
Design, Synthesis, In Vitro and In Silico Studies of New Thiazolylhydrazine-Piperazine Derivatives as Selective MAO-A Inhibitors

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
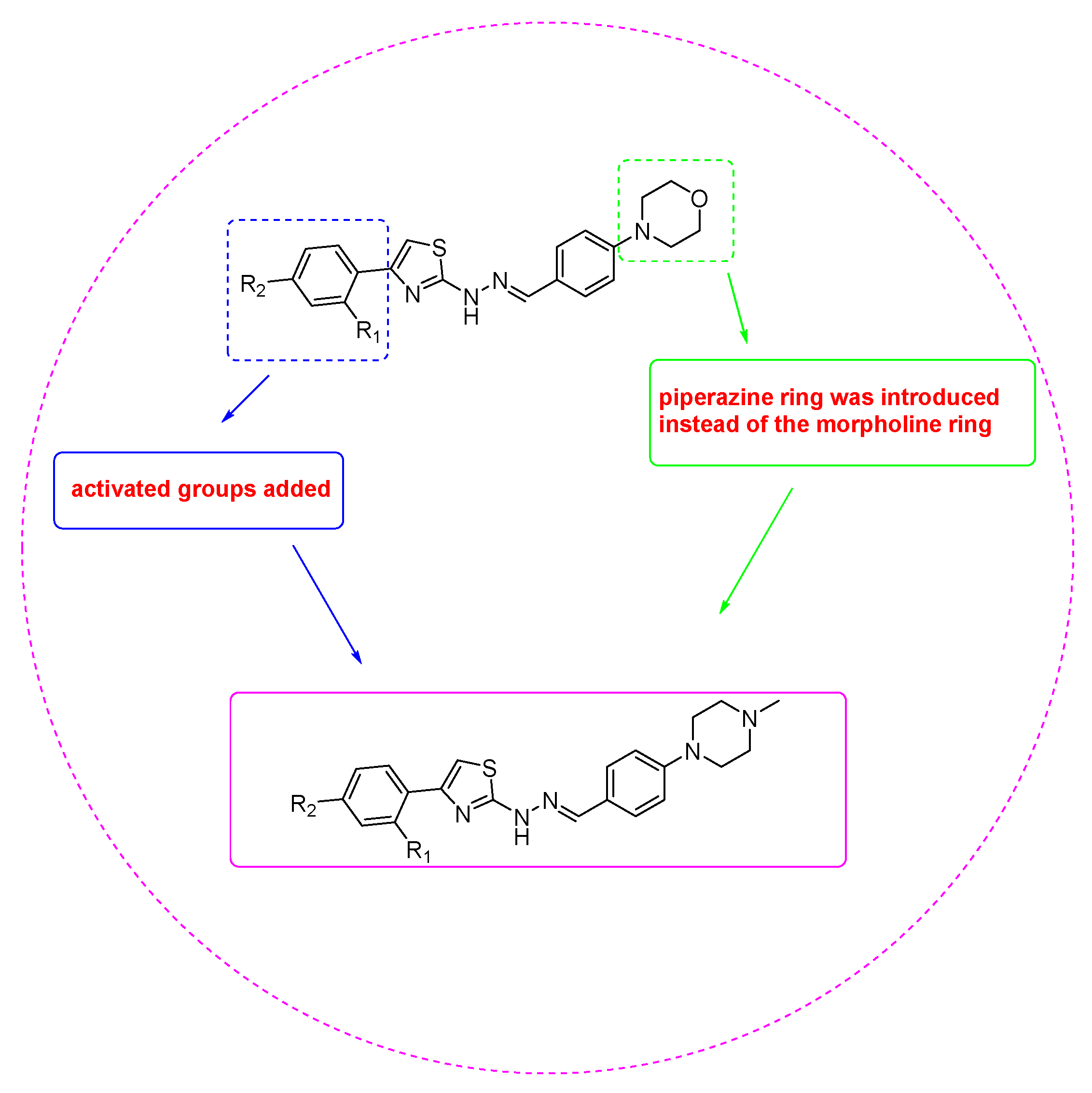
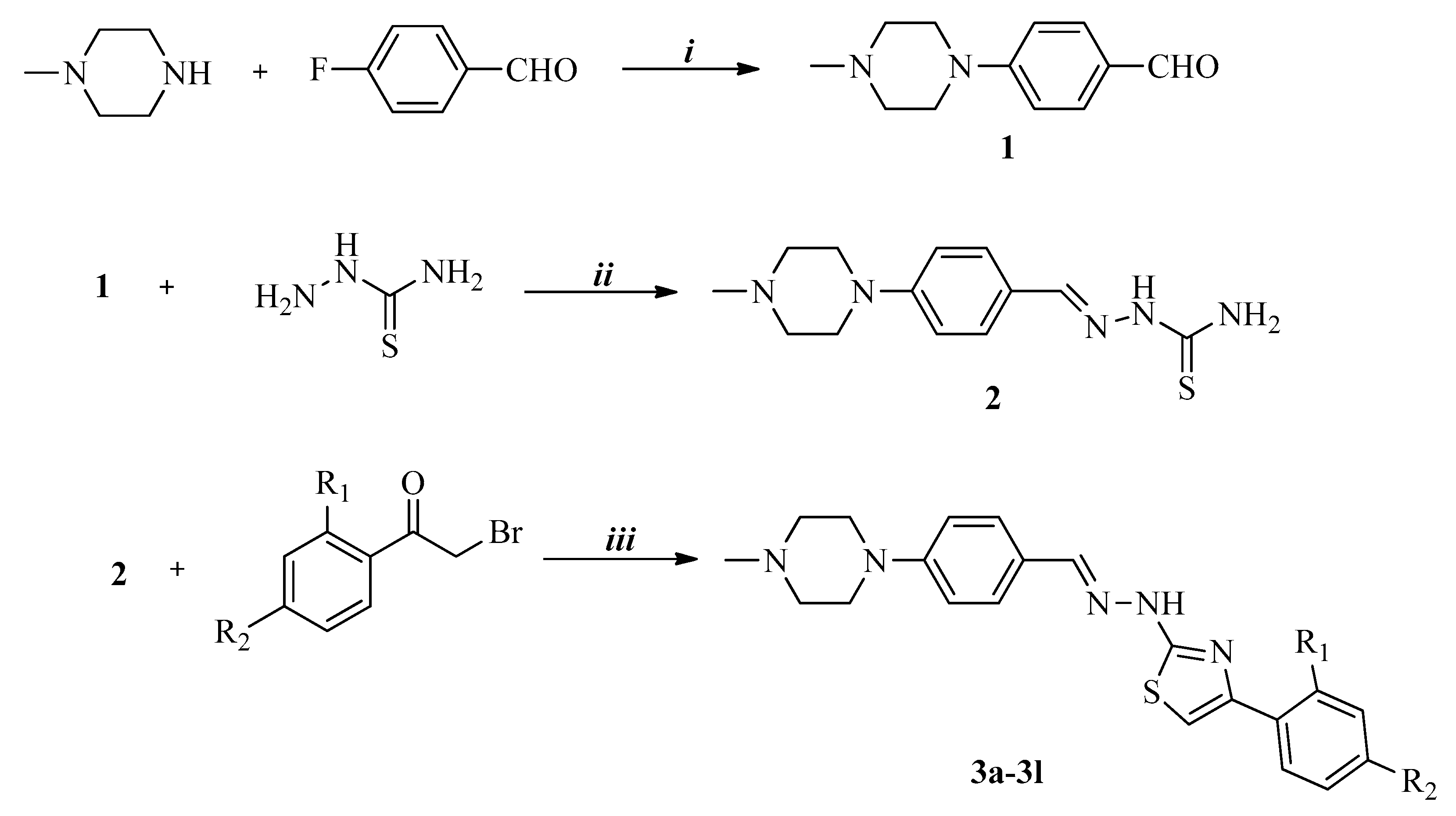
2.1. Chemistry
2.1.1. General Procedure for the Synthesis of the Compounds
Synthesis of 4-(4-Methylpiperazin-1-yl)benzaldehyde (1)
Synthesis of 2-[4-(4-Methylpiperazin-1-yl)benzylidene]hydrazinecarbothioamide (2)
Synthesis of 4-(2,4-Disubstitutedphenyl)-2-{2-[4-(4-methylpiperazin-1-yl)benzylidene]hydrazinyl} thiazoles (3a–3l)
2.2. In Vitro MAO-A and MAO-B Inhibition Assay
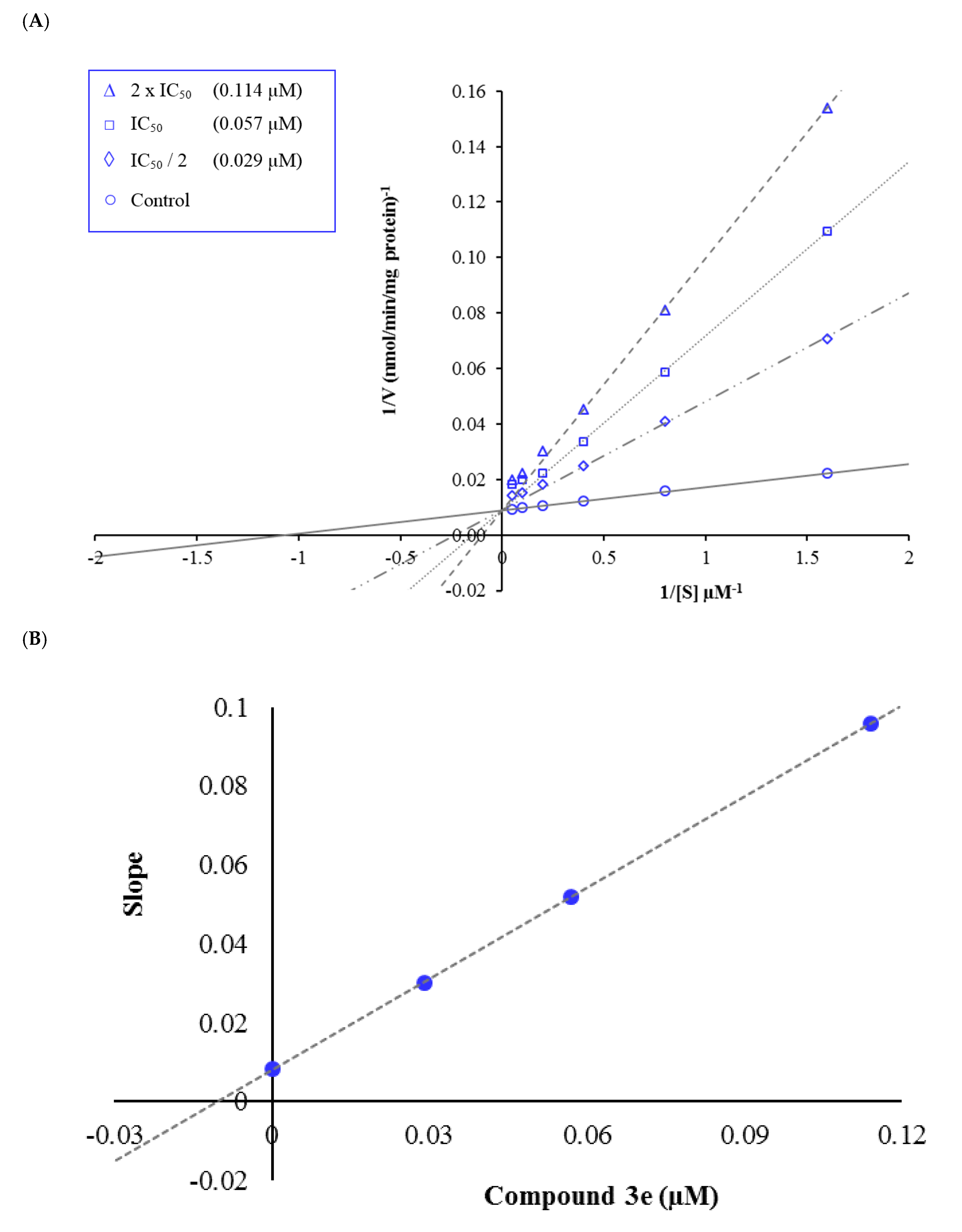
2.3. Enzyme Kinetic Studies
2.4. Cytotoxicity Assay
2.5. Prediction of ADME Parameters
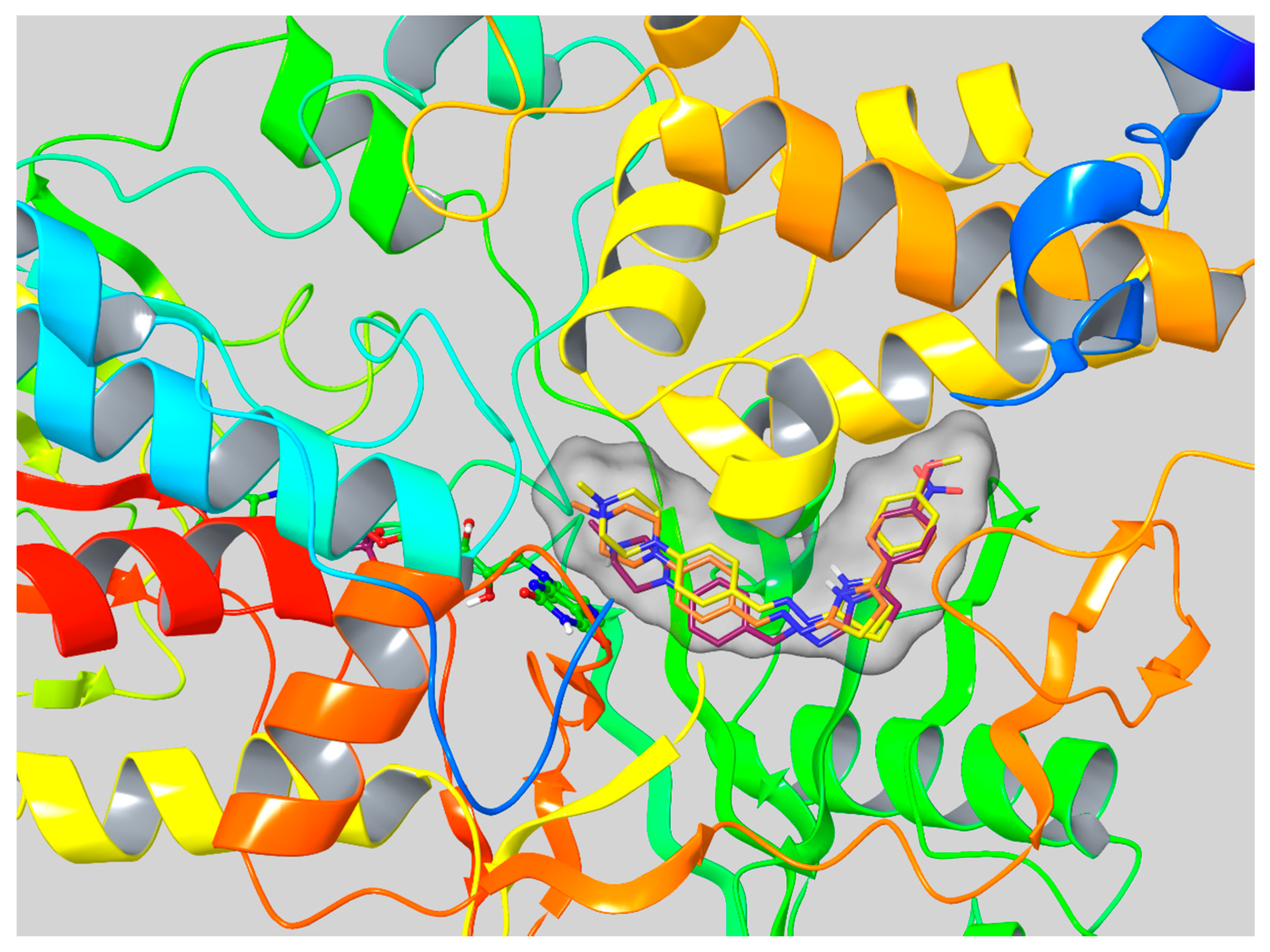
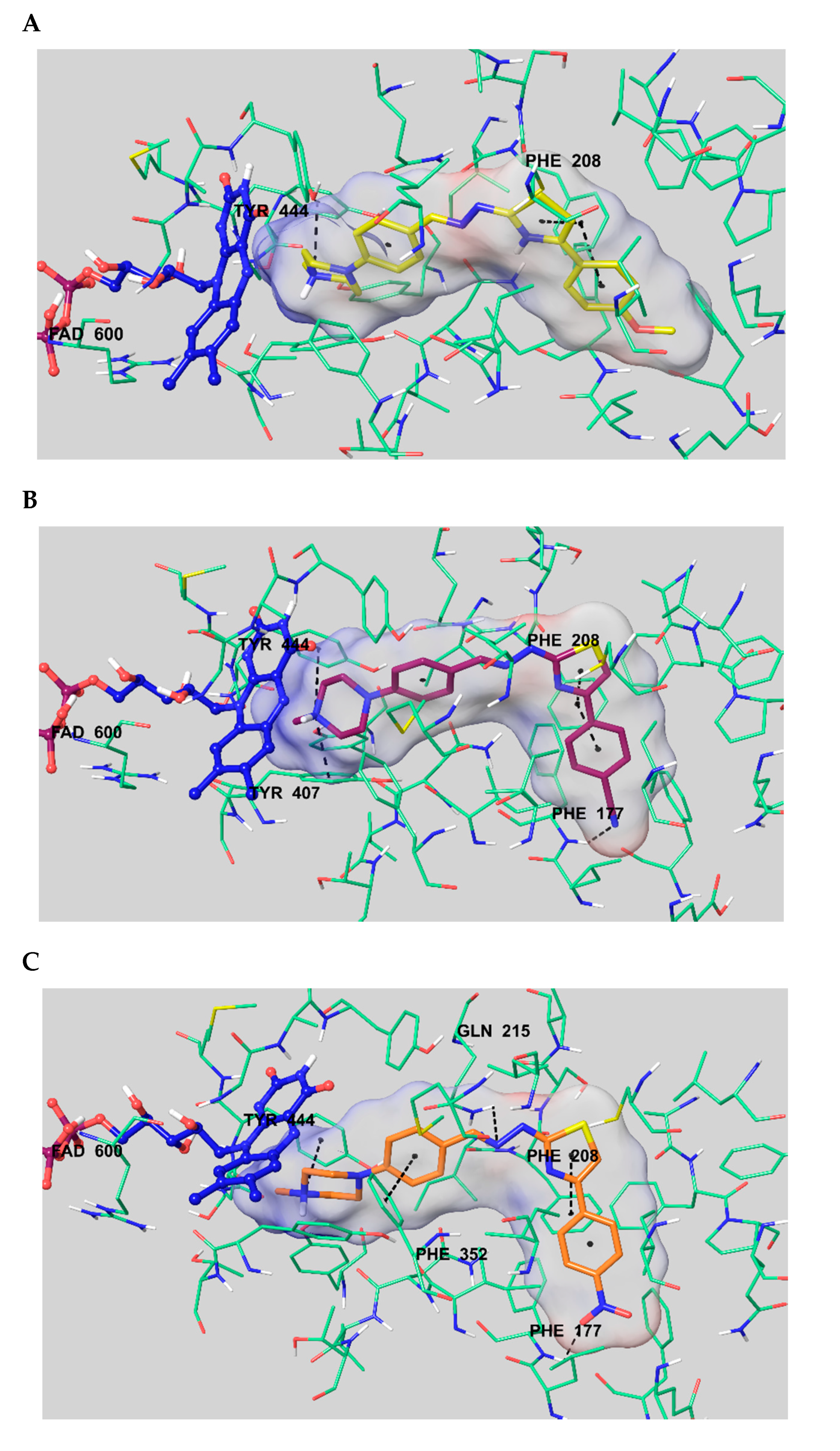
2.6. Molecular Docking Studies
3. Results and Discussion
3.1. Chemistry
3.2. MAO Inhibition Assay
3.3. Kinetic Studies of Enzyme Inhibition
3.4. Cytotoxicity Assay
3.5. Prediction of ADME Parameters
3.6. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sang, Z.; Wang, K.; Wang, H.; Yu, L.; Wang, H.; Ma, Q.; Ye, M.; Han, X.; Liu, W. Design, synthesis and biological evaluation of phthalimide-alkylamine derivatives as balanced multifunctional cholinesterase and monoamine oxidase-B inhibitors for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2017, 27, 5053–5059. [Google Scholar] [CrossRef]
- Li, S.; Lv, X.; Cheng, K.; Tian, Y.; Huang, X.; Kong, H.; Duan, Y.; Han, J.; Liao, C.; Xie, Z. Discovery of novel 2, 3-dihydro-1H-inden-1-amine derivatives as selective monoamine oxidase B inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Shetnev, A.; Shlenev, R.; Efimova, J.; Ivanovskii, S.; Tarasov, A.; Petzer, A.; Petzer, J.P. 1,3,4-Oxadiazol-2-ylbenzenesulfonamides as privileged structures for the inhibition of monoamine oxidase B. Bioorganic Med. Chem. Lett. 2019, 29, 126677. [Google Scholar] [CrossRef] [PubMed]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef]
- Desideri, N.; Monaco, L.P.; Fioravanti, R.; Biava, M.; Yáñez, M.; Alcaro, S.; Ortuso, F. (E)-3-Heteroarylidenechroman-4-ones as potent and selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 117, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xiong, J.; Guan, H.D.; Wang, C.H.; Lei, X.; Hu, J.F. Discovery, synthesis, biological evaluation and molecular docking study of (R)-5-methylmellein and its analogs as selective monoamine oxidase A inhibitors. Bioorganic Med. Chem. 2019, 27, 2027–2040. [Google Scholar] [CrossRef]
- Pignatello, R.; Mazzone, S.; Castelli, F.; Mazzone, P.; Raciti, G.; Mazzone, G. MAOI activity of thiosemicarbazides and related 2-thiazolylhydrazines. Die Pharm. 1994, 49, 272–276. [Google Scholar]
- Raciti, G.; Mazzone, P.; Raudino, A.; Mazzone, G.; Cambria, A. Inhibition of rat liver mitochondrial monoamine oxidase by hydrazine-thiazole derivatives: Structure-activity relationships. Bioorganic Med. Chem. 1995, 3, 1485–1491. [Google Scholar] [CrossRef]
- Secci, D.; Bolasco, A.; Carradori, S.; D’Ascenzio, M.; Nescatelli, R.; Yáñez, M. Recent advances in the development of selective human MAO-B inhibitors: (Hetero) arylidene-(4-substituted-thiazol-2-yl) hydrazines. Eur. J. Med. Chem. 2012, 58, 405–417. [Google Scholar] [CrossRef]
- Chimenti, F.; Bolasco, A.; Secci, D.; Chimenti, P.; Granese, A.; Carradori, S.; Yáñez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Investigations on the 2-thiazolylhydrazyne scaffold: Synthesis and molecular modeling of selective human monoamine oxidase inhibitors. Bioorganic Med. Chem. 2010, 18, 5715–5723. [Google Scholar] [CrossRef]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; Maccioni, E.; Cardia, M.C.; Yáñez, M.; Orallo, F.; et al. Synthesis, semipreparative HPLC separation, biological evaluation, and 3D-QSAR of hydrazothiazole derivatives as human monoamine oxidase B inhibitors. Bioorganic Med. Chem. 2010, 18, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Maccioni, E.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Befani, O.; Turini, P.; Alcaro, S.; Ortuso, F.; et al. Selective inhibitory activity against MAO and molecular modeling studies of 2-thiazolylhydrazone derivatives. J. Med. Chem. 2007, 50, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Gritsch, S.; Guccione, S.; Hoffmann, R.; Cambria, A.; Raciti, G.; Langer, T. A 3D QSAR study of monoamino oxidase-B inhibitors using the chemical function based pharmacophore generation approach. J. Enzy. Inh. 2011, 16, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Sağlık, B.N.; Çavuşoğlu, B.K.; Osmaniye, D.; Levent, S.; Çevik, U.A.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A.; Öztürk, Y. In vitro and in silico evaluation of new thiazole compounds as monoamine oxidase inhibitors. Bioorganic Chem. 2019, 85, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; Korkut, B.; Atlı, Ö.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis and biological assessment of new thiazolylhydrazine derivatives as selective and reversible hMAO-A inhibitors. Eur. J. Med. Chem. 2018, 144, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; İnci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of new hydrazone derivatives for MAO enzymes inhibitory activity. Molecules 2017, 22, 1381. [Google Scholar] [CrossRef]
- Ilgın, S.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; Acar Çevik, U.; Kaya Çavuşoğlu, B.; Özkay, Y.; Kaplancıklı, Z.A. Design and synthesis of new benzothiazole compounds as selective hMAO-B inhibitors. Molecules 2017, 22, 2187. [Google Scholar] [CrossRef]
- Can, Ö.D.; Osmaniye, D.; Demir Özkay, Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2017, 131, 92–106. [Google Scholar] [CrossRef]
- Sağlık, B.N.; Ilgın, S.; Özkay, Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole–piperazines. Bioorganic Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef]
- Patel, S.; Ghewala, N.; Suthar, A.; Shah, A. In-vitro cytotoxicity activity of Solanum nigrum extract against Hela cell line and Vero cell line. Int. J. Pharm. Pharm. Sci. 2009, 1, 38–46. [Google Scholar]
- QikProp, version 4.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2 Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 10.6; Schrödinger, LLC: New York, NY, USA, 2016.
- LigPrep, version 3.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Induced Fit Docking Protocol; Schrödinger Release 2016–2; Glide; Schrödinger, LLC: New York, NY, USA; Prime; Schrödinger, LLC: New York, NY, USA, 2016.
- Çavuşoğlu, B.K.; Sağlık, B.N.; Osmaniye, D.; Levent, S.; Acar Çevik, U.; Karaduman, A.B.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and biological evaluation of new thiosemicarbazone derivative Schiff bases as monoamine oxidase inhibitory agents. Molecules 2018, 23, 60. [Google Scholar] [CrossRef] [PubMed]
- Morcoss, M.M.; Abdelhafez, E.S.M.N.; Ibrahem, R.A.; Abdel-Rahman, H.M.; Abdel-Aziz, M.; Abou El-Ella, D.A. Design, synthesis, mechanistic studies and in silico ADME predictions of benzimidazole derivatives as novel antifungal agents. Bioorg. Chem. 2020, 101, 103956. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. ADME-SAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Franco, L.; Dominy Beryl, W.; Feeney Paul, J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swiss ADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Taylor, R. Organic fluorine hardly ever accepts hydrogen bonds. Chem-A Eur. J. Med. Chem. 1997, 3, 89–98. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–3l are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | Compound | R1 | R2 |
|---|---|---|---|---|---|
| 3a | -H | -H | 3g | -H | -Cl |
| 3b | -H | -CH3 | 3h | -H | -Br |
| 3c | -H | -OCH3 | 3i | -H | -Phenyl |
| 3d | -H | -CN | 3j | -CH3 | -CH3 |
| 3e | -H | -NO2 | 3k | -F | -F |
| 3f | -H | -F | 3l | -Cl | -Cl |
| Compounds | MAO-A % Inhibition | MAO-B % Inhibition | ||
|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−3 M | 10−4 M | |
| 3a | 68.137 ± 1.026 | 34.297 ± 0.851 | 32.258 ± 0.985 | 25.011 ± 0.721 |
| 3b | 70.957 ± 1.114 | 31.456 ± 0.732 | 34.553 ± 0.886 | 27.591 ± 0.649 |
| 3c | 92.075 ± 2.218 | 87.671 ± 1.874 | 30.336 ± 0.812 | 21.474 ± 0.879 |
| 3d | 90.192 ± 2.035 | 84.369 ± 1.808 | 36.648 ± 0.903 | 26.044 ± 0.836 |
| 3e | 95.314 ± 1.895 | 90.788 ± 1.728 | 39.102 ± 0.810 | 28.163 ± 0.791 |
| 3f | 75.942 ± 1.235 | 40.666 ± 0.980 | 30.655 ± 0.912 | 24.718 ± 0.854 |
| 3g | 73.661 ± 1.108 | 33.503 ± 0.833 | 31.250 ± 0.789 | 23.952 ± 0.623 |
| 3h | 70.753 ± 1.317 | 37.905 ± 0.796 | 34.362 ± 0.824 | 22.104 ± 0.796 |
| 3i | 66.197 ± 0.972 | 41.499 ± 0.870 | 30.589 ± 0.836 | 21.475 ± 0.809 |
| 3j | 72.308 ± 1.033 | 38.122 ± 0.798 | 28.143 ± 0.901 | 20.034 ± 0.792 |
| 3k | 79.991 ± 1.299 | 35.134 ± 0.833 | 29.573 ± 0.782 | 23.194 ± 0.876 |
| 3l | 73.521 ± 1.180 | 40.578 ± 0.914 | 33.667 ± 0.991 | 21.373 ± 0.769 |
| Moclobemide | 94.121 ± 2.760 | 82.143 ± 2.691 | - | - |
| Clorgiline | 96.940 ± 1.250 | 91.308 ± 1.305 | - | - |
| Selegiline | - | - | 98.258 ± 1.052 | 96.107 ± 1.165 |
| Compounds | MAO-A % Inhibition | IC50 (µM) | ||||||
|---|---|---|---|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | ||
| 3c | 92.075 ± 2.218 | 87.671 ± 1.874 | 75.428 ± 1.425 | 70.985 ± 1.095 | 48.336 ± 0.869 | 30.910 ± 0.711 | 21.785 ± 0.638 | 0.188 ± 0.008 |
| 3d | 90.192 ± 2.035 | 84.369 ± 1.808 | 81.059 ± 1.937 | 78.997 ± 1.247 | 49.589 ± 0.811 | 30.637 ± 0.699 | 18.107 ± 0.593 | 0.117 ± 0.004 |
| 3e | 95.314 ± 1.895 | 90.788 ± 1.728 | 85.025 ± 1.027 | 82.367 ± 1.392 | 63.942 ± 0.893 | 42.570 ± 0.835 | 23.018 ± 0.715 | 0.057 ± 0.002 |
| Moclobemide | 94.121 ± 2.760 | 82.143 ± 2.691 | 60.458 ± 2.559 | 36.151 ± 1.984 | 22.135 ± 0.337 | 18.166 ± 0.812 | 14.128 ± 0.725 | 6.061 ± 0.262 |
| Clorgiline | 96.940 ± 1.250 | 91.308 ± 1.305 | 87.635 ± 1.456 | 78.498 ± 1.024 | 65.235 ± 0.997 | 34.198 ± 0.841 | 22.477 ± 0.736 | 0.062 ± 0.002 |
| Compound | IC50 (µM) NIH/3T3 Cell Line | IC50 (µM) MAO-A Enzyme |
|---|---|---|
| 3e | >1000 | 0.057 ± 0.002 |
| Compound | MW | RB | DM | MV | DHB | AHB | PSA | logP | logS | PCaco | logBB | PMDCK | CNS | PM | %HOA | VRF | VRT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 377.506 | 4 | 2.979 | 1263.957 | 1 | 7 | 46.680 | 4.092 | −5.338 | 957.697 | 0.369 | 1032.177 | 1 | 2 | 100 | 0 | 0 |
| 3b | 391.533 | 4 | 2.503 | 1322.897 | 1 | 7 | 46.680 | 4.393 | −5.895 | 957.686 | 0.357 | 1032.164 | 1 | 3 | 100 | 0 | 1 |
| 3c | 407.532 | 5 | 3.620 | 1331.741 | 1 | 7.750 | 55.161 | 4.126 | −5.444 | 957.690 | 0.301 | 1032.168 | 1 | 3 | 100 | 0 | 0 |
| 3d | 402.516 | 5 | 9.298 | 1330.658 | 1 | 8.500 | 72.475 | 3.323 | −6.273 | 198.052 | −0.544 | 187.908 | 1 | 2 | 87.508 | 0 | 1 |
| 3e | 422.504 | 5 | 12.959 | 1346.359 | 1 | 8 | 95.346 | 3.384 | −5.568 | 98.431 | −0.912 | 88.255 | 0 | 3 | 82.432 | 0 | 0 |
| 3f | 395.497 | 4 | 5.390 | 1280.070 | 1 | 7 | 46.684 | 4.326 | −5.702 | 957.638 | 0.480 | 1866.285 | 2 | 2 | 100 | 0 | 1 |
| 3g | 411.951 | 4 | 5.330 | 1308.076 | 1 | 7 | 46.682 | 4.583 | −6.075 | 957.661 | 0.535 | 2546.410 | 2 | 2 | 100 | 0 | 1 |
| 3h | 456.402 | 4 | 5.015 | 1316.988 | 1 | 7 | 46.682 | 4.660 | −6.190 | 957.675 | 0.547 | 2737.950 | 2 | 2 | 100 | 0 | 1 |
| 3i | 453.604 | 5 | 2.742 | 1496.172 | 1 | 7 | 46.680 | 5.687 | −7.304 | 957.700 | 0.269 | 1032.180 | 1 | 2 | 100 | 1 | 1 |
| 3j | 405.560 | 4 | 3.108 | 1347.245 | 1 | 7 | 41.791 | 4.557 | −5.748 | 1152.894 | 0.472 | 1261.337 | 2 | 4 | 100 | 0 | 1 |
| 3k | 413.487 | 4 | 4.457 | 1289.708 | 1 | 7 | 45.242 | 4.487 | −5.929 | 994.545 | 0.573 | 2862.946 | 2 | 2 | 100 | 0 | 1 |
| 3l | 446.396 | 4 | 4.564 | 1316.883 | 1 | 7 | 43.269 | 4.759 | −5.928 | 1060.430 | 0.695 | 4373.785 | 2 | 2 | 100 | 0 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sağlık, B.N.; Cebeci, O.; Acar Çevik, U.; Osmaniye, D.; Levent, S.; Kaya Çavuşoğlu, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, In Vitro and In Silico Studies of New Thiazolylhydrazine-Piperazine Derivatives as Selective MAO-A Inhibitors. Molecules 2020, 25, 4342. https://doi.org/10.3390/molecules25184342
Sağlık BN, Cebeci O, Acar Çevik U, Osmaniye D, Levent S, Kaya Çavuşoğlu B, Ilgın S, Özkay Y, Kaplancıklı ZA. Design, Synthesis, In Vitro and In Silico Studies of New Thiazolylhydrazine-Piperazine Derivatives as Selective MAO-A Inhibitors. Molecules. 2020; 25(18):4342. https://doi.org/10.3390/molecules25184342
Chicago/Turabian StyleSağlık, Begüm Nurpelin, Osman Cebeci, Ulviye Acar Çevik, Derya Osmaniye, Serkan Levent, Betül Kaya Çavuşoğlu, Sinem Ilgın, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2020. "Design, Synthesis, In Vitro and In Silico Studies of New Thiazolylhydrazine-Piperazine Derivatives as Selective MAO-A Inhibitors" Molecules 25, no. 18: 4342. https://doi.org/10.3390/molecules25184342
APA StyleSağlık, B. N., Cebeci, O., Acar Çevik, U., Osmaniye, D., Levent, S., Kaya Çavuşoğlu, B., Ilgın, S., Özkay, Y., & Kaplancıklı, Z. A. (2020). Design, Synthesis, In Vitro and In Silico Studies of New Thiazolylhydrazine-Piperazine Derivatives as Selective MAO-A Inhibitors. Molecules, 25(18), 4342. https://doi.org/10.3390/molecules25184342