
Biased Opioid Ligands


Abstract
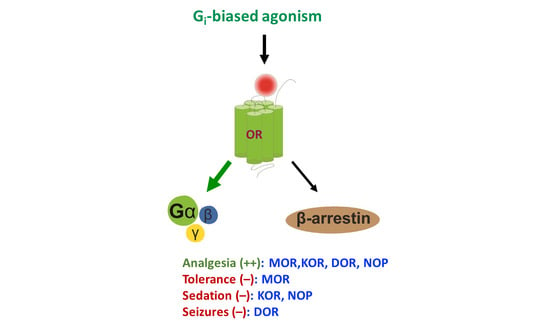
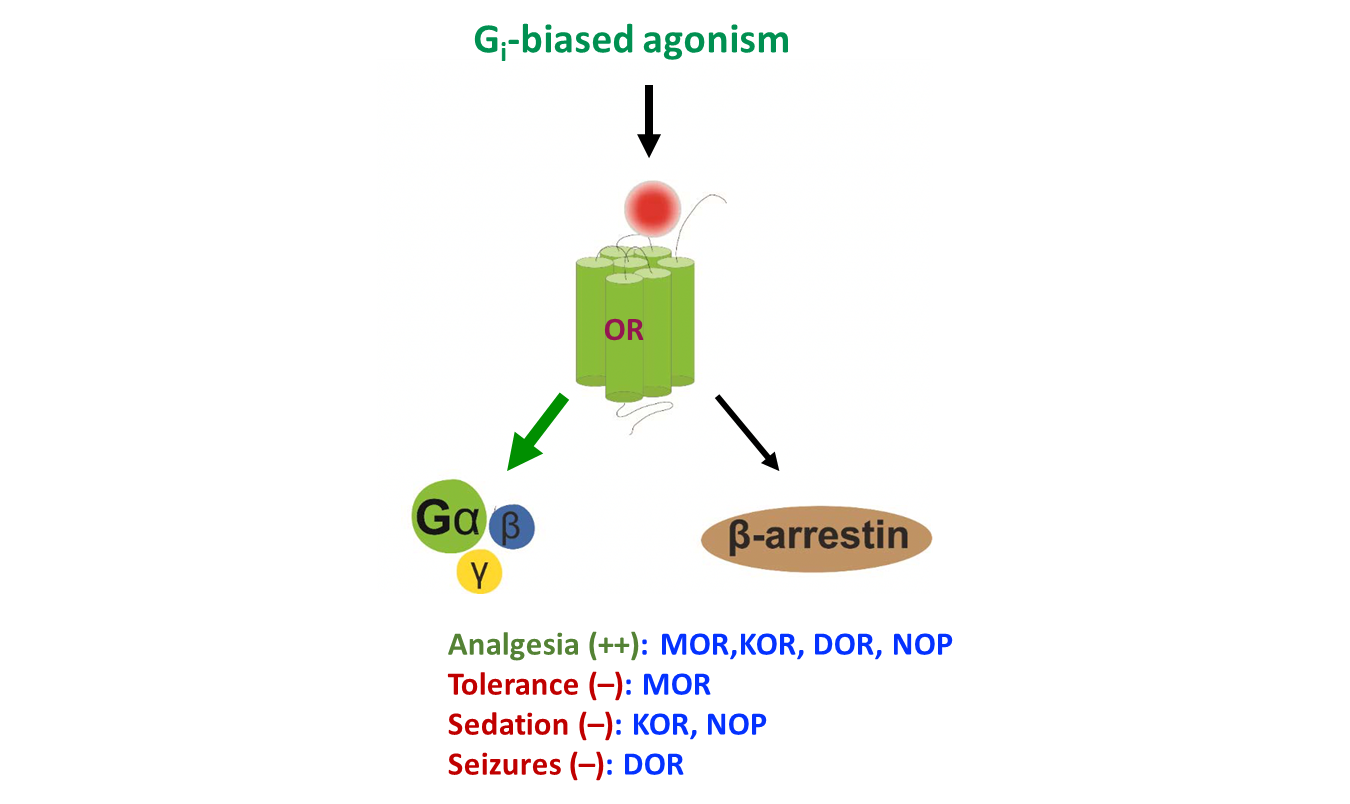
1. Introduction
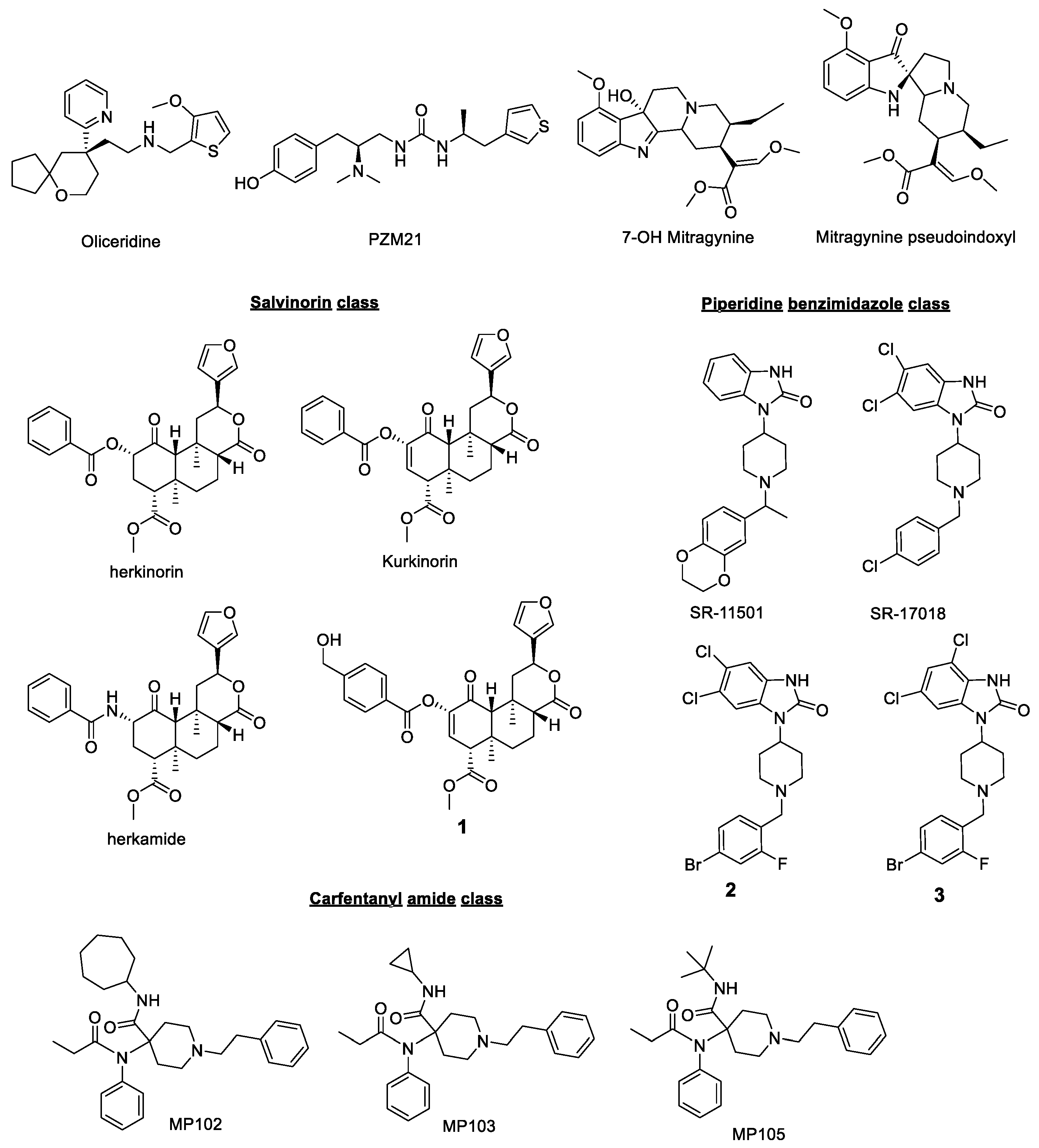
2. MOR Biased Agonism
- Oliceridine/TRV130
- PZM21
- 7-OH mitragynine/Mitragynine pseudoindoxyl
- Herkinorin/Kurkinorin
- Piperidine benzimidazoles
- Carfentanyl amides
- Controversy on biased agonists of the MOR
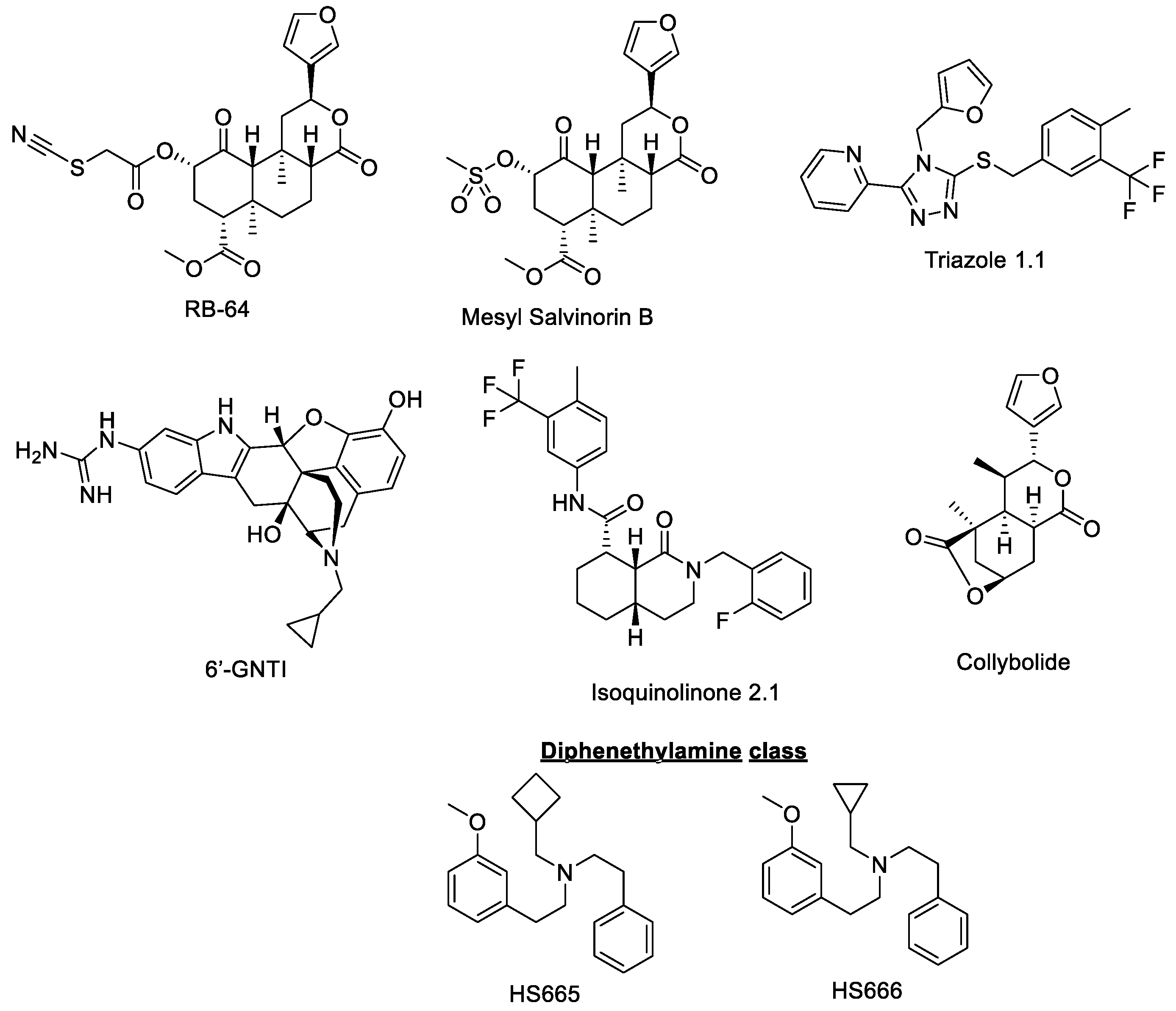
3. KOR Biased Agonism
- RB64
- Mesyl salvinorin B
- Triazole 1.1
- Diphenethylamines HS665 and HS666
- 6′-GNTI
- Isoquinolinone 2.1
- Collybolide
4. Biased Agonism on DOR
- DOR and biased signaling: mechanistic overview
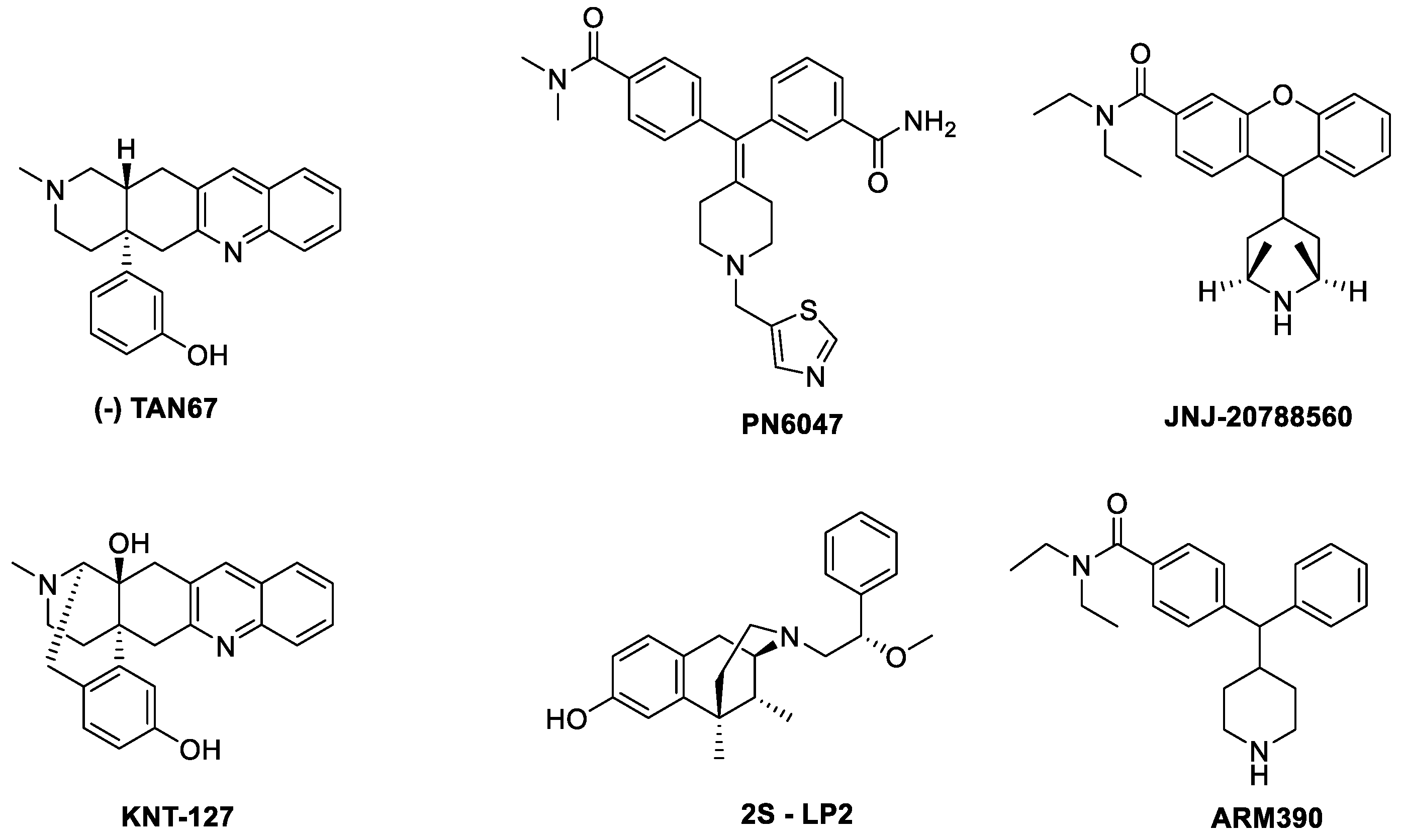
- TAN67
- PN6047
- KNT127
- JNJ 20788560
- 2S-LP2
- ARM390
5. Biased Agonism on NOP Receptor
- Mechanism of biased signaling on NOP receptor
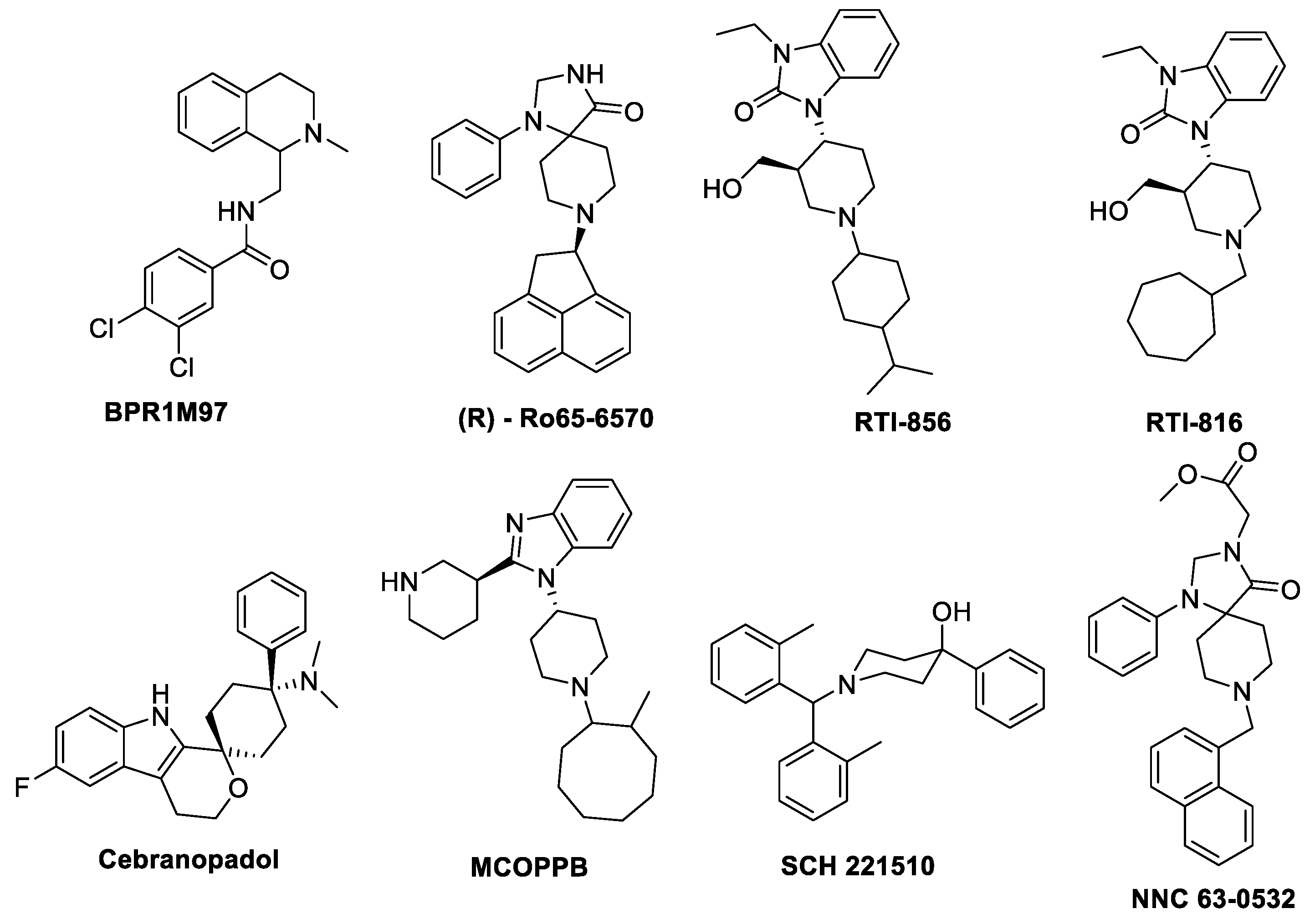
- BPR1M97
- Ro 65-6570
- SCH221510
- Cebranopadol
- MCOPPB
- NNC 63-0532
- RTI–819 and RTI–856
6. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.K.S.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zemmel, R. Value of novelty? Nat. Rev. Drug Discov. 2002, 1, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Roth, B.L. New Technologies for Elucidating Opioid Receptor Function. Trends Pharmacol. Sci. 2016, 37, 279–289. [Google Scholar] [CrossRef] [PubMed]
- DeWeerdt, S. Tracing the US opioid crisis to its roots. Nature 2019, 573, S10–S12. [Google Scholar] [CrossRef]
- Deupi, X.; Kobilka, B. Activation of G Protein–Coupled Receptors. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 2007; Volume 74, pp. 137–166. ISBN 978-0-12-034288-4. [Google Scholar]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional Selectivity at the -Opioid Receptor: Implications for Understanding Opioid Analgesia and Tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef]
- Franco, R.; Aguinaga, D.; Jiménez, J.; Lillo, J.; Martínez-Pinilla, E.; Navarro, G. Biased receptor functionality versus biased agonism in G-protein-coupled receptors. Biomol. Concepts 2018, 9, 143–154. [Google Scholar] [CrossRef]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Stanczyk, M.A.; Kandasamy, R. Biased agonism: The quest for the analgesic holy grail. PAIN Rep. 2018, 3, e650. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Mores, K.L.; Cassell, R.J.; van Rijn, R.M. Arrestin recruitment and signaling by G protein-coupled receptor heteromers. Neuropharmacology 2019, 152, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Fields, H.L. The Doctor’s Dilemma: Opiate Analgesics and Chronic Pain. Neuron 2011, 69, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Charlton, S.J. Biased Agonism in Drug Discovery—Is It Too Soon to Choose a Path? Mol. Pharmacol. 2018, 93, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M. Morphine Side Effects in -Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Liu, C.; Kang, J.; Yang, J.; Pei, G.; Wu, C. Improvement of Morphine-Mediated Analgesia by Inhibition of β-Arrestin 2 Expression in Mice Periaqueductal Gray Matter. Int. J. Mol. Sci. 2009, 10, 954–963. [Google Scholar] [CrossRef]
- Yang, C.-H.; Huang, H.-W.; Chen, K.-H.; Chen, Y.-S.; Sheen-Chen, S.-M.; Lin, C.-R. Antinociceptive potentiation and attenuation of tolerance by intrathecal β-arrestin 2 small interfering RNA in rats. Br. J. Anaesth. 2011, 107, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; DeWire, S.M.; Crombie, A.L.; et al. Structure–Activity Relationships and Discovery of a G Protein Biased μ Opioid Receptor Ligand, [(3-Methoxythiophen-2-yl)methyl]({2-[(9 R )-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine (TRV130), for the Treatment of Acute Severe Pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef]
- Gutridge, A.M.; Robins, M.T.; Cassell, R.J.; Uprety, R.; Mores, K.L.; Ko, M.J.; Pasternak, G.W.; Majumdar, S.; van Rijn, R.M. G protein-biased kratom-alkaloids and synthetic carfentanil-amide opioids as potential treatments for alcohol use disorder. Br. J. Pharmacol. 2020, 177, 1497–1513. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception: PZM21 depresses respiration. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef]
- Váradi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabó, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef]
- Tidgewell, K.; Groer, C.E.; Harding, W.W.; Lozama, A.; Schmidt, M.; Marquam, A.; Hiemstra, J.; Partilla, J.S.; Dersch, C.M.; Rothman, R.B.; et al. Herkinorin Analogues with Differential β-Arrestin-2 Interactions. J. Med. Chem. 2008, 51, 2421–2431. [Google Scholar] [CrossRef]
- Crowley, R.S.; Riley, A.P.; Sherwood, A.M.; Groer, C.E.; Shivaperumal, N.; Biscaia, M.; Paton, K.; Schneider, S.; Provasi, D.; Kivell, B.M.; et al. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting μ Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem. 2016, 59, 11027–11038. [Google Scholar] [CrossRef]
- Crowley, R.S.; Riley, A.P.; Alder, A.F.; Anderson, R.J.; Luo, D.; Kaska, S.; Maynez, P.; Kivell, B.M.; Prisinzano, T.E. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Design, Synthesis, and Evaluation of Analogues with Improved Potency and G-protein Activation Bias at the μ-Opioid Receptor. ACS Chem. Neurosci. 2020, 11, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, N.M.; Schmid, C.L.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Chen, Y.T.; Cameron, M.D.; Bohn, L.M.; Bannister, T.D. Optimization of a Series of Mu Opioid Receptor (MOR) Agonists with High G Protein Signaling Bias. J. Med. Chem. 2018, 61, 8895–8907. [Google Scholar] [CrossRef] [PubMed]
- Commissioner, O. FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings (accessed on 7 September 2020).
- Pedersen, M.F.; Wróbel, T.M.; Märcher-Rørsted, E.; Pedersen, D.S.; Møller, T.C.; Gabriele, F.; Pedersen, H.; Matosiuk, D.; Foster, S.R.; Bouvier, M.; et al. Biased agonism of clinically approved μ-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology 2020, 166, 107718. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-Y.; Li, W.-W.; Nwaneshiudu, C.; Irvine, K.-A.; Clark, J.D. Pharmacological Characters of Oliceridine, a μ-Opioid Receptor G-Protein–Biased Ligand in Mice. Anesth. Analg. 2019, 129, 1414–1421. [Google Scholar] [CrossRef]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef]
- Kudla, L.; Bugno, R.; Skupio, U.; Wiktorowska, L.; Solecki, W.; Wojtas, A.; Golembiowska, K.; Zádor, F.; Benyhe, S.; Buda, S.; et al. Functional characterization of a novel opioid, PZM21, and its effects on the behavioural responses to morphine. Br. J. Pharmacol. 2019, 176, 4434–4445. [Google Scholar] [CrossRef]
- Yaksh, T.L.; Eddinger, K.A.; Kokubu, S.; Wang, Z.; DiNardo, A.; Ramachandran, R.; Zhu, Y.; He, Y.; Weren, F.; Quang, D.; et al. Mast Cell Degranulation and Fibroblast Activation in the Morphine-induced Spinal Mass: Role of Mas-related G Protein-coupled Receptor Signaling. Anesthesiology 2019, 131, 132–147. [Google Scholar] [CrossRef]
- Hassan, Z.; Muzaimi, M.; Navaratnam, V.; Yusoff, N.H.M.; Suhaimi, F.W.; Vadivelu, R.; Vicknasingam, B.K.; Amato, D.; von Hörsten, S.; Ismail, N.I.W.; et al. From Kratom to mitragynine and its derivatives: Physiological and behavioural effects related to use, abuse, and addiction. Neurosci. Biobehav. Rev. 2013, 37, 138–151. [Google Scholar] [CrossRef]
- Matsumoto, K.; Horie, S.; Ishikawa, H.; Takayama, H.; Aimi, N.; Ponglux, D.; Watanabe, K. Antinociceptive effect of 7-hydroxymitragynine in mice: Discovery of an orally active opioid analgesic from the Thai medicinal herb Mitragyna speciosa. Life Sci. 2004, 74, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Horie, S.; Takayama, H.; Ishikawa, H.; Aimi, N.; Ponglux, D.; Murayama, T.; Watanabe, K. Antinociception, tolerance and withdrawal symptoms induced by 7-hydroxymitragynine, an alkaloid from the Thai medicinal herb Mitragyna speciosa. Life Sci. 2005, 78, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Hatori, Y.; Murayama, T.; Tashima, K.; Wongseripipatana, S.; Misawa, K.; Kitajima, M.; Takayama, H.; Horie, S. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur. J. Pharmacol. 2006, 549, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, A.C.; Uprety, R.; Grinnell, S.G.; Langreck, C.; Pekarskaya, E.A.; Le Rouzic, V.; Ansonoff, M.; Gassaway, M.M.; Pintar, J.E.; Pasternak, G.W.; et al. 7-Hydroxymitragynine Is an Active Metabolite of Mitragynine and a Key Mediator of Its Analgesic Effects. ACS Cent. Sci. 2019, 5, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Hemby, S.E.; McIntosh, S.; Leon, F.; Cutler, S.J.; McCurdy, C.R. Abuse liability and therapeutic potential of the Mitragyna speciosa (kratom) alkaloids mitragynine and 7-hydroxymitragynine: Kratom abuse liability. Addict. Biol. 2018. [Google Scholar] [CrossRef]
- Groer, C.E.; Tidgewell, K.; Moyer, R.A.; Harding, W.W.; Rothman, R.B.; Prisinzano, T.E.; Bohn, L.M. An Opioid Agonist that Does Not Induce μ-Opioid Receptor—Arrestin Interactions or Receptor Internalization. Mol. Pharmacol. 2007, 71, 549–557. [Google Scholar] [CrossRef]
- Butelman, E.R.; Kreek, M.J. Salvinorin A, a kappa-opioid receptor agonist hallucinogen: Pharmacology and potential template for novel pharmacotherapeutic agents in neuropsychiatric disorders. Front. Pharmacol. 2015, 6, 190. [Google Scholar] [CrossRef]
- Wu, Z.; Hruby, V.J. Toward a Universal μ-Agonist Template for Template-Based Alignment Modeling of Opioid Ligands. ACS Omega 2019, 4, 17457–17476. [Google Scholar] [CrossRef]
- Majumdar, S.; Devi, L.A. Strategy for making safer opioids bolstered. Nature 2018, 553, 286–288. [Google Scholar] [CrossRef]
- Váradi, A.; Palmer, T.C.; Haselton, N.; Afonin, D.; Subrath, J.J.; Le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Marrone, G.F.; Borics, A.; et al. Synthesis of Carfentanil Amide Opioids Using the Ugi Multicomponent Reaction. ACS Chem. Neurosci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmiedel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020, 177, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Carr, G.V.; Mague, S.D. p38: The Link between the κ-Opioid Receptor and Dysphoria. J. Neurosci. 2008, 28, 2299–2300. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, J.M.; Messinger, D.I.; Knakal, C.R.; Kuhar, J.R.; Schattauer, S.S.; Bruchas, M.R.; Zweifel, L.S.; Kieffer, B.L.; Phillips, P.E.M.; Chavkin, C. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J. Neurosci. 2015, 35, 12917–12931. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.; Lotts, T.; Antal, A.; Bernhard, J.; Ständer, S. Systemic Kappa Opioid Receptor Agonists in the Treatment of Chronic Pruritus: A Literature Review. Acta Derm. Venereol. 2012, 92, 555–560. [Google Scholar] [CrossRef]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G Protein-Biased κ-Opioid Receptor Agonist RB-64 is Analgesic with a Unique Spectrum of Activities In Vivo. J. Pharmacol. Exp. Ther. 2014, 352, 98–109. [Google Scholar] [CrossRef]
- Kivell, B.; Paton, K.; Kumar, N.; Morani, A.; Culverhouse, A.; Shepherd, A.; Welsh, S.; Biggerstaff, A.; Crowley, R.; Prisinzano, T. Kappa Opioid Receptor Agonist Mesyl Sal B Attenuates Behavioral Sensitization to Cocaine with Fewer Aversive Side effects than Salvinorin A in Rodents. Molecules 2018, 23, 2602. [Google Scholar] [CrossRef]
- Brust, T.F.; Morgenweck, J.; Kim, S.A.; Rose, J.H.; Locke, J.L.; Schmid, C.L.; Zhou, L.; Stahl, E.L.; Cameron, M.D.; Scarry, S.M.; et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 2016, 9, ra117. [Google Scholar] [CrossRef]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice: HS666 produces analgesia without causing aversion. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef]
- Rives, M.-L.; Rossillo, M.; Liu-Chen, L.-Y.; Javitch, J.A. 6′-Guanidinonaltrindole (6′-GNTI) Is a G Protein-biased κ-Opioid Receptor Agonist That Inhibits Arrestin Recruitment. J. Biol. Chem. 2012, 287, 27050–27054. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Streicher, J.M.; Groer, C.E.; Munro, T.A.; Zhou, L.; Bohn, L.M. Functional Selectivity of 6′-Guanidinonaltrindole (6′-GNTI) at κ-Opioid Receptors in Striatal Neurons. J. Biol. Chem. 2013, 288, 22387–22398. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lovell, K.M.; Frankowski, K.J.; Slauson, S.R.; Phillips, A.M.; Streicher, J.M.; Stahl, E.; Schmid, C.L.; Hodder, P.; Madoux, F.; et al. Development of Functionally Selective, Small Molecule Agonists at Kappa Opioid Receptors. J. Biol. Chem. 2013, 288, 36703–36716. [Google Scholar] [CrossRef]
- Gupta, A.; Gomes, I.; Bobeck, E.N.; Fakira, A.K.; Massaro, N.P.; Sharma, I.; Cavé, A.; Hamm, H.E.; Parello, J.; Devi, L.A. Collybolide is a novel biased agonist of κ-opioid receptors with potent antipruritic activity. Proc. Natl. Acad. Sci. USA 2016, 113, 6041–6046. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Crowley, R.; Prisinzano, T.; Kreek, M.J. Effects of mesyl salvinorin B alone and in combination with naltrexone on alcohol deprivation effect in male and female mice. Neurosci. Lett. 2018, 673, 19–23. [Google Scholar] [CrossRef]
- Lovell, K.M.; Frankowski, K.J.; Stahl, E.L.; Slauson, S.R.; Yoo, E.; Prisinzano, T.E.; Aubé, J.; Bohn, L.M. Structure–Activity Relationship Studies of Functionally Selective Kappa Opioid Receptor Agonists that Modulate ERK 1/2 Phosphorylation While Preserving G Protein Over βArrestin2 Signaling Bias. ACS Chem. Neurosci. 2015, 6, 1411–1419. [Google Scholar] [CrossRef]
- Shim, W.-S.; Oh, U. Histamine-Induced Itch and its Relationship with Pain. Mol. Pain 2008, 4. [Google Scholar] [CrossRef]
- Huskinson, S.L.; Platt, D.M.; Brasfield, M.; Follett, M.E.; Prisinzano, T.E.; Blough, B.E.; Freeman, K.B. Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys. Psychopharmacology 2020, 237, 2075–2087. [Google Scholar] [CrossRef]
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and Pharmacological Evaluation of a Diphenethylamine Derivative (HS665), a Highly Potent and Selective κ Opioid Receptor Agonist. J. Med. Chem. 2012, 55, 10302–10306. [Google Scholar] [CrossRef]
- Fortin, M.; Degryse, M.; Petit, F.; Hunt, P.F. The dopamine D2 agonists RU 24213 and RU 24926 are also KAPPA-opioid receptor antagonists. Neuropharmacology 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Erli, F.; Guerrieri, E.; Ben Haddou, T.; Lantero, A.; Mairegger, M.; Schmidhammer, H.; Spetea, M. Highly Potent and Selective New Diphenethylamines Interacting with the κ-Opioid Receptor: Synthesis, Pharmacology, and Structure–Activity Relationships. J. Med. Chem. 2017, 60, 7579–7590. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.; Reed, B.; Erazo, J.; Ben-Ezra, A.; Kreek, M.J. Signaling properties of structurally diverse kappa opioid receptor ligands: Towards in vitro models of in vivo responses. ACS Chem. Neurosci. 2019, 9b00195. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. USA 2005, 102, 9050–9055. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Scopton, A.P.; Rives, M.-L.; Bikbulatov, R.V.; Polepally, P.R.; Brown, P.J.; Kenakin, T.; Javitch, J.A.; Zjawiony, J.K.; Roth, B.L. Identification of Novel Functionally Selective κ-Opioid Receptor Scaffolds. Mol. Pharmacol. 2014, 85, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zangrandi, L.; Burtscher, J.; MacKay, J.P.; Colmers, W.F.; Schwarzer, C. The G-protein biased partial κ opioid receptor agonist 6′-GNTI blocks hippocampal paroxysmal discharges without inducing aversion: G-protein biased κ-receptor agonists in epilepsy. Br. J. Pharmacol. 2016, 173, 1756–1767. [Google Scholar] [CrossRef]
- Lu, K.; Luo, T.; Xiang, Z.; You, Z.; Fathi, R.; Chen, J.; Yang, Z. A Concise and Diversity-Oriented Strategy for the Synthesis of Benzofurans and Indoles via Ugi and Diels−Alder Reactions. J. Comb. Chem. 2005, 7, 958–967. [Google Scholar] [CrossRef]
- Frankowski, K.J.; Ghosh, P.; Setola, V.; Tran, T.B.; Roth, B.L.; Aubé, J. N-Alkyl-octahydroisoquinolin-1-one-8-carboxamides: Selective and Nonbasic κ-Opioid Receptor Ligands. ACS Med. Chem. Lett. 2010, 1, 189–193. [Google Scholar] [CrossRef]
- Bui, A.-M.; Cavé, A.; Janot, M.-M.; Parello, J.; Potier, P.; Scheidegger, U. Isolement et analyse structurale du collybolide, nouveau sesquiterpene extrait de Collybia maculata alb. et sch. ex fries (basidiomycetes). Tetrahedron 1974, 30, 1327–1336. [Google Scholar] [CrossRef]
- Erspamer, V.; Melchiorri, P.; Falconieri-Erspamer, G.; Negri, L.; Corsi, R.; Severini, C.; Barra, D.; Simmaco, M.; Kreil, G. Deltorphins: A family of naturally occurring peptides with high affinity and selectivity for 6 opioid binding sites. Proc. Natl. Acad. Sci. USA 1989, 86, 5188–5192. [Google Scholar] [CrossRef]
- Gavériaux-Ruff, C.; Karchewski, L.A.; Hever, X.; Matifas, A.; Kieffer, B.L. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur. J. Neurosci. 2008, 27, 2558–2567. [Google Scholar] [CrossRef]
- Pradhan, A.A.A.; Walwyn, W.; Nozaki, C.; Filliol, D.; Erbs, E.; Matifas, A.; Evans, C.; Kieffer, B.L. Ligand-Directed Trafficking of the δ-Opioid Receptor In Vivo: Two Paths Toward Analgesic Tolerance. J. Neurosci. 2010, 30, 16459–16468. [Google Scholar] [CrossRef] [PubMed]
- Filliol, D.; Ghozland, S.; Chluba, J.; Martin, M.; Matthes, H.W.D.; Simonin, F.; Befort, K.; Gavériaux-Ruff, C.; Dierich, A.; LeMeur, M.; et al. Mice deficient for δ- and μ-opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet. 2000, 25, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.A.; Befort, K.; Nozaki, C.; Gavériaux-Ruff, C.; Kieffer, B.L. The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends Pharmacol. Sci. 2011, 32, 581–590. [Google Scholar] [CrossRef]
- Cahill, C.M.; Morinville, A.; Hoffert, C.; O’Donnell, D.; Beaudet, A. Up-regulation and trafficking of d opioid receptor in a model of chronic inflammation: Implications for pain control. Pain 2003, 101, 199–208. [Google Scholar] [CrossRef]
- Henry, A.G.; White, I.J.; Marsh, M.; von Zastrow, M.; Hislop, J.N. The Role of Ubiquitination in Lysosomal Trafficking of δ-Opioid Receptors. Traffic 2011, 12, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Codd, E.E.; Carson, J.R.; Colburn, R.W.; Stone, D.J.; Van Besien, C.R.; Zhang, S.-P.; Wade, P.R.; Gallantine, E.L.; Meert, T.F.; Molino, L.; et al. JNJ-20788560 [9-(8-Azabicyclo[3.2.1]oct-3-ylidene)-9 H-xanthene-3-carboxylic Acid Diethylamide], a Selective Delta Opioid Receptor Agonist, is a Potent and Efficacious Antihyperalgesic Agent That Does Not Produce Respiratory Depression, Pharmacologic Tolerance, or Physical Dependence. J. Pharmacol. Exp. Ther. 2009, 329, 241–251. [Google Scholar] [CrossRef]
- Broom, D.; Jutkiewicz, E.; Folk, J.; Traynor, J.; Rice, K.; Woods, J. Convulsant activity of a non-peptidic δ-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology 2002, 164, 42–48. [Google Scholar] [CrossRef]
- Nozaki, C.; Nagase, H.; Nemoto, T.; Matifas, A.; Kieffer, B.L.; Gaveriaux-Ruff, C. In vivo properties of KNT-127, a novel δ opioid receptor agonist: Receptor internalization, antihyperalgesia and antidepressant effects in mice: KNT-127 ligand-biased agonism at δ opioid receptor. Br. J. Pharmacol. 2014, 171, 5376–5386. [Google Scholar] [CrossRef]
- Gendron, L.; Cahill, C.M.; von Zastrow, M.; Schiller, P.W.; Pineyro, G. Molecular Pharmacology of δ-Opioid Receptors. Pharmacol. Rev. 2016, 68, 631–700. [Google Scholar] [CrossRef]
- Tian, X.; Guo, J.; Zhu, M.; Li, M.; Wu, G.; Xia, Y. δ-Opioid Receptor Activation Rescues the Functional TrkB Receptor and Protects the Brain from Ischemia-Reperfusion Injury in the Rat. PLoS ONE 2013, 8, e69252. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, F.A.; Zelnik, J.C.; Traynor, J.R. G Protein independent phosphorylation and internalization of the δ-opioid receptor. J. Neurochem. 2009, 109, 1526–1535. [Google Scholar] [CrossRef]
- Xu, C.; Hong, M.-H.; Zhang, L.-S.; Hou, Y.-Y.; Wang, Y.-H.; Wang, F.-F.; Chen, Y.-J.; Xu, X.-J.; Chen, J.; Xie, X.; et al. Serine 363 of the δ-opioid receptor is crucial for adopting distinct pathways to activate ERK1/2 in response to stimulation with different ligands. J. Cell Sci. 2010, 123, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Loh, H.H.; Law, P.-Y. Phosphorylation of the δ-Opioid Receptor Regulates Its β-Arrestins Selectivity and Subsequent Receptor Internalization and Adenylyl Cyclase Desensitization. J. Biol. Chem. 2007, 282, 22315–22323. [Google Scholar] [CrossRef]
- Aguila, B.; Coulbault, L.; Davis, A.; Marie, N.; Hasbi, A.; Le bras, F.; Tóth, G.; Borsodi, A.; Gurevich, V.V.; Jauzac, P.; et al. ßarrestin1-biased agonism at human δ-opioid receptor by peptidic and alkaloid ligands. Cell. Signal. 2012, 24, 699–707. [Google Scholar] [CrossRef][Green Version]
- Cen, B.; Xiong, Y.; Ma, L.; Pei, G. Direct and differential interaction of beta-arrestins with the intracellular domains of different opioid receptors. Mol. Pharmacol. 2001, 59, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.A.; Perroy, J.; Walwyn, W.M.; Smith, M.L.; Vicente-Sanchez, A.; Segura, L.; Bana, A.; Kieffer, B.L.; Evans, C.J. Agonist-Specific Recruitment of Arrestin Isoforms Differentially Modify Delta Opioid Receptor Function. J. Neurosci. 2016, 36, 3541–3551. [Google Scholar] [CrossRef]
- Vicente-Sanchez, A.; Dripps, I.J.; Tipton, A.F.; Akbari, H.; Akbari, A.; Jutkiewicz, E.M.; Pradhan, A.A. Tolerance to high-internalizing δ opioid receptor agonist is critically mediated by arrestin 2. Br. J. Pharmacol. 2018, 175, 3050–3059. [Google Scholar] [CrossRef]
- Dripps, I.J.; Boyer, B.T.; Neubig, R.R.; Rice, K.C.; Traynor, J.R.; Jutkiewicz, E.M. Role of signalling molecules in behaviours mediated by the δ opioid receptor agonist SNC80: Signalling bias and δ-receptor activity. In Vivo Br. J. Pharmacol. 2018, 175, 891–901. [Google Scholar] [CrossRef]
- Pradhan, A.A.; Smith, M.L.; Kieffer, B.L.; Evans, C.J. Ligand-directed signalling within the opioid receptor family: Ligand-directed signalling at opioid receptors. Br. J. Pharmacol. 2012, 167, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Vargas, N.N.; Gong, J.; Wisdom, M.J.; Jensen, D.D.; Latorre, R.; Hegron, A.; Teng, S.; DiCello, J.J.; Rajasekhar, P.; Veldhuis, N.A.; et al. Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. Proc. Natl. Acad. Sci. USA 2020, 117, 15281–15292. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.E.; Asghar, J.; Hill, R.; Henderson, G.; Borbely, E.; Tekus, V.; Helyes, Z.; Palandri, J.; Bailey, C.; Starke, I.; et al. A Novel G Protein–Biased Agonist at the δ Opioid Receptor with Analgesic Efficacy in Models of Chronic Pain. J. Pharmacol. Exp. Ther. 2020, 372, 224–236. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Calò, G.; Pappalardo, F.; Ferrari, F.; Russo, G.; Arena, E.; Montenegro, L.; Chiechio, S.; Prezzavento, O.; et al. (2S)-N-2-methoxy-2-phenylethyl-6,7-benzomorphan compound (2S-LP2): Discovery of a biased mu/delta opioid receptor agonist. Eur. J. Med. Chem. 2019, 168, 189–198. [Google Scholar] [CrossRef]
- Chiang, T.; Sansuk, K.; van Rijn, R.M. β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake: Biased δ receptor agonists for treating alcohol use disorders. Br. J. Pharmacol. 2016, 173, 332–343. [Google Scholar] [CrossRef]
- Nagase, H.; Yajima, Y.; Fujii, H.; Kawamura, K.; Narita, M.; Kamei, J.; Suzuki, T. The pharmacological profile of delta opioid receptor ligands, (+) and (-) TAN-67 on pain modulation. Life Sci. 2001, 68, 2227–2231. [Google Scholar] [CrossRef]
- van Rijn, R.M.; Brissett, D.I.; Whistler, J.L. Dual Efficacy of Delta Opioid Receptor-Selective Ligands for Ethanol Drinking and Anxiety. J. Pharmacol. Exp. Ther. 2010, 335, 133–139. [Google Scholar] [CrossRef]
- Nagase, H.; Nemoto, T.; Matsubara, A.; Saito, M.; Yamamoto, N.; Osa, Y.; Hirayama, S.; Nakajima, M.; Nakao, K.; Mochizuki, H.; et al. Design and synthesis of KNT-127, a δ-opioid receptor agonist effective by systemic administration. Bioorg. Med. Chem. Lett. 2010, 20, 6302–6305. [Google Scholar] [CrossRef]
- Saitoh, A.; Sugiyama, A.; Nemoto, T.; Fujii, H.; Wada, K.; Oka, J.-I.; Nagase, H.; Yamada, M. The novel δ opioid receptor agonist KNT-127 produces antidepressant-like and antinociceptive effects in mice without producing convulsions. Behav. Brain Res. 2011, 223, 271–279. [Google Scholar] [CrossRef]
- Saitoh, A.; Yamada, M. Antidepressant-like Effects of δ Opioid Receptor Agonists in Animal Models. Curr. Neuropharmacol. 2012, 10, 231–238. [Google Scholar] [CrossRef]
- Pradhan, A.A.A.; Becker, J.A.J.; Scherrer, G.; Tryoen-Toth, P.; Filliol, D.; Matifas, A.; Massotte, D.; Gavériaux-Ruff, C.; Kieffer, B.L. In Vivo Delta Opioid Receptor Internalization Controls Behavioral Effects of Agonists. PLoS ONE 2009, 4, e5425. [Google Scholar] [CrossRef] [PubMed]
- Marie, N.; Lecoq, I.; Jauzac, P.; Allouche, S. Differential Sorting of Human δ-Opioid Receptors after Internalization by Peptide and Alkaloid Agonists. J. Biol. Chem. 2003, 278, 22795–22804. [Google Scholar] [CrossRef] [PubMed]
- Witkin, J.M.; Statnick, M.A.; Rorick-Kehn, L.M.; Pintar, J.E.; Ansonoff, M.; Chen, Y.; Tucker, R.C.; Ciccocioppo, R. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol. Ther. 2014, 141, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Schröder, W.; Lambert, D.G.; Ko, M.C.; Koch, T. Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists: The N/OFQ-NOP receptor system in analgesia. Br. J. Pharmacol. 2014, 171, 3777–3800. [Google Scholar] [CrossRef] [PubMed]
- Byford, A.J.; Anderson, A.; Jones, P.S.; Palin, R.; Houghton, A.K. The Hypnotic, Electroencephalographic, and Antinociceptive Properties of Nonpeptide ORL1 Receptor Agonists after Intravenous Injection in Rodents. Anesth. Analg. 2007, 104, 174–179. [Google Scholar] [CrossRef]
- Linz, K.; Schröder, W.; Frosch, S.; Christoph, T. Opioid-type Respiratory Depressant Side Effects of Cebranopadol in Rats Are Limited by Its Nociceptin/Orphanin FQ Peptide Receptor Agonist Activity. Anesthesiology 2017, 126, 708–715. [Google Scholar] [CrossRef]
- Ding, H.; Kiguchi, N.; Yasuda, D.; Daga, P.R.; Polgar, W.E.; Lu, J.J.; Czoty, P.W.; Kishioka, S.; Zaveri, N.T.; Ko, M.-C. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci. Transl. Med. 2018, 10, eaar3483. [Google Scholar] [CrossRef]
- Shoblock, J.R. The Pharmacology of Ro 64-6198, a Systemically Active, Nonpeptide NOP Receptor (Opiate Receptor-Like 1, ORL-1) Agonist with Diverse Preclinical Therapeutic Activity. CNS Drug Rev. 2007, 13, 107–136. [Google Scholar] [CrossRef]
- Gavioli, E.C.; Calo’, G. Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol. Ther. 2013, 140, 10–25. [Google Scholar] [CrossRef]
- Vitale, G.; Filaferro, M.; Micioni Di Bonaventura, M.V.; Ruggieri, V.; Cifani, C.; Guerrini, R.; Simonato, M.; Zucchini, S. Effects of [Nphe1, Arg14, Lys15] N/OFQ-NH2 (UFP-101), a potent NOP receptor antagonist, on molecular, cellular and behavioural alterations associated with chronic mild stress. J. Psychopharmacol. 2017, 31, 691–703. [Google Scholar] [CrossRef]
- Asth, L.; Ruzza, C.; Malfacini, D.; Medeiros, I.; Guerrini, R.; Zaveri, N.T.; Gavioli, E.C.; Calo’, G. Beta-arrestin 2 rather than G protein efficacy determines the anxiolytic-versus antidepressant-like effects of nociceptin/orphanin FQ receptor ligands. Neuropharmacology 2016, 105, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Angeletti, S.; Sanna, P.P.; Weiss, F.; Massi, M. Effect of nociceptin/orphanin FQ on the rewarding properties of morphine. Eur. J. Pharmacol. 2000, 404, 153–159. [Google Scholar] [CrossRef]
- Di Giannuario, A.; Pieretti, S. Nociceptin differentially affects morphine-induced dopamine release from the nucleus accumbens and nucleus caudate in rats. Peptides 2000, 21, 1125–1130. [Google Scholar] [CrossRef]
- Ko, M.-C.; Caló, G. (Eds.) The Nociceptin/Orphanin FQ Peptide Receptor. In Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2019; Volume 254, ISBN 978-3-030-20185-2. [Google Scholar]
- Chang, S.D.; Mascarella, S.W.; Spangler, S.M.; Gurevich, V.V.; Navarro, H.A.; Carroll, F.I.; Bruchas, M.R. Quantitative Signaling and Structure-Activity Analyses Demonstrate Functional Selectivity at the Nociceptin/Orphanin FQ Opioid Receptor. Mol. Pharmacol. 2015, 88, 502–511. [Google Scholar] [CrossRef]
- Zhang, N.R.; Planer, W.; Siuda, E.R.; Zhao, H.-C.; Stickler, L.; Chang, S.D.; Baird, M.A.; Cao, Y.-Q.; Bruchas, M.R. Serine 363 Is Required for Nociceptin/Orphanin FQ Opioid Receptor (NOPR) Desensitization, Internalization, and Arrestin Signaling. J. Biol. Chem. 2012, 287, 42019–42030. [Google Scholar] [CrossRef]
- Mann, A.; Moulédous, L.; Froment, C.; O’Neill, P.R.; Dasgupta, P.; Günther, T.; Brunori, G.; Kieffer, B.L.; Toll, L.; Bruchas, M.R.; et al. Agonist-selective NOP receptor phosphorylation correlates in vitro and in vivo and reveals differential post-activation signaling by chemically diverse agonists. Sci. Signal. 2019, 12, eaau8072. [Google Scholar] [CrossRef]
- Malfacini, D.; Ambrosio, C.; Gro’, M.C.; Sbraccia, M.; Trapella, C.; Guerrini, R.; Bonora, M.; Pinton, P.; Costa, T.; Calo’, G. Pharmacological Profile of Nociceptin/Orphanin FQ Receptors Interacting with G-Proteins and β-Arrestins 2. PLoS ONE 2015, 10, e0132865. [Google Scholar] [CrossRef]
- Donica, C.L.; Awwad, H.O.; Thakker, D.R.; Standifer, K.M. Cellular Mechanisms of Nociceptin/Orphanin FQ (N/OFQ) Peptide (NOP) Receptor Regulation and Heterologous Regulation by N/OFQ. Mol. Pharmacol. 2013, 83, 907–918. [Google Scholar] [CrossRef]
- Vezzi, V.; Onaran, H.O.; Molinari, P.; Guerrini, R.; Balboni, G.; Calò, G.; Costa, T. Ligands Raise the Constraint That Limits Constitutive Activation in G Protein-coupled Opioid Receptors. J. Biol. Chem. 2013, 288, 23964–23978. [Google Scholar] [CrossRef]
- Rizzi, A.; Cerlesi, M.C.; Ruzza, C.; Malfacini, D.; Ferrari, F.; Bianco, S.; Costa, T.; Guerrini, R.; Trapella, C.; Calo’, G. Pharmacological characterization of cebranopadol a novel analgesic acting as mixed nociceptin/orphanin FQ and opioid receptor agonist. Pharmacol. Res. Perspect. 2016, 4, e00247. [Google Scholar] [CrossRef]
- Chao, P.-K.; Chang, H.-F.; Chang, W.-T.; Yeh, T.-K.; Ou, L.-C.; Chuang, J.-Y.; Tsu-An Hsu, J.; Tao, P.-L.; Loh, H.H.; Shih, C.; et al. BPR1M97, a dual mu opioid receptor/nociceptin-orphanin FQ peptide receptor agonist, produces potent antinociceptive effects with safer properties than morphine. Neuropharmacology 2020, 166, 107678. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-R.; Ke, Y.-Y.; Yeh, T.-K.; Lin, S.-Y.; Ou, L.-C.; Chen, S.-C.; Chang, W.-T.; Chang, H.-F.; Wu, Z.-H.; Hsieh, C.-C.; et al. Discovery, structure-activity relationship studies, and anti-nociceptive effects of N-(1,2,3,4-tetrahydro-1-isoquinolinylmethyl)benzamides as novel opioid receptor agonists. Eur. J. Med. Chem. 2017, 126, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, J.; Adam, G.; Röver, S.; Cesura, A.M.; Dautzenberg, F.M.; Jenck, F. 8-Acenaphthen-1-yl-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one derivatives as orphanin FQ receptor agonists. Bioorg. Med. Chem. Lett. 1999, 9, 2343–2348. [Google Scholar] [CrossRef]
- Holanda, V.A.D.; Pacifico, S.; Azevedo Neto, J.; Finetti, L.; Lobão-Soares, B.; Calo, G.; Gavioli, E.C.; Ruzza, C. Modulation of the NOP receptor signaling affects resilience to acute stress. J. Psychopharmacol. 2019, 33, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Kotlińska, J.; Wichmann, J.; Legowska, A.; Rolka, K.; Silberring, J. Orphanin FQ/nociceptin but not Ro 65-6570 inhibits the expression of cocaine-induced conditioned place preference. Behav. Pharmacol. 2002, 13, 229–235. [Google Scholar] [CrossRef]
- Rutten, K.; De Vry, J.; Bruckmann, W.; Tzschentke, T.M. Effects of the NOP receptor agonist Ro65-6570 on the acquisition of opiate- and psychostimulant-induced conditioned place preference in rats. Eur. J. Pharmacol. 2010, 645, 119–126. [Google Scholar] [CrossRef]
- Varty, G.B.; Lu, S.X.; Morgan, C.A.; Cohen-Williams, M.E.; Hodgson, R.A.; Smith-Torhan, A.; Zhang, H.; Fawzi, A.B.; Graziano, M.P.; Ho, G.D.; et al. The Anxiolytic-Like Effects of the Novel, Orally Active Nociceptin Opioid Receptor Agonist 8-[bis(2-Methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510). J. Pharmacol. Exp. Ther. 2008, 326, 672–682. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Kögel, B.Y.; Frosch, S.; Linz, K. Limited potential of cebranopadol to produce opioid-type physical dependence in rodents: Weak cebranopadol dependence. Addict. Biol. 2018, 23, 1010–1019. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Linz, K.; Frosch, S.; Christoph, T. Antihyperalgesic, Antiallodynic, and Antinociceptive Effects of Cebranopadol, a Novel Potent Nociceptin/Orphanin FQ and Opioid Receptor Agonist, after Peripheral and Central Administration in Rodent Models of Neuropathic Pain. Pain Pract. 2017, 17, 1032–1041. [Google Scholar] [CrossRef]
- Hirao, A.; Imai, A.; Sugie, Y.; Yamada, Y.; Hayashi, S.; Toide, K. Pharmacological Characterization of the Newly Synthesized Nociceptin/Orphanin FQ–Receptor Agonist 1-[1-(1-Methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole as an Anxiolytic Agent. J. Pharmacol. Sci. 2008, 106, 361–368. [Google Scholar] [CrossRef]
- Thomsen, C.; Hohlweg, R. (8-Naphthalen-1-ylmethyl-4-oxo-1-phenyl-1,3,8-triaza-spiro[4.5]dec-3-yl)-acetic acid methyl ester (NNC 63-0532) is a novel potent nociceptin receptor agonist. Br. J. Pharmacol. 2000, 131, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Sreenivasan, V.; Christie, M.J. Intrinsic efficacy of opioid ligands and its importance for apparent bias, operational analysis and therapeutic window. Mol. Pharmacol. 2020, mol.119.119214. [Google Scholar] [CrossRef] [PubMed]
- Grim, T.W.; Acevedo-Canabal, A.; Bohn, L.M. Toward Directing Opioid Receptor Signaling to Refine Opioid Therapeutics. Biol. Psychiatry 2020, 87, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Burgman, M.; Haselton, N.; Grinnell, S.; Ocampo, J.; Pasternak, A.R.; Pasternak, G.W. Generation of novel radiolabeled opiates through site-selective iodination. Bioorg. Med. Chem. Lett. 2011, 21, 4001–4004. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Z.; Narayan, A.; Le Rouzic, V.P.; Xu, M.; Hunkele, A.; Brown, T.G.; Hoefer, W.F.; Rossi, G.C.; Rice, R.C.; et al. Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. J. Clin. Investig. 2017, 127, 1561–1573. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Functional | G-Protein | Emax | β-Arrestin2 | Emax | PubChem | Ref |
|---|---|---|---|---|---|---|---|
| Selectivity | EC50 (nM) | EC50 (nM) | ID | ||||
| TRV130 | G-protein | 8.1 (cAMP) | 84 | 7.3 (PathHunter) | 15 | 66553195 | [23] |
| Morphine | Balanced | 7.4 (cAMP) | 100 | 6.3 (PathHunter) | 100 | 5288826 | [23] |
| agonist | |||||||
| TRV130 | G-protein | 7.97 (Glosensor) | 75 | 8.02 (BRET) | 26 | 66553195 | [24] |
| DAMGO | Balanced | 8.58 (Glosensor) | 100 | 8.3 (BRET) | 100 | 5462471 | [24] |
| agonist | |||||||
| TRV130 | G-protein | 7.9 (cAMP) | 86 | Inactive (PathHunter) | NQ | 66553195 | [25] |
| DAMGO | Balanced | 8.4 (cAMP) | 100 | 6.7 (PathHunter) | 100 | 5462471 | [25] |
| agonist | |||||||
| TRV130 | G-protein | 8.66 (cAMP) | 86 | 7.71 (BRET) | 58 | 66553195 | [26] |
| partial agonist | |||||||
| DAMGO | Balanced | 8.48 (cAMP) | 100 | 7.55 (BRET) | 100 | 5462471 | [26] |
| agonist | |||||||
| PZM21 | G-protein | 7.73 (Glosensor) | 83 | 7.68 (BRET) | 32 | 121596705 | [24] |
| DAMGO | Balanced | 8.58 (Glosensor) | 100 | 8.3 (BRET) | 100 | 5462471 | [24] |
| agonist | |||||||
| PZM21 | G-protein | 110 (BRET) | 39 | 450 (BRET) | 18 | 121596705 | [27] |
| DAMGO | Balanced | 390 (BRET) | 100 | 1200 (BRET) | 100 | 5462471 | [27] |
| agonist | |||||||
| PZM21 | G-protein | 8.64 (cAMP) | 84 | 7.56 (BRET) | 59 | 121596705 | [26] |
| partial agonist | |||||||
| DAMGO | Balanced | 8.48 (cAMP) | 100 | 1200 (BRET) | 100 | 5462471 | [26] |
| agonist | |||||||
| 7-OH | G-protein | 34.5 (BRET) | 47 | Inactive (BRET) | NQ | 44301524 | [28] |
| DAMGO | Balanced | 1 (BRET) | 100 | NA (BRET) | 100 | 5462471 | [28] |
| agonist | |||||||
| 7-OH | G-protein | 53 (GTPγS) | 77 | Inactive (PathHunter) | NQ | 44301524 | [29] |
| DAMGO | Balanced | 19 (GTPγS) | 100 | 106 (PathHunter) | 100 | 5462471 | [29] |
| agonist | |||||||
| 7OH | G-protein | 7.8 (cAMP) | 84 | Inactive (PathHunter) | NQ | 44301524 | [25] |
| DAMGO | Balanced | 8.4 (cAMP) | 100 | 6.7 (PathHunter ) | 100 | 5462471 | [25] |
| agonist | |||||||
| Selectivity | EC50 (nM) | EC50 (nM) | ID | ||||
| Mitragynine | G-protein | 1.7 (CHO) | 82 | Inactive | NQ | 44301701 | [29] |
| pseudoindoxyl | PathHunter | ||||||
| DAMGO | Balanced | 19 (GTPγS) | 100 | 106 (PathHunter) | 100 | 5462471 | [29] |
| agonist | |||||||
| Herkinorin | G-protein | 500 (CHO) | 130 | No internalization of | NQ | 11431898 | [30] |
| βarr2-GFP | |||||||
| Herkamide | Balanced | 360 (CHO) | 134 | Internalization of | NQ | NA | [30] |
| agonist | βarr2-GFP seen | ||||||
| DAMGO | Balanced | 40 (CHO) | 100 | Internalization of | NQ | 5462471 | [30] |
| agonist | βarr2-GFP seen | ||||||
| Herkinorin | Balanced | 7.08 (Glosensor) | 104 | 7.15 (BRET) | 104 | 11431898 | [24] |
| agonist | |||||||
| DAMGO | Balanced | 8.58 (Glosensor) | 100 | 8.3 (BRET) | 100 | 5462471 | [24] |
| agonist | |||||||
| Kurkinorin | G-protein | 1.2 (cAMP) | 100 | 140 (PathHunter) | 96 | 132079904 | [31] |
| DAMGO | Balanced | 0.6 (cAMP) | 100 | 42 (PathHunter) | 100 | 5462471 | [31] |
| agonist | |||||||
| 1 | G-protein | 0.03 (cAMP) | 100 | 14 (PathHunter) | 81 | NA | [32] |
| DAMGO | Balanced | 0.6 (cAMP) | 100 | 42 (PathHunter) | 100 | 5462471 | [32] |
| agonist | |||||||
| SR-11501 | β-arrestin2 | 7.9(cAMP) | 98 | 374 (PathHunter) | 59 | 146025598 | [33] |
| SR-17018 | G-protein | 76 (cAMP) | 105 | >10,000 (PathHunter) | 10 | 130431397 | [33] |
| DAMGO | Balanced | 5.2 (cAMP) | 100 | 229 (PathHunter) | 100 | 5462471 | [33] |
| agonist | |||||||
| SR-11501 | β-arrestin2 | 133(GTPγS) | 98 | 374 (PathHunter) | 59 | 146025598 | [33] |
| SR-17018 | G-protein | 193 (GTPγS) | 72 | >10,000 (PathHunter) | 10 | 130431397 | [33] |
| DAMGO | Balanced | 34 (GTPγS) | 100 | 229 (PathHunter) | 100 | 5462471 | [33] |
| agonist | |||||||
| SR-17018 | G-protein | 7.67 (cAMP) | 62 | 6.48(BRET) | 49 | 130431397 | [26] |
| partial agonist | |||||||
| DAMGO | Balanced | 8.48 (cAMP) | 100 | 1200 (BRET) | 100 | 5462471 | [26] |
| agonist | |||||||
| 2 | G-protein | 91(GTPγS) | 74 | >10,000 (PathHunter) | 66 | NA | [34] |
| 3 | G-protein | 153 (GTPγS) | 91 | >10,000 (PathHunter) | 12 | NA | [34] |
| DAMGO | Balanced | 34 (GTPγS) | 100 | 229 (PathHunter) | 100 | 5462471 | [34] |
| agonist | |||||||
| DAMGO | Balanced | 8.4 (cAMP) | 100 | 6.7 (PathHunter) | 100 | 5462471 | [25] |
| agonist | |||||||
| MP102 | G-protein | 5.4 (cAMP) | 88 | 5.2 (PathHunter) | 16 | NA | [25] |
| MP103 | Balanced | 6.5 (cAMP) | 90 | 6.3 (PathHunter) | 63 | 146025824 | [25] |
| agonist | |||||||
| MP105 | Balanced | 6.7 (cAMP) | 87 | 6.6 (PathHunter) | 54 | 146025825 | [25] |
| agonist |
| Ligand | Functional | G-Protein | Emax | β-Arrestin2 | Emax | PubChem | Ref |
|---|---|---|---|---|---|---|---|
| Selectivity | EC50 (nM) | EC50 (nM) | ID | ||||
| RB64 | G-protein | 5.2 (cAMP) | 99 | 1130 (Tango) | 126 | 73347341 | [59] |
| Salvinorin A | Balanced | 4.73 (cAMP) | 100 | 10.5 (Tango) | 100 | 128563 | [59] |
| agonist | |||||||
| Mesyl Salvinorin B | G-protein | 0.12 (cAMP) | 101 | 236 (PathHunter) | 90 | 11271318 | [60] |
| U50,488H | Balanced | 0.23 (cAMP) | 100 | 162 (PathHunter) | 100 | 3036289 | [60] |
| agonist | |||||||
| Triazole 1.1 | G-protein | 77 (GTPγS) | 101 | 4995 (PathHunter) | 98 | 46245518 | [61] |
| U50,488H | Balanced | 24 (GTPγS) | 100 | 52.7 (PathHunter) | 100 | 3036289 | [61] |
| agonist | |||||||
| HS665 | G-protein | 4.98 (GTPγS) | 88 | 463 (PathHunter) | 55 | 71452041 | [62] |
| HS666 | G-protein | 35.7 (GTPγS) | 50 | 449 (PathHunter) | 24 | 71452040 | [62] |
| U69,693 | Balanced | 18.2 (GTPγS) | 100 | 67.7 (PathHunter) | 100 | 105104 | [62] |
| agonist | |||||||
| 6’GNTI | G-protein | 1.6 (BRET) | 64 | Inactive (BRET) | NQ | 146673012 | [63] |
| U50,488H | Balanced | 43 (BRET) | 100 | 2000 (BRET) | 100 | 3036289 | [63] |
| agonist | |||||||
| 6’GNTI | G-protein | 2.1 (GTPγS) | 37 | 5.9 (PathHunter) | 12 | 146673012 | [64] |
| U50,488H | Balanced | 69 (GTPγS) | 100 | 59 (PathHunter) | 100 | 3036289 | [64] |
| agonist | |||||||
| Isoquinolinone 2.1 | G-protein | 84.7 (GTPγS) | 89 | Inactive (PathHunter) | NQ | 121231409 | [65] |
| U69,693 | Balanced | 51 (GTPγS) | 100 | 131 (PathHunter) | 100 | 105104 | [65] |
| agonist | |||||||
| Collybolide | G-protein | 2 (GTPγS) | 124 * | NA | NA | 21669398 | [66] |
| Salvinorin A | Balanced | 0.2 (GTPγS) | 136 * | NA | NA | 128563 | [66] |
| agonist |
| Ligand | Functional | G-Protein | Emax | β-Arrestin2 | Emax | PubChem | Ref |
|---|---|---|---|---|---|---|---|
| Selectivity | EC50 (nM) | EC50 (nM) | ID | ||||
| PN6047 | G-protein | 8.9 (BRET) | 128 | 145 (BRET) | 115 | 121430051 | [105] |
| DADLE | Balanced | 2.5 (BRET) | 100 | 69 (BRET) | 100 | 6917707 | [105] |
| agonist | |||||||
| 2S-LP2 | G-protein | 32 (BRET) | 93 | 1862 (BRET) | 72 | 146025789 | [106] |
| DADLE | Balanced | 59 (BRET) | 100 | 20 (BRET) | 100 | 6917707 | [106] |
| agonist | |||||||
| TAN-67 | G-protein | 2.5 (cAMP) | 100 | 12.6 (PathHunter) | 41 | 9950038 | [107] |
| KNT-127 | G-protein | 2 (cAMP) | 100 | 3.2 (PathHunter) | 71 | 275705784 | [107] |
| ARM390 | G-protein | 126 (cAMP) | 100 | 316 (PathHunter) | 103 | 9841259 | [107] |
| DPDPE | Balanced | 6.3 (cAMP) | 100 | 25.1 (PathHunter) | 100 | 104787 | [107] |
| agonist | |||||||
| ARM390 | G-protein | 110 (BRET) | 120 | 832 (BRET) | 137 | 9841259 | [105] |
| DADLE | Balanced | 2.5 (BRET) | 100 | 69 (BRET) | 100 | 6917707 | [105] |
| agonist | |||||||
| JNJ20788560 | G-protein | 5.6 (GTPγS) | 92 | NA | NA | 46911863 | [88] |
| SNC80 | Balanced | 5.4 (GTPγS) | 100 | NA | NA | 123924 | [88] |
| Ligand | Functional | G-Protein | Emax | β-Arrestin2 | Emax | PubChem | Ref |
|---|---|---|---|---|---|---|---|
| Selectivity | EC50 (nM) | EC50 (nM) | ID | ||||
| Ro 65-6570 | G-protein | 17 (BRET) | 96 | 427 (BRET) | 84 | 15512229 | [130] |
| OFQ/N | Balanced | 3.6 (BRET) | 100 | 9.6 (BRET) | 100 | 6324645 | [130] |
| agonist | |||||||
| Ro 65-6570 | G-protein | 6.8 (BRET) | 92 | 102 (BRET) | 64 | 15512229 | [133] |
| Cebranopadol | G-protein | 3.2 (BRET) | 86 | Inactive (BRET) | NQ | 11848225 | [133] |
| OFQ/N | Balanced | 6.9 (BRET) | 100 | 6.6 (BRET) | 100 | 6324645 | [133] |
| agonist | |||||||
| MCOPPB | G-protein | 0.025 (cAMP) | 105 | 1585 (BRET) | 99 | 24800108 | [127] |
| SCH221,510 | G-protein | 4.3 (cAMP) | 103 | 4266 (BRET) | 87 | 9887077 | [127] |
| NNC 63-0532 | G-protein | 26.3 (cAMP) | 74 | Inactive (BRET) | NQ | 9803475 | [127] |
| RTI-819 | G-protein | 72.4 (cAMP) | 75 | Inactive (BRET) | NQ | 146034954 | [127] |
| RTI-856 | G-protein | 7.24 (cAMP) | 77 | Inactive (BRET) | NQ | 146034955 | [127] |
| OFQ/N | Balanced | 0.2 (cAMP) | 100 | 204 (BRET) | 100 | 6324645 | [127] |
| agonist | |||||||
| BPR1M97 | G-protein | 1.8 (cAMP) | 109 | 5100 (PathHunter) | 14 | 137541784 | [134] |
| OFQ/N | Balanced | 0.4 (cAMP) | 100 | 3 (PathHunter) | 100 | 6324645 | [134] |
| agonist |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. https://doi.org/10.3390/molecules25184257
Faouzi A, Varga BR, Majumdar S. Biased Opioid Ligands. Molecules. 2020; 25(18):4257. https://doi.org/10.3390/molecules25184257
Chicago/Turabian StyleFaouzi, Abdelfattah, Balazs R. Varga, and Susruta Majumdar. 2020. "Biased Opioid Ligands" Molecules 25, no. 18: 4257. https://doi.org/10.3390/molecules25184257
APA StyleFaouzi, A., Varga, B. R., & Majumdar, S. (2020). Biased Opioid Ligands. Molecules, 25(18), 4257. https://doi.org/10.3390/molecules25184257