–7485. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Rao, Y.; Liu, Y.; Jiang, L.; Xiong, J.; Fan, Y.J.; Shen, Z.; Sessler, J.L.; Zhang, J.L. Aromaticity versus Regioisomeric Effect of β-Substituents in Porphyrinoids. Phys. Chem. Chem. Phys. 2019, 21, 10152–10162. [Google Scholar] [CrossRef] [PubMed]
- Hewage, N.; Daddario, P.; Lau, K.S.F.; Guberman-Pfeffer, M.J.; Gascón, J.A.; Zeller, M.; Lee, C.O.; Khalil, G.E.; Gouterman, M.; Brückner, C. Bacterio- and Isobacteriodilactones by Stepwise or Direct Oxidations of meso-Tetrakis(pentafluorophenyl)porphyrin. J. Org. Chem. 2019, 84, 239–256. [Google Scholar] [CrossRef]
- Yu, Y.; Czepukojc, B.; Jacob, C.; Jiang, Y.; Zeller, M.; Brückner, C.; Zhang, J.-L. Porphothionolactones: Synthesis, Structure, Physical, and Chemical Properties of a Chemidosimeter for Hypochlorite. Org. Biomol. Chem. 2013, 11, 4613–4621. [Google Scholar] [CrossRef]
- Buchler, J.W. Synthesis and Properties of Metalloporphyrins. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 1, pp. 389–483. [Google Scholar]
- Khalil, G.; Gouterman, M.; Ching, S.; Costin, C.; Coyle, L.; Gouin, S.; Green, E.; Sadilek, M.; Wan, R.; Yearyean, J.; et al. Synthesis and Spectroscopic Characterization of Ni, Zn, Pd and Pt Tetra(pentafluorophenyl)porpholactone with Comparison to Mg, Zn, Y, Pd and Pt Metal Complexes of Tetra(pentafluorophenyl)porphine. J. Porphyr. Phthalocyanines 2002, 6, 135–145. [Google Scholar] [CrossRef]
- Ke, X.S.; Yang, B.Y.; Cheng, X.; Chan Sharon, L.F.; Zhang, J.L. Ytterbium(III) Porpholactones: β-Lactonization of Porphyrin Ligands Enhances Sensitization Efficiency of Lanthanide Near-Infrared Luminescence. Chem. Eur. J. 2014, 20, 4324–4333. [Google Scholar] [CrossRef]
- Ke, X.S.; Ning, Y.; Tang, J.; Hu, J.Y.; Yin, H.Y.; Wang, G.X.; Yang, Z.S.; Jie, J.; Liu, K.; Meng, Z.S.; et al. Gadolinium(III) Porpholactones as Efficient and Robust Singlet Oxygen Photosensitizers. Chem. Eur. J. 2016, 22, 9676–9686. [Google Scholar] [CrossRef]
- Liang, L.; Lv, H.; Yu, Y.; Wang, P.; Zhang, J.-L. Iron(III) Tetrakis(pentafluorophenyl)porpholactone Catalyzes Nitrogen Atom Transfer to C=C and C-H Bonds with Organic Azides. Dalton Trans. 2012, 41, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Cetin, A.; Ziegler, C.J. Structure and Catalytic Activity of a Manganese(III) Tetraphenylporpholactone. Dalton Trans. 2005, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Nurttila Sandra, S.; Dzik Wojciech, I.; Becker, R.; Rodgers, J.; Reek Joost, N.H. Tuning the Porphyrin Building Block in Self-Assembled Cages for Branched-Selective Hydroformylation of Propene. Chem. Eur. J. 2017, 23, 14769–14777. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.; Tehrani, A.A.; Fard, M.A.; Sadegh, B.M.M.; Khavasi, H.R. First Catalytic Application of Metal Complexes of Porpholactone and Dihydroxychlorin in the Sulfoxidation Reaction. Catal. Commun. 2009, 11, 232–235. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Wang, T.; Meng, Y.-S.; Rao, Y.; Wang, B.-W.; Zheng, J.; Gao, S.; Zhang, J.-L. Enhancing the Reactivity of Nickel(II) in Hydrogen Evolution Reactions (HERs) by β-Hydrogenation of Porphyrinoid Ligands. Chem. Sci. 2017, 8, 5953–5961. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Xue, H.; Wang, T.; Guo, Y.; Meng, Y.-S.; Li, X.; Zheng, J.; Brückner, C.; Rao, G.; Britt, R.D.; et al. Mimicking of Tunichlorin: Deciphering the Importance of a β-Hydroxyl Substituent on Boosting the Hydrogen Evolution Reaction. ACS Catal. 2020, 2177–2188. [Google Scholar] [CrossRef]
- To, W.-P.; Liu, Y.; Lau, T.-C.; Che, C.-M. A Robust Palladium(II)–Porphyrin Complex as Catalyst for Visible Light Induced Oxidative C–H Functionalization. Chem. Eur. J. 2013, 19, 5654–5664. [Google Scholar] [CrossRef]
- Yang, Z.-S.; Yao, Y.; Sedgwick, A.C.; Li, C.; Xia, Y.; Wang, Y.; Kang, L.; Wang, B.-W.; Su, H.; Gao, S.; et al. Rational Design of an “All-in-one” Phototheranostic. Chem. Sci. 2020, 11, 8204–8213. [Google Scholar] [CrossRef]
- Gouterman, M.; Callis, J.; Dalton, L.; Khalil, G.; Mebarki, Y.; Cooper, K.R.; Grenier, M. Dual Luminophor Pressure-sensitive Paint III. Application to Automotive Model Testing. Meas. Sci. Technol. 2004, 15, 1986–1994. [Google Scholar] [CrossRef]
- Khalil, G.E.; Costin, C.; Crafton, J.; Jones, G.; Grenoble, S.; Gouterman, M.; Callis, J.B.; Dalton, L.R. Dual-luminophor Pressure-sensitive Paint, I. Ratio of Reference to Sensor Giving a Small Temperature Dependency. Sens. Actuators B 2004, 97, 13–21. [Google Scholar] [CrossRef]
- Zelelow, B.; Khalil, G.E.; Phelan, G.; Carlson, B.; Gouterman, M.; Callis, J.B.; Dalton, L.R. Dual Luminophor Pressure Sensitive Paint II. Lifetime Based Measurement of Pressure and Temperature. Sens. Actuators B 2003, 96, 304–314. [Google Scholar] [CrossRef]
- Worlinsky, J.L.; Zarate, G.; Zeller, M.; Ghandehari, M.; Khalil, G.; Brückner, C. Tuning the Dynamic High pH Sensing Range of [meso-Tetraarylporpholactonato]M(II) Complexes by Variation of the Central Metal Ion, the Aryl Substituents, and Introduction of a β-Nitro Group. J. Porphyr. Phthalocyanines 2013, 17, 836–849. [Google Scholar] [CrossRef]
- Khalil, G.E.; Daddario, P.; Lau, K.S.F.; Imtiaz, S.; King, M.; Gouterman, M.; Sidelev, A.; Puran, N.; Ghandehari, M.; Brückner, C. meso-Tetraarylporpholactones as High pH Sensors. Analyst 2010, 135, 2125–2131. [Google Scholar] [CrossRef]
- Liu, E.; Ghandehari, M.; Brückner, C.; Khalil, G.; Worlinsky, J.; Jin, W.; Sidelev, A.; Hyland, M.A. Mapping High pH Levels in Hydrated Calcium Silicates. Cement Concrete Res. 2017, 95, 232–239. [Google Scholar] [CrossRef]
- Tang, J.; Chen, J.-J.; Jing, J.; Chen, J.-Z.; Lv, H.; Yu, Y.; Xub, P.; Zhang, J.-L. β-Lactonization of Fluorinated Porphyrin Enhances LDL Binding Affinity, Cellular Uptake with Selective Intracellular Localization. Chem. Sci. 2014, 5, 558–566. [Google Scholar] [CrossRef]
- Wan, J.R.; Gouterman, M.; Green, E.; Khalil, G.E. High performance Liquid Chromatography Separation and Analysis of Metallotetra(pentafluorophenyl)porpholactone. J. Liq. Chromatogr. 1994, 17, 2045–2056. [Google Scholar] [CrossRef]
- Köpke, T.; Pink, M.; Zaleski, J.M. Elucidation of the Extraordinary 4-Membered Pyrrole Ring-contracted Azeteoporphyrinoid as an Intermediate in Chlorin Oxidation. Chem. Commun. 2006, 4940–4942. [Google Scholar] [CrossRef]
- Brückner, C. The Breaking and Mending of meso-Tetraarylporphyrins: Transmuting the Pyrrolic Building Blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef]
- Yu, Y.; Lv, H.; Ke, X.; Yang, B.; Zhang, J.-L. Ruthenium-Catalyzed Oxidation of the Porphyrin ß-ß’-Pyrrolic Ring: A General and Efficient Approach to Porpholactones. Adv. Synth. Catal. 2012, 354, 3509–3516. [Google Scholar] [CrossRef]
- Spellane, P.J.; Gouterman, M.; Antipas, A.; Kim, S.; Liu, Y.C. Electronic Spectra and Four-orbital Energies of Free-base, Zinc, Copper, and Palladium Tetrakis(perfluorophenyl)porphyrins. Inorg. Chem. 1980, 19, 386–391. [Google Scholar] [CrossRef]
- Available through standard chemical supply houses but also suppliers specialized on porphyrins, such as Frontier Scientific. Available online: https://www.frontiersci.com (accessed on 21 September 2020).
- Available through standard chemical supply houses but also suppliers specialized on porphyrins, such as PorphyChem. Available online: https://www.porphychem.com (accessed on 21 September 2020).
- Dean, M.L.; Schmink, J.R.; Leadbeater, N.E.; Brückner, C. Microwave-promoted Insertion of Group 10 Metals into Free Base Porphyrins and Chlorins: Scope and Limitations. Dalton Trans. 2008, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yang, B.; Jing, J.; Yu, Y.; Zhang, J.; Zhang, J.-L. Dual Facet of Gold(III) in the Reactions of Gold(III) and Porphyrins. Dalton Trans. 2012, 41, 3116–3118. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A Simplified Synthesis for TPP. J. Org. Chem. 1967, 32, 476. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo Reactions Revisited: Synthesis of Tetraphenylporphyrins under Equilibrium Conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthesis of meso-substituted porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Buchler, J.W.; Dreher, C.; Künzel, F.M. Synthesis and Coordination Chemistry of Noble Metal Porphyrins. Struct. Bond. 1995, 84, 1–69. [Google Scholar]
- Brückner, C.; Rettig, S.J.; Dolphin, D. Formation of a meso-Tetraphenylsecochlorin and a Homoporphyrin with a Twist. J. Org. Chem. 1998, 63, 2094–2098. [Google Scholar] [CrossRef]
- Samankumara, L.P.; Zeller, M.; Krause, J.A.; Brückner, C. Syntheses, Structures, Modification, and Optical Properties of meso-Tetraaryl-2,3-dimethoxychlorin, and Two Isomeric meso-Tetraaryl-2,3,12,13-tetrahydroxybacteriochlorins. Org. Biomol. Chem. 2010, 8, 1951–1965. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Jenkins, H.A.; Brückner, C. Free Base meso-Tetraaryl-morpholinochlorins and Porpholactone from meso-Tetraaryl-2,3-dihydroxychlorin. Org. Lett. 2003, 5, 19–22. [Google Scholar] [CrossRef]
- Hewage, N.; Zeller, M.; Brückner, C. Oxidations of Chromene-annulated Chlorins. Org. Biomol. Chem. 2017, 15, 396–407. [Google Scholar] [CrossRef]
- Ogikubo, J.; Meehan, E.; Engle, J.T.; Ziegler, C.J.; Brückner, C. meso-Tetraphenyl-2-oxabacteriochlorins and meso-Tetraphenyl-2,12/13-dioxabacteriochlorins. J. Org. Chem. 2013, 78, 2840–2852. [Google Scholar] [CrossRef]
- Ogikubo, J.; Meehan, E.; Engle, J.T.; Ziegler, C.; Brückner, C. meso-Aryl-3-alkyl-2-oxachlorins. J. Org. Chem. 2012, 77, 6199–6207. [Google Scholar] [CrossRef] [PubMed]
- Ogikubo, J.; Brückner, C. Tunable meso-Tetraphenylalkyloxazolo-chlorins and -bacteriochlorins. Org. Lett. 2011, 13, 2380–2383. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.R.; Perez, M.J.; Brückner, C.; Weissleder, R. A Polymeric Nanoparticle Preparation that Eradicates Tumors. Nano Lett. 2005, 5, 2552–2556. [Google Scholar] [CrossRef]
- Akhigbe, J.; Haskoor, J.; Zeller, M.; Brückner, C. Porpholactams and Chlorolactams: Replacement of a β,β-Double Bond in meso-Tetraphenyl-porphyrins and -chlorins by a Lactam Moiety. Chem. Commun. 2011, 47, 8599–8601. [Google Scholar] [CrossRef] [PubMed]
- Akhigbe, J.; Haskoor, J.P.; Krause, J.A.; Zeller, M.; Brückner, C. Formation, Structure and Reactivity of meso-Tetraaryl-chlorolactones, -porpholactams, and chlorolactams, Porphyrin and Chlorin Analogues Incorporating Oxazolone or Imidazolone Moieties. Org. Biomol. Chem. 2013, 11, 3616–3628. [Google Scholar] [CrossRef]
- Luciano, M.P.; Akhigbe, J.; Ding, J.; Thuita, D.; Hamchand, R.; Zeller, M.; Brückner, C. An Alternate Route of Transforming meso-Tetraarylporphyrins to Porpholactams, and Their Conversion to Amine-Functionalized Imidazoloporphyrins. J. Org. Chem. 2018, 83, 9619–9630. [Google Scholar] [CrossRef]
- Brückner, C.; Atoyebi, A.O.; Girouard, D.; Lau, K.S.F.; Akhigbe, J.; Samankumara, L.; Damunupola, D.; Khalil, G.E.; Gouterman, M.; Krause, J.A.; et al. Stepwise Preparation of meso-Tetraphenyl- and meso-Tetrakis(4-trifluoromethylphenyl)bacteriodilactones and their Zinc(II) and Palladium(II) Complexes. Eur. J. Org. Chem. 2020, 2020, 475–482. [Google Scholar] [CrossRef]
Sample Availability: Samples of all porpholactones described are available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thuita, D.; Damunupola, D.; Brückner, C. Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes. Molecules 2020, 25, 4351. https://doi.org/10.3390/molecules25184351
Thuita D, Damunupola D, Brückner C. Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes. Molecules. 2020; 25(18):4351. https://doi.org/10.3390/molecules25184351
Chicago/Turabian StyleThuita, Damaris, Dinusha Damunupola, and Christian Brückner. 2020. "Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes" Molecules 25, no. 18: 4351. https://doi.org/10.3390/molecules25184351
APA StyleThuita, D., Damunupola, D., & Brückner, C. (2020). Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes. Molecules, 25(18), 4351. https://doi.org/10.3390/molecules25184351