
Methylation of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic, Crystallographic, and Molecular Docking Studies

 ,
, 
 ,
,
Abstract
1. Introduction
2. Results and Discussion
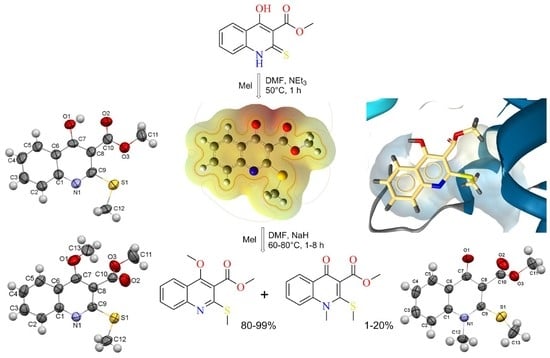
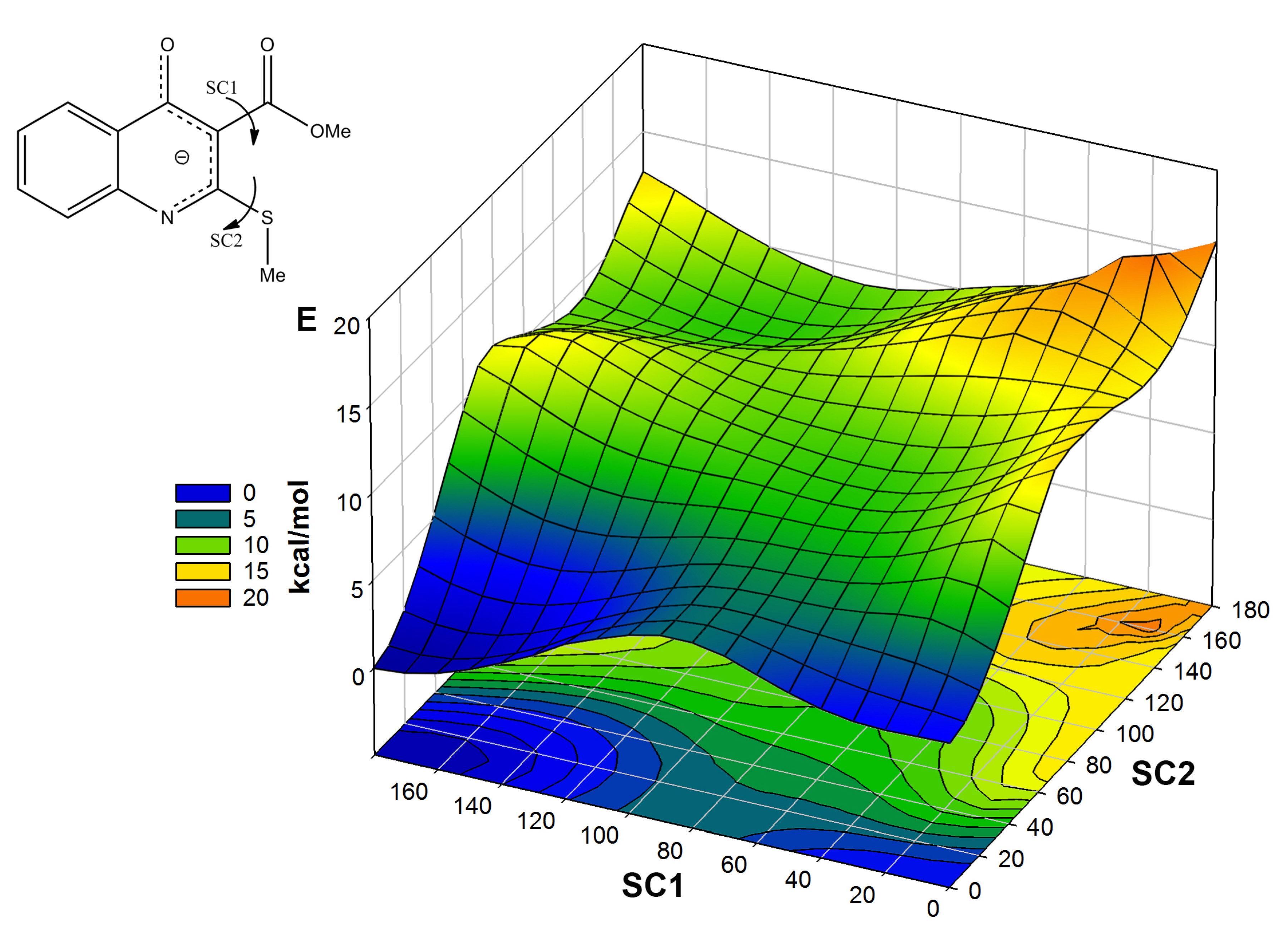
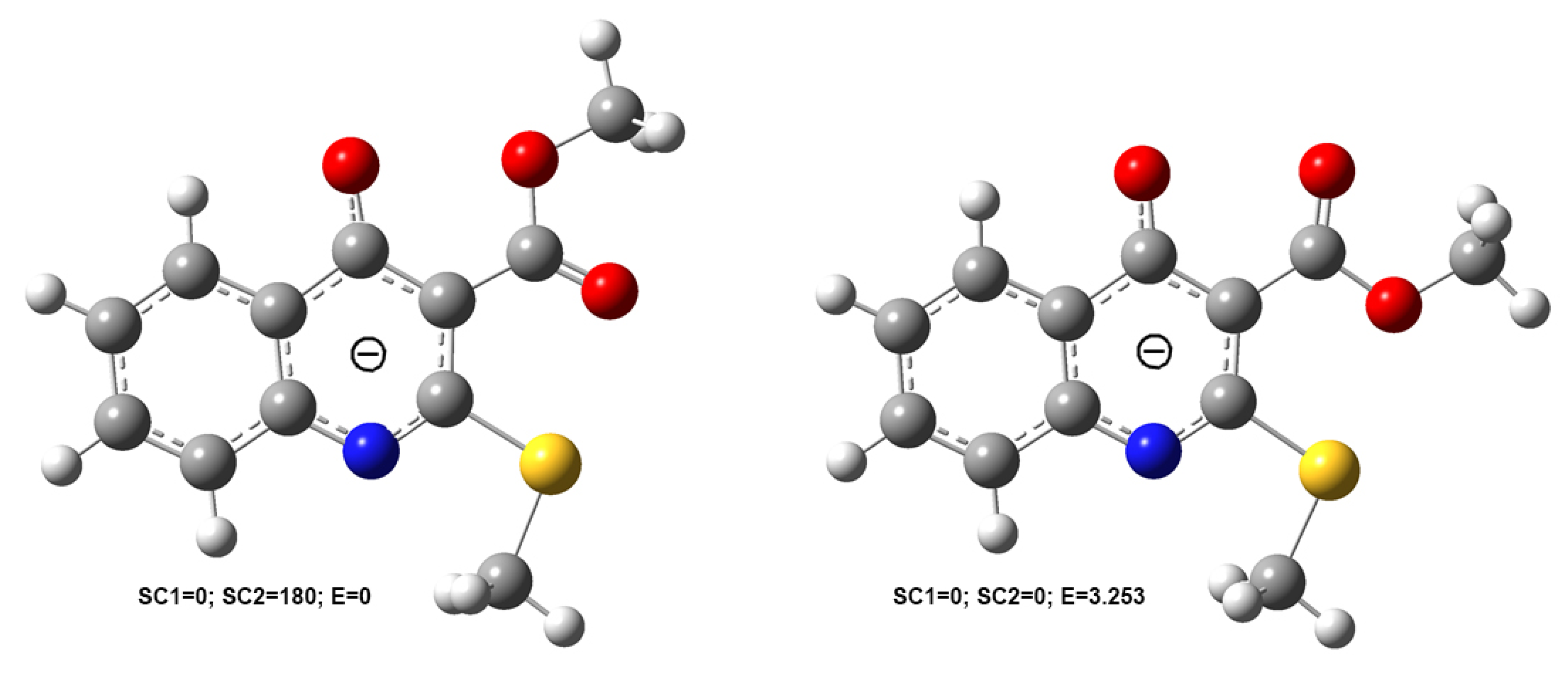
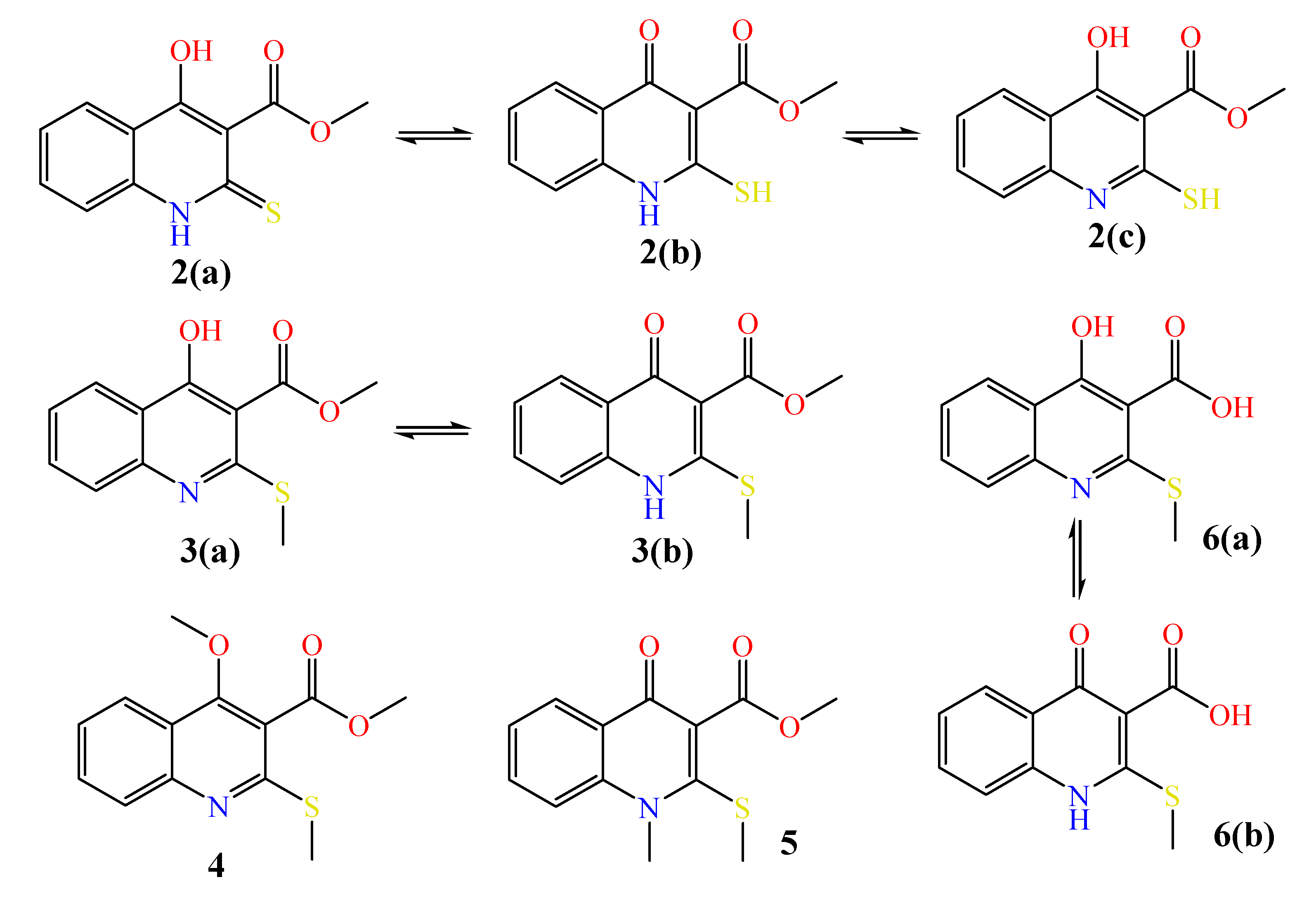
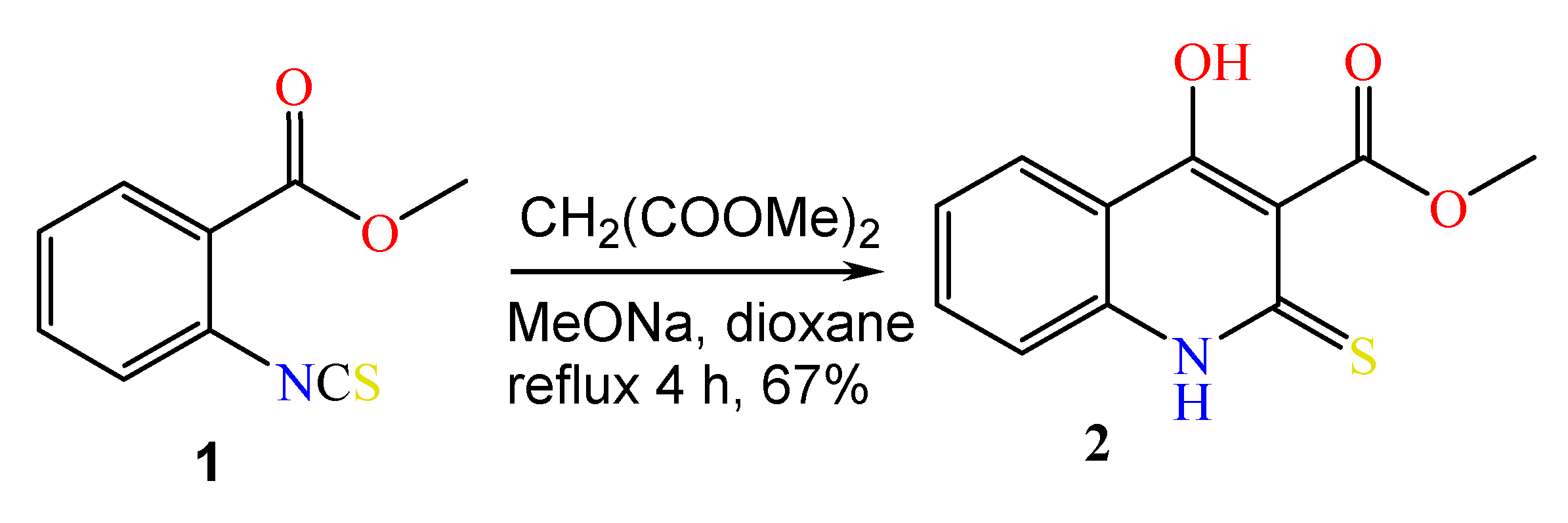
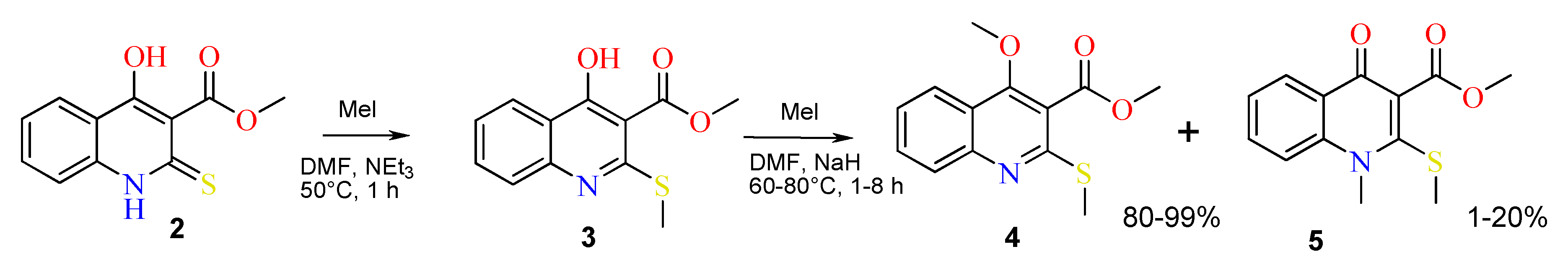
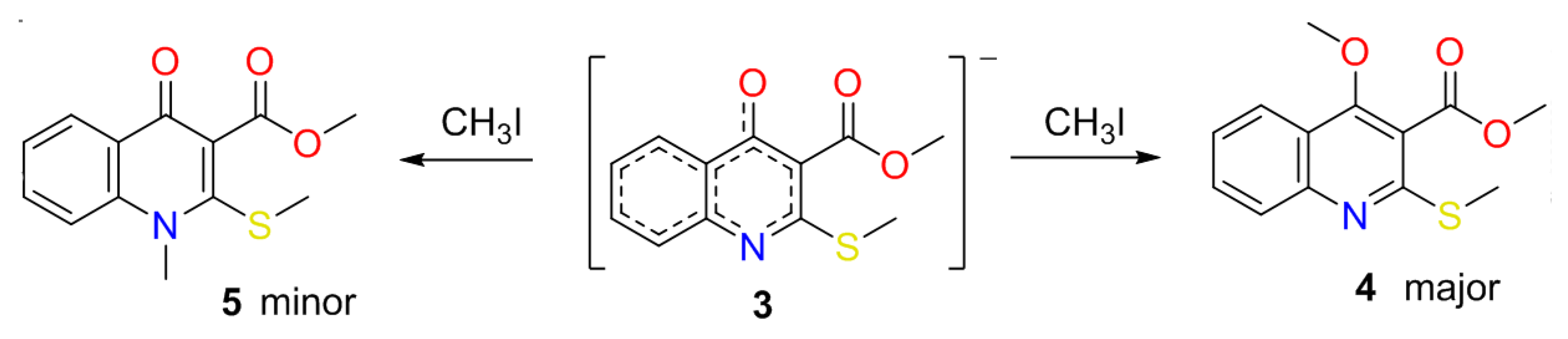
2.1. The Reaction of Alkylation of 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate 2 by CH3I
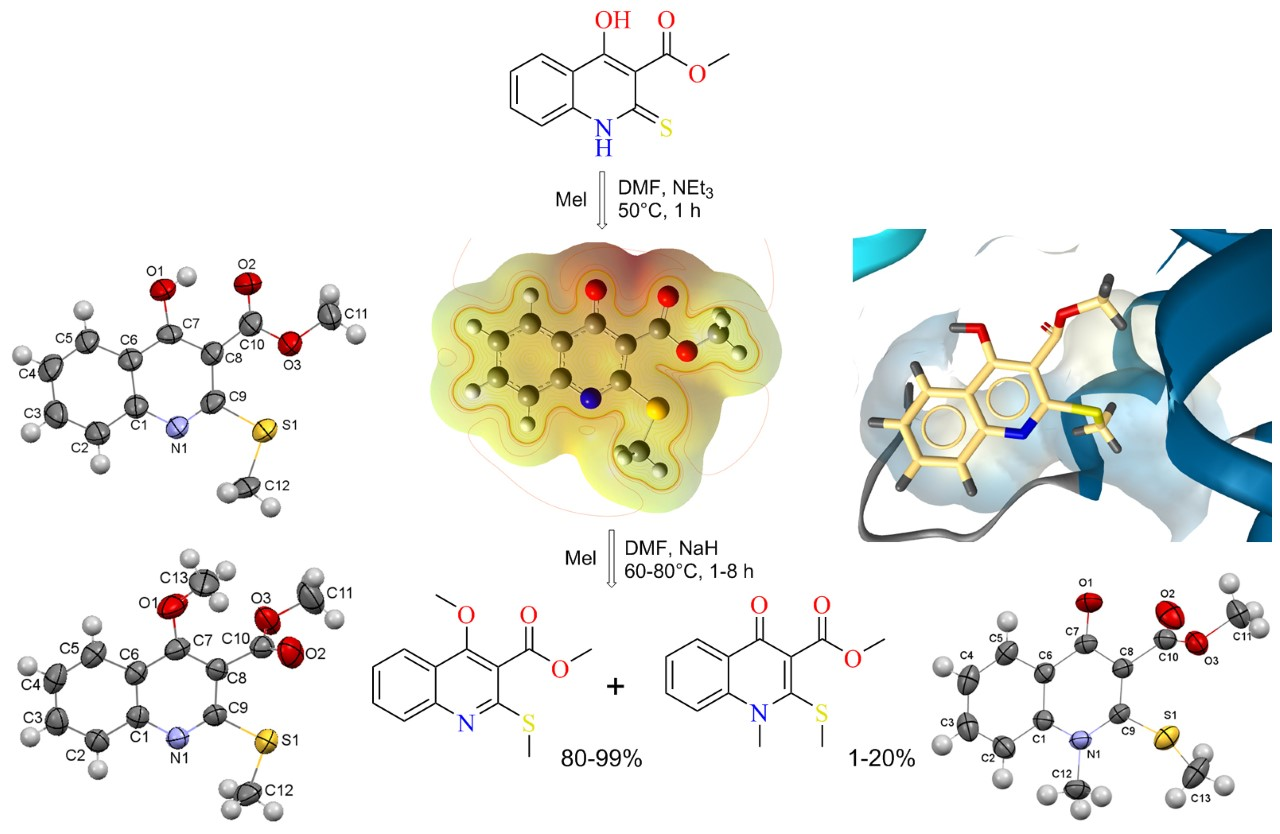
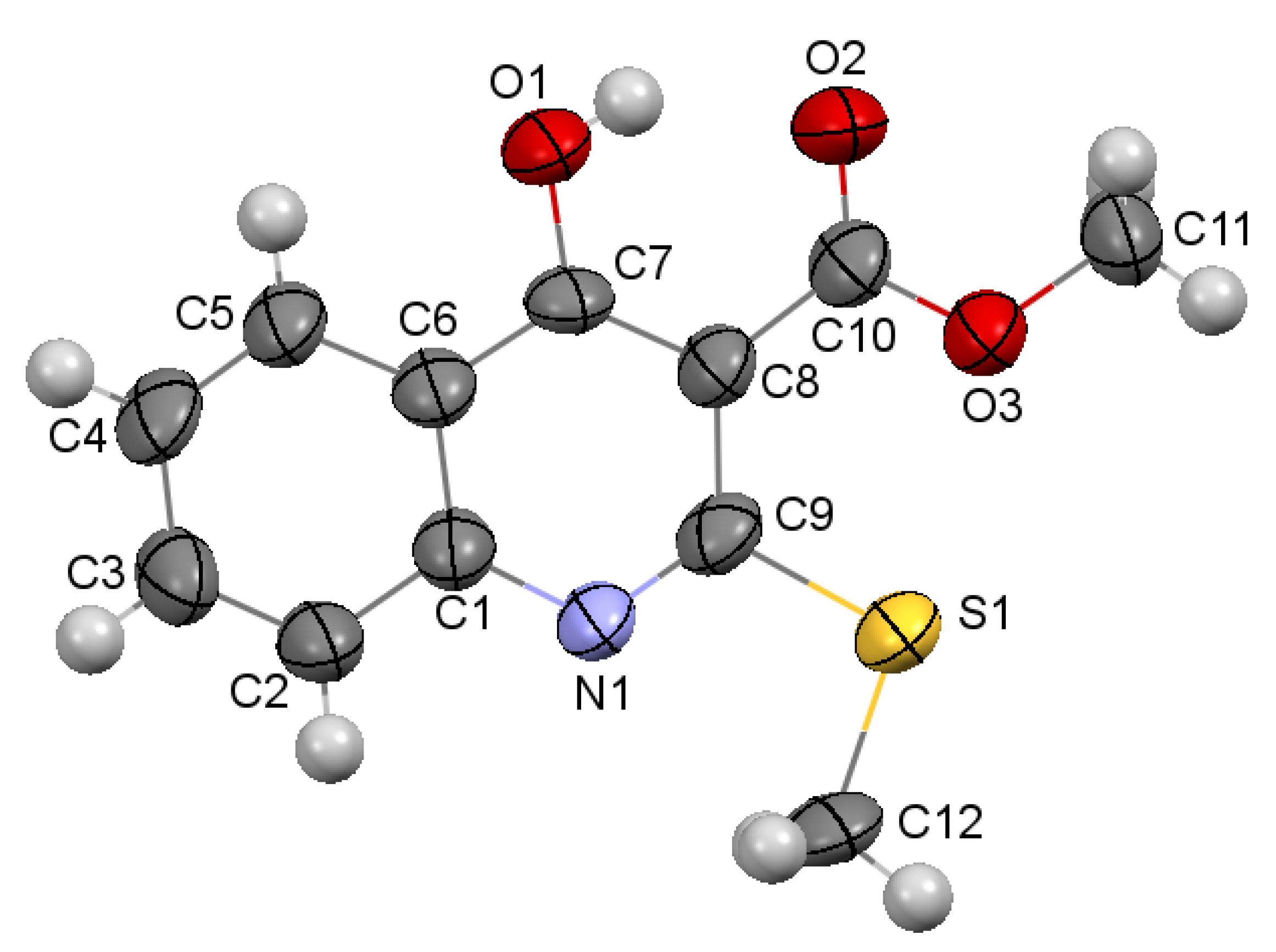
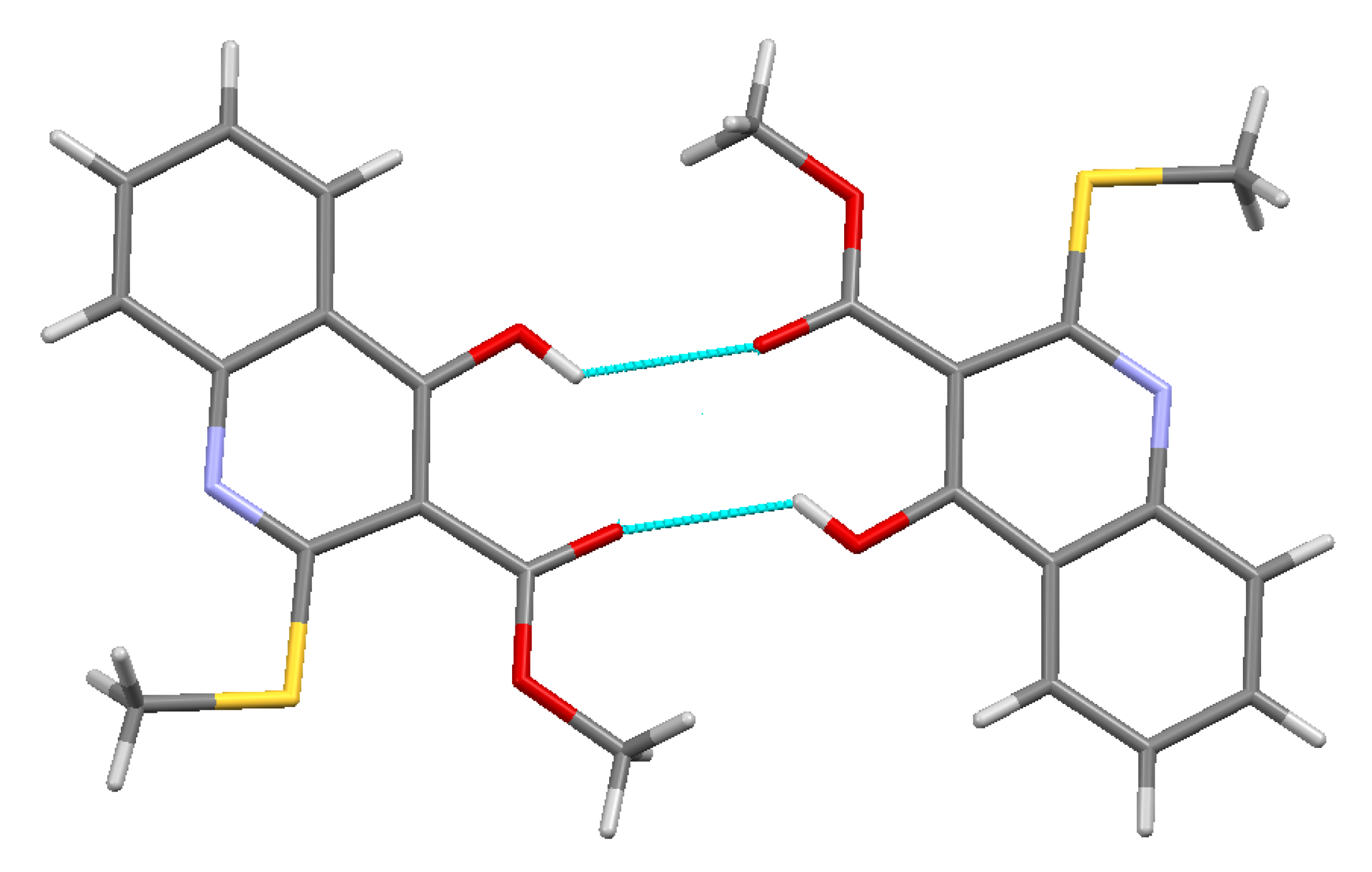
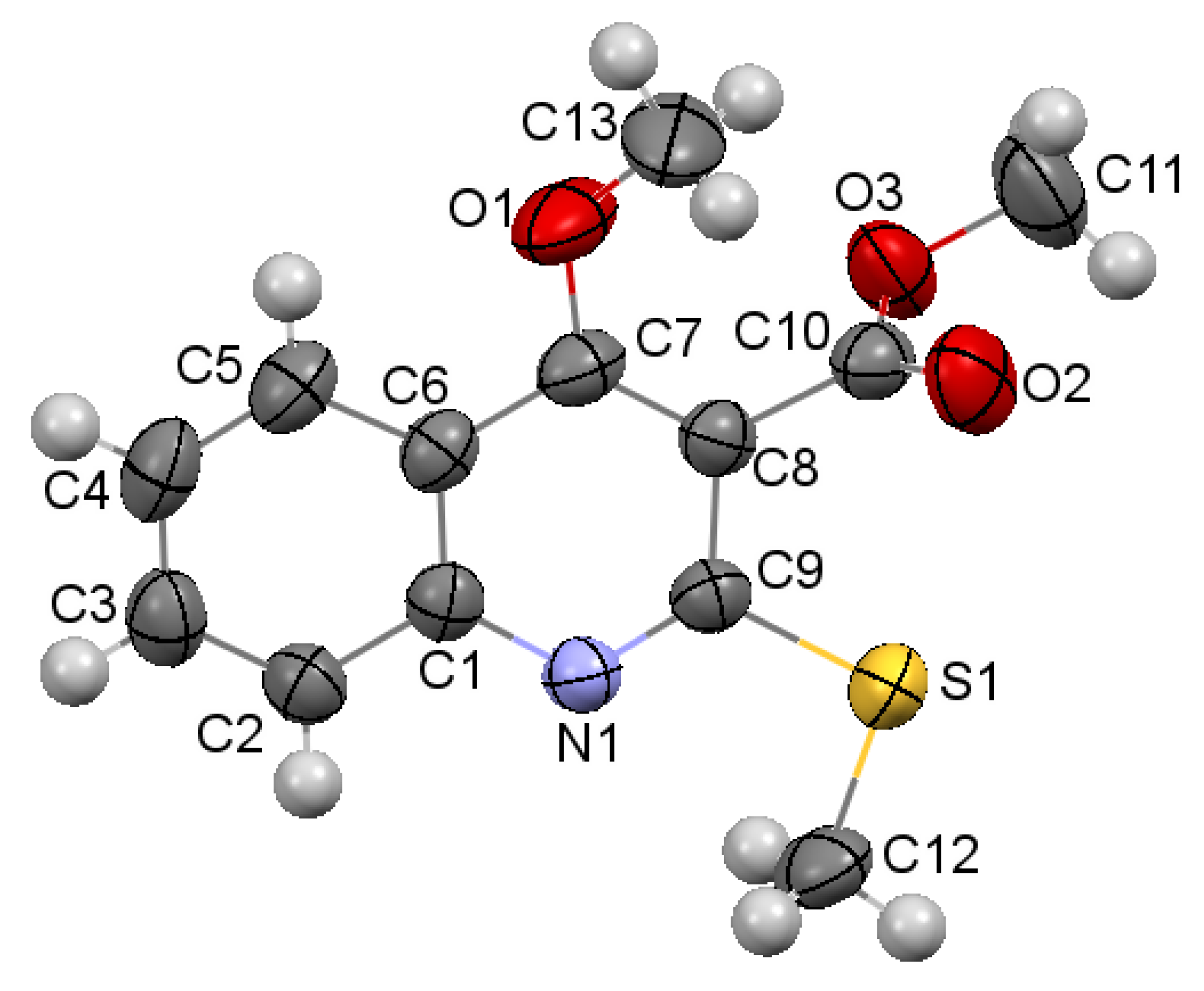
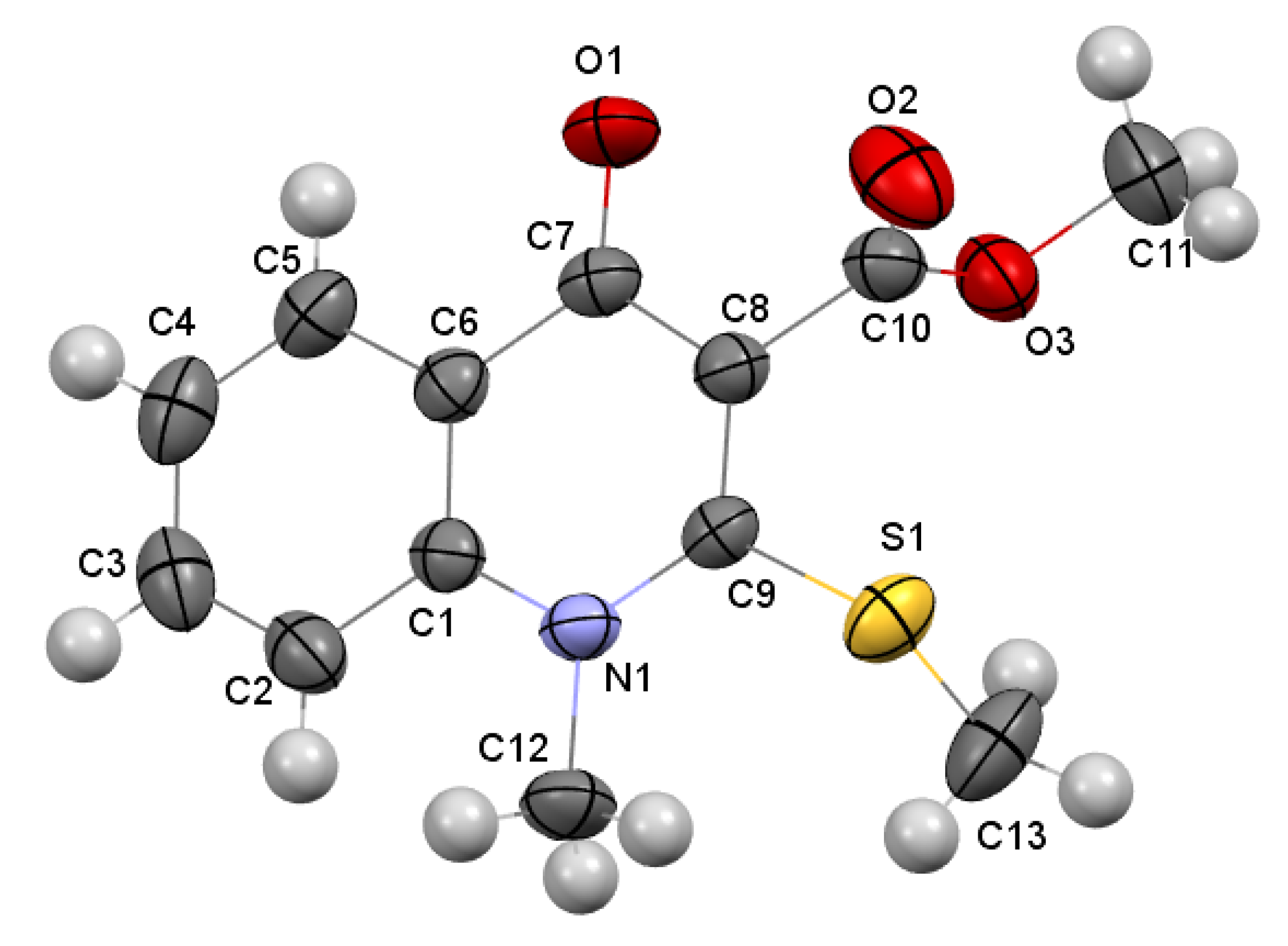
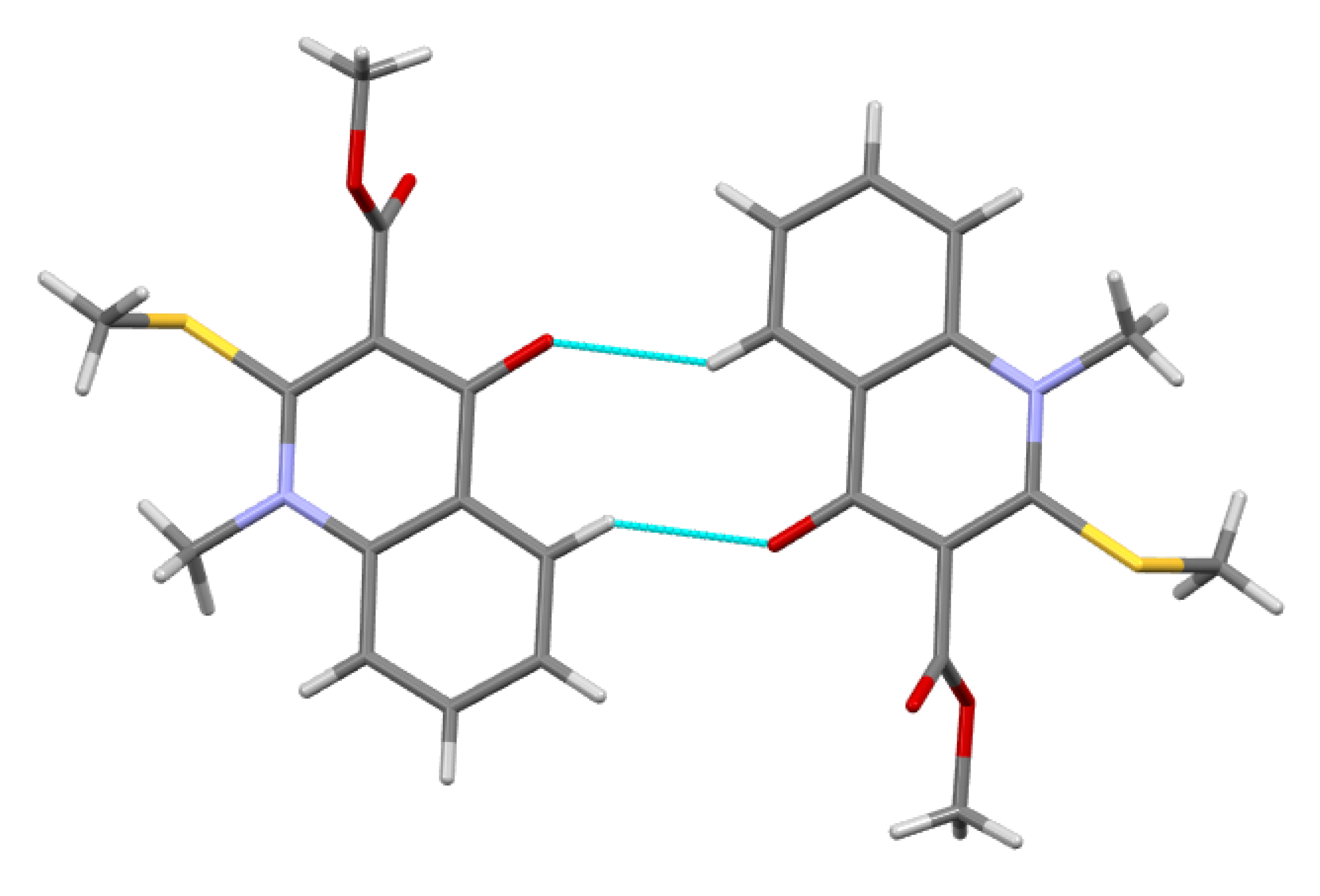
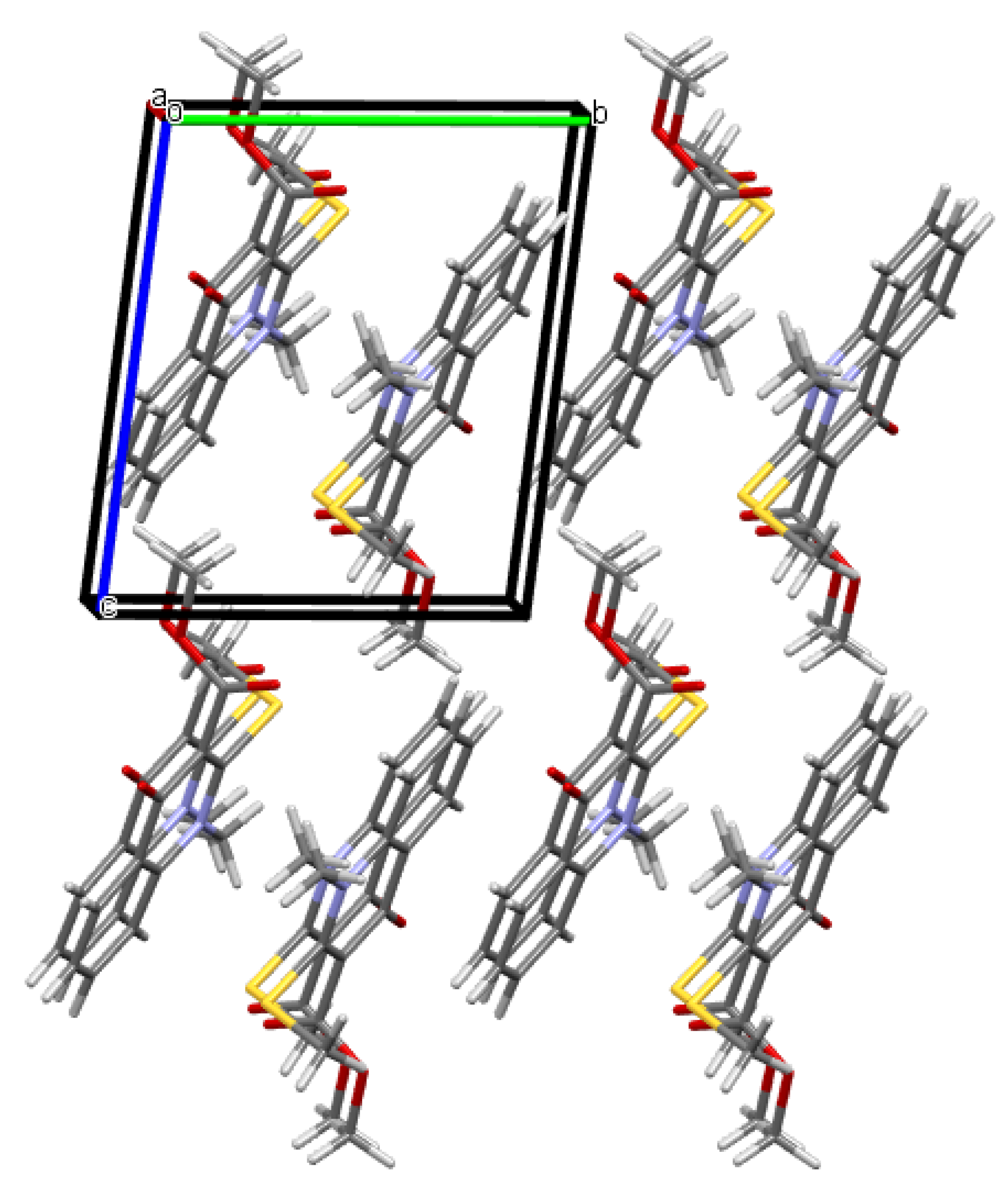
2.2. X-ray Analysis of the Reaction Products
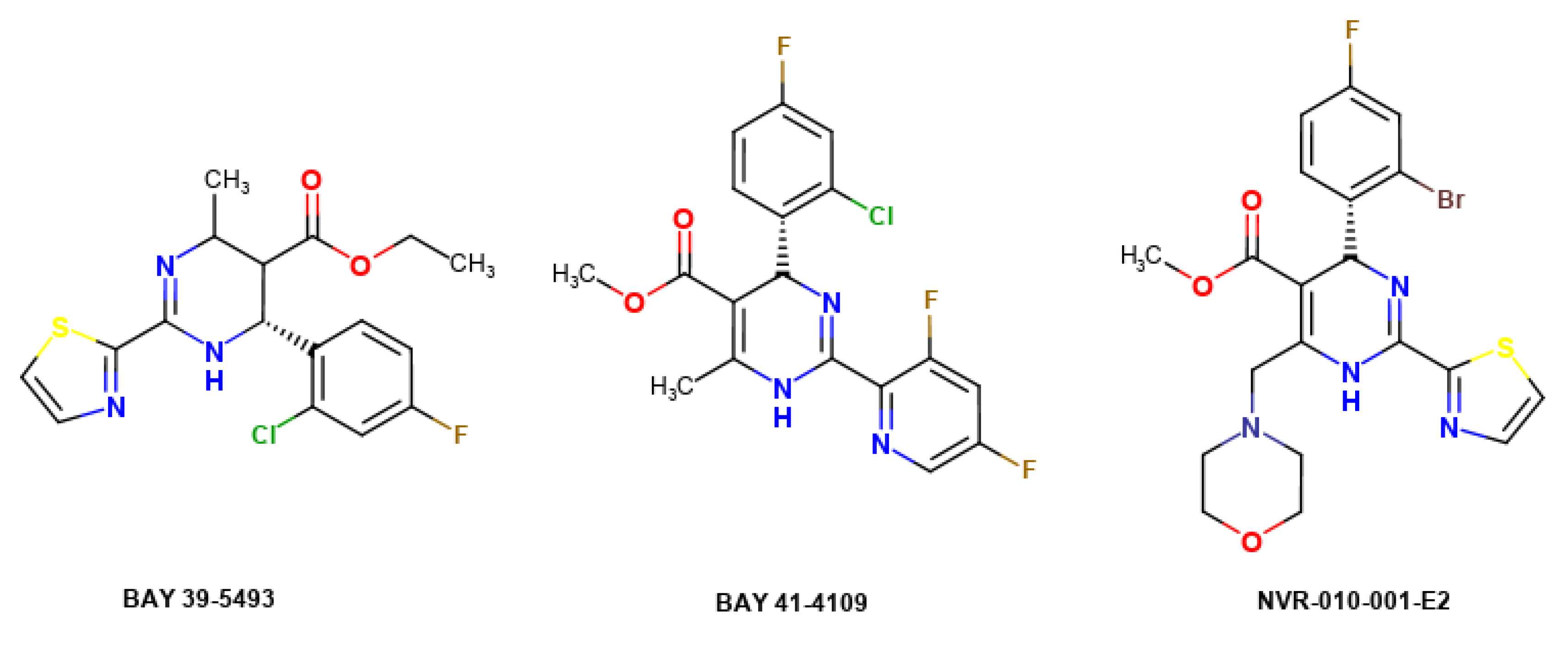
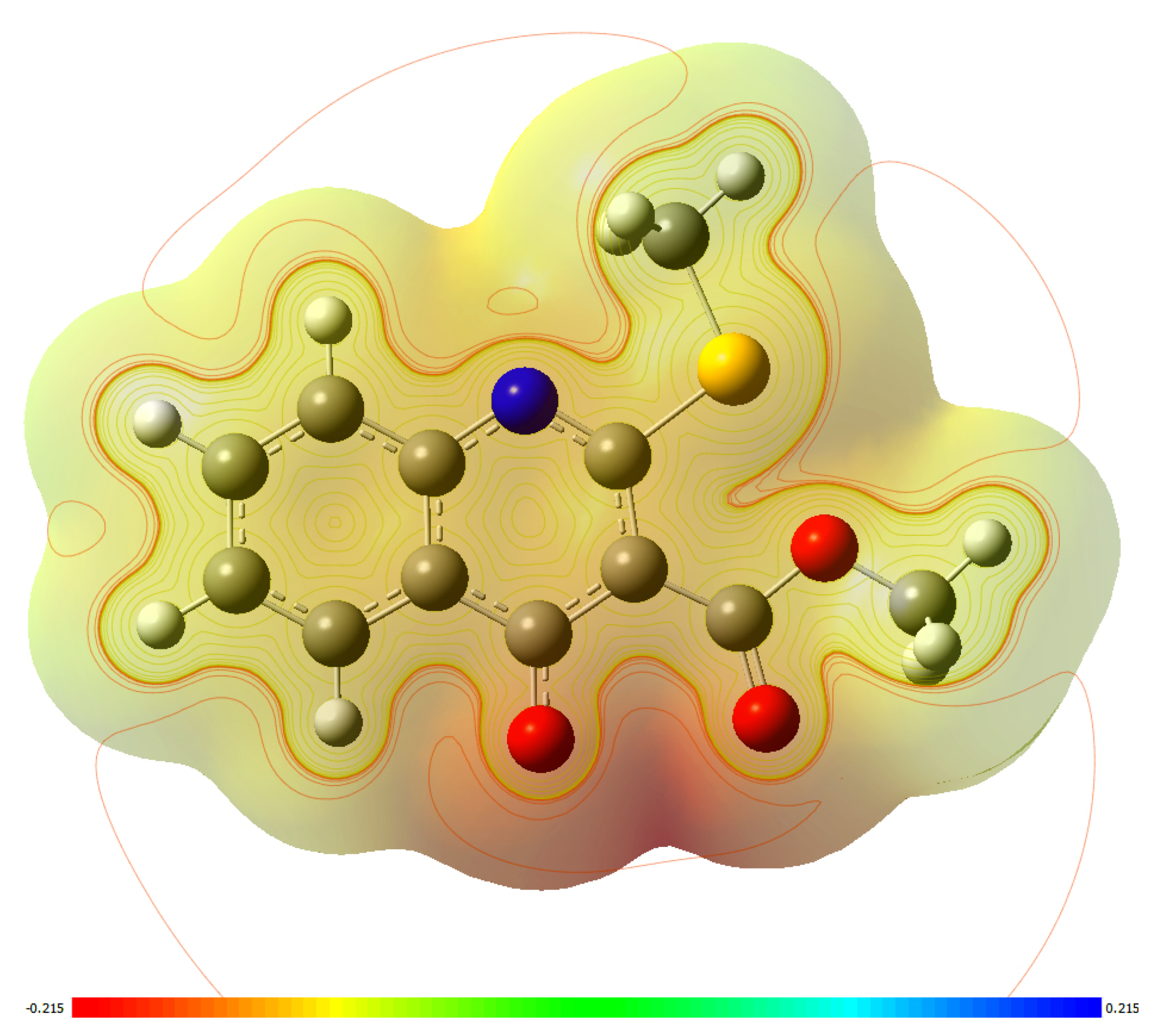
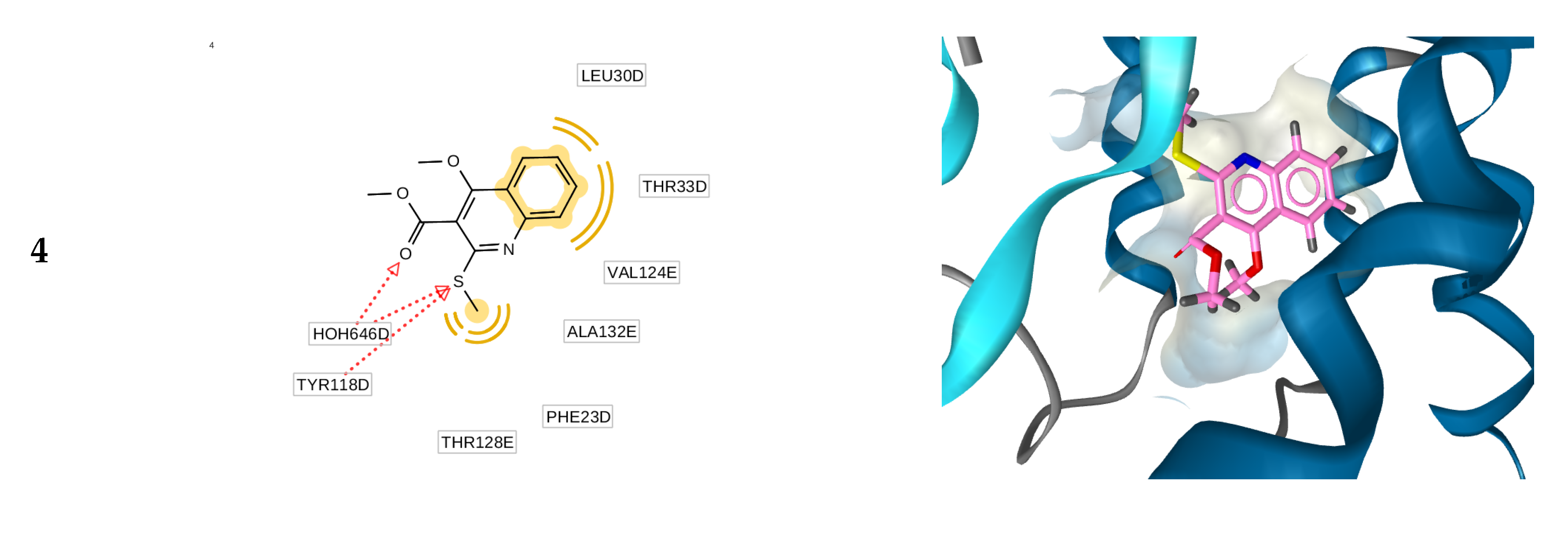
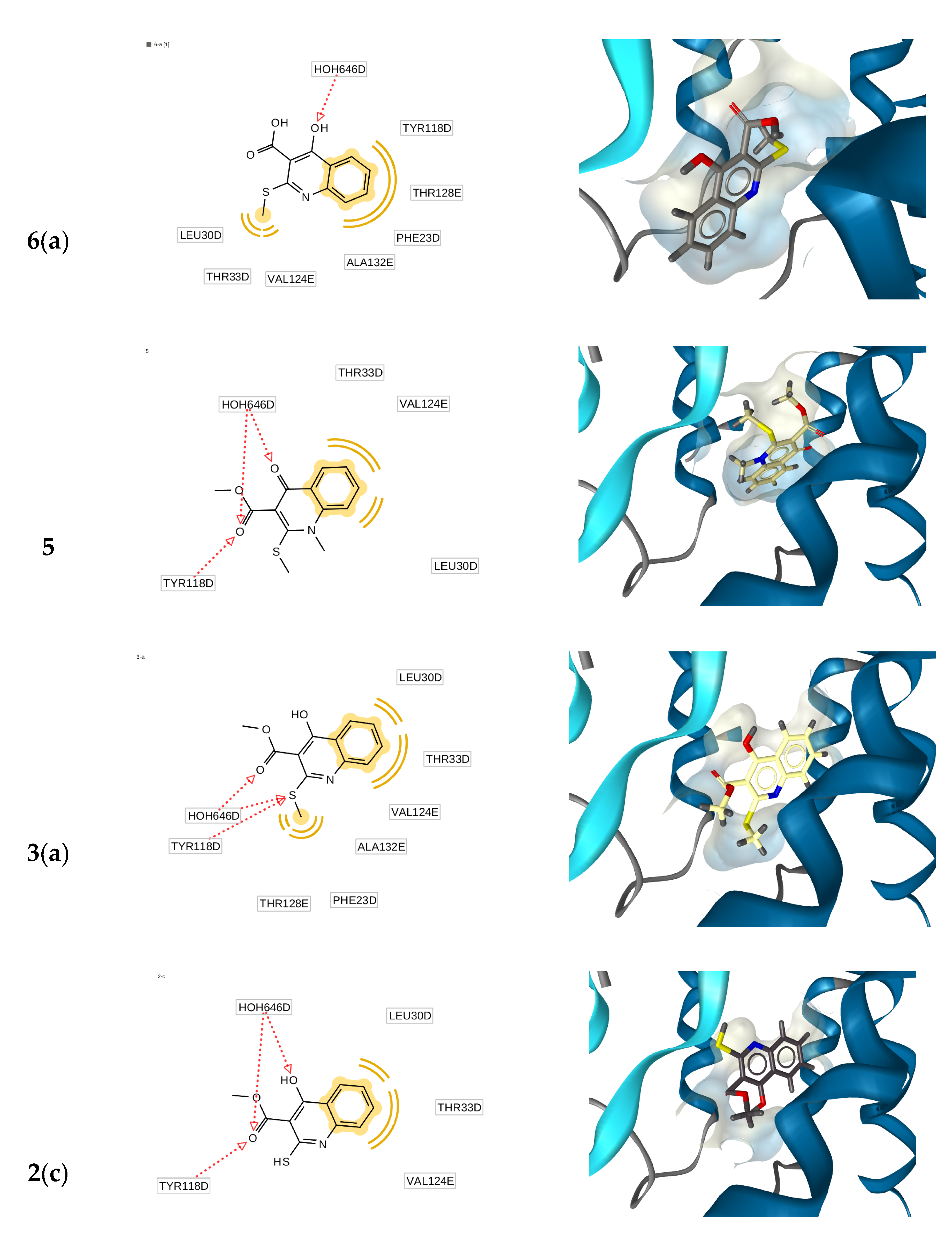
2.3. Molecular Docking Simulations
2.4. Anti-Hepatitis B Virus (HBV) Activity
3. Materials and Methods
3.1. General Information
3.2. Synthesis
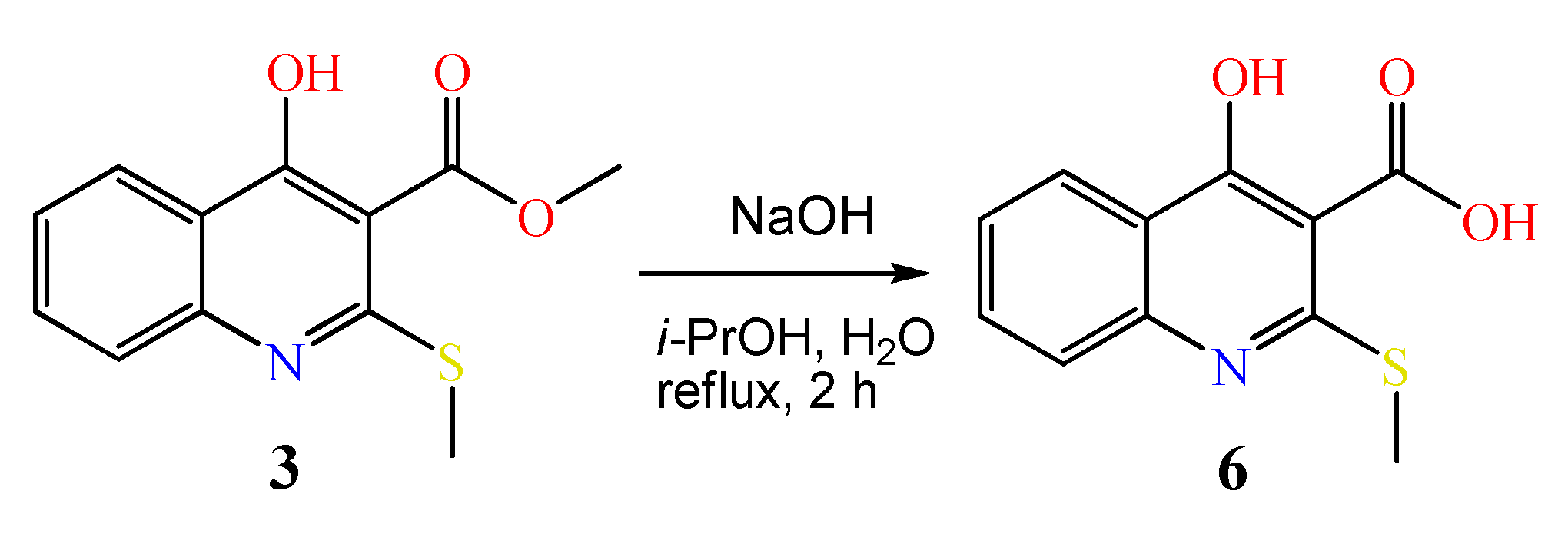
3.2.1. Synthesis of Methyl 4-Hydroxy-2-(methylthio)quinoline-3-carboxylate 3
3.2.2. Reaction of Methyl 4-Hydroxy-2-(methylthio)quinoline-3-carboxylate 3 with CH3I with the Presence of Sodium Hydride
3.2.3. Reaction of Methyl 4-Hydroxy-2-(methylthio)quinoline-3-carboxylate 3 with CH3I with the Presence of K2CO3
3.2.4. Synthesis of 4-Hydroxy-2-(methylthio)quinoline-3-carboxylic acid 6
3.3. X-ray Diffraction Study
3.3.1. Methyl 4-Hydroxy-2-(methylthio)quinoline-3-carboxylate 3
3.3.2. Methyl 4-Methoxy-2-(methylthio)quinoline-3-carboxylate 4
3.3.3. Methyl 1-Methyl-2-(methylthio)-4-oxo-1,4-dihydroquinoline-3-carboxylate 5
3.4. Anti-Hepatitis B Virus (HBV) Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gleckman, R.; Alvarez, S.; Joubert, D.W.; Matthews, S.J. Drug therapy reviews: Oxolinic acid. Am. J. Hosp. Pharm. 1979, 36, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Garcia de Mateos-Verchere, J.; Vaugeois, J.M.; Naudin, B.; Costentin, J. Behavioural and neurochemical evidence that the antimicrobial agent oxolinic acid is a dopamine uptake inhibitor. Eur. Neuropsychopharmacol. 1998, 8, 255–259. [Google Scholar] [CrossRef]
- Kazuno, K.; Kise, M.; Kitano, M.; Ozaki, M.; Segawa, J.; Shirahase, I.; Tomii, Y. Preparation of Oxothiazetoquinolinecarboxylates as Bactericides. GB Patent 2190376B, 18 November 1987. [Google Scholar]
- Taguchi, M.; Kondo, H.; Inoue, Y.; Kawahata, Y.; Tsukamoto, G. Epoxymethanothiazoloquinolone carboxylic Acid Derivatives as Medical Bactericides, Their Preparation, and Formulations Containing Them. EPA 286089A1, 12 October 1988. [Google Scholar]
- Kise, M.; Kitano, M.; Ozaki, M.; Kazuno, K.; Matsuda, M.; Shirahase, I.; Segawa, J. Preparation and Testing of 6-Fluoro-7-piperazino-4-oxo-4H-[1,3]thiazeto[3,2-a]quinolinecarboxylate Derivatives as Antibactericides. EPA 315828A1, 17 May 1989. [Google Scholar]
- Segawa, J.; Kitano, M.; Kazuno, K.; Tsuda, M.; Shirahase, I.; Ozaki, M.; Matsuda, M.; Kise, M. Studies on pyridonecarboxylic acids. 2. Synthesis and antibacterial activity of 8-substituted 7-fluoro-5-oxo-5H-thiazolo[3,2-a]quinoline-4-carboxylic acids. J. Heterocycl. Chem. 1992, 29, 1117–1123. [Google Scholar] [CrossRef]
- Segawa, J.; Kazuno, K.; Matsuoka, M.; Shirahase, I.; Ozaki, M.; Matsuda, M.; Tomii, Y.; Kitano, M.; Kise, M. Studies on pyridonecarboxylic acids. III. Synthesis and antibacterial activity evaluation of 1,8-disubstituted 6-fluoro-4-oxo-7-piperazinyl-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid derivatives. Chem. Pharm. Bull. 1995, 43, 63–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahmed, A.; Daneshtalab, M. Polycyclic quinolones (part 1) - Thieno[2,3-b]benzo[h]quinoline derivatives: Design, synthesis, preliminary in vitro and in silico studies. Heterocycles 2012, 85, 103–122. [Google Scholar] [CrossRef]
- Segawa, J. Process for Producing Quinolinecarboxylic Acid Derivative. WO 9404506A1, 3 March 1994. [Google Scholar]
- Zankel, T.C.; Isbell, S.L. Use of Thiol Compounds to Treat Neurological Diseases. WO 2018200527A1, 1 November 2018. [Google Scholar]
- Lim, H.J.; Park, S.J.; Jeong, K.C.; Seo, H.K.; Ahn, K.O.; Lee, S.J.; Lee, E.S. Preparation of Quinolinones Inhibiting Formation of c-Myc/Max/DNA Complex. WO 2018021810 A1, 1 February 2018. [Google Scholar]
- Jeong, K.C.; Lim, H.J.; Park, S.J.; Seo, H.K.; Ahn, K.O.; Lee, S.J.; Lee, E.S. Anticancer Pharmaceutical Composition. WO 2018021849 A1, 1 February 2018. [Google Scholar]
- Endres, D.; Miyahara, M.; Moisant, P.; Zlotnick, A. A reaction landscape identifies the intermediates critical for self-assembly of virus capsids and other polyhedral structures. Protein Sci. 2005, 14, 1518–1525. [Google Scholar] [CrossRef]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. 2006, 19, 542–548. [Google Scholar] [CrossRef]
- Choi, I.G.; Yu, Y.G. Interaction and assembly of HBV structural proteins: Novel target sites of anti-HBV agents. Infect. Disord. Drug Targets 2007, 7, 251–256. [Google Scholar] [CrossRef]
- Arsianti, A.; Yanuar, A. Molecular Docking and Dynamic Simulation Benzoylated Emodin into HBV Core Protein. J. Young Pharm. 2018, 10, S20–S24. [Google Scholar] [CrossRef]
- Ivashchenko, A.; Mitkin, O.; Kravchenko, D.; Kuznetsova, I.; Kovalenko, S.; Bunyatyan, N.; Langer, T. Synthesis, X-ray crystal structure, Hirshfeld surface analysis, and molecular docking study of novel hepatitis B (HBV) inhibitor: 8-Fluoro-5-(4-fluorobenzyl)-3-(2-methoxybenzyl)-3,5-dihydro-4H-pyrimido[5,4-b]indol-4-one. Crystals 2019, 9, 379. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Mitkin, O.D.; Kravchenko, D.V.; Kovalenko, S.M.; Shishkina, S.V.; Bunyatyan, N.D.; Konovalova, I.S.; Dmitrieva, I.G.; Ivanov, V.V.; Langer, T. Synthesis, X-ray crystal structure, Hirshfeld surface analysis, and molecular docking study of novel inhibitor of hepatitis B: Methyl 4-fluoro-3-(morpholinosulfonyl)benzo[b]thiophene-2-carboxylate. Heliyon 2019, 5, e02738. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, S.M.; Drushlyak, O.G.; Konovalova, I.S.; Mariutsa, I.O.; Kravchenko, D.V.; Ivachtchenko, A.V.; Mitkin, O.D. Novel One-Pot Synthesis of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic and Crystallographic Studies. Molbank 2019, 2019, M1085. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian-09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zefirov, Y.V.; Zorky, P.M. New applications of van der Waals radii in chemistry. Russ. Chem. Rev. 1995, 64, 415–428. [Google Scholar] [CrossRef]
- Burgi, H.-B.; Dunitz, J.D. Structure Correlation; VCH: Weinheim, Germany, 1994; Volume 2, p. 741. [Google Scholar]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D with Features for Chemicals, Crystals, Materials and Biomolecules. Available online: http://jmol.sourceforge.net/ (accessed on 1 October 2008).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein Data Bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from proteinbound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cuconati, A. Hepatitis B Virus: Methods and Protocols. In Methods in Molecular Biology; Springer Science + Business Media LLC: Berlin/Heidelberg, Germany, 2017; volume 1540, pp. 27–36. [Google Scholar] [CrossRef]
- Donkers, J.M.; Zehnder, B.; van Westen, G.J.P.; Kwakkenbos, M.J.; Jzerman, A.P.; Oude Elferink, R.P.J.; Beuers, U.; Urban, S.; van de Graaf, F.J. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta. Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Solvent | Base | Temperature (°C) | Reaction Time (h) | Yield of 4 (%), Estimated by 1H-NMR | Yield of 5 (%), Estimated by 1H-NMR |
|---|---|---|---|---|---|---|
| 1 | DMF | NEt3 | 80 | 8 | 3 1 | 0 |
| 2 | DMF | DBU | 80 | 8 | 7 1 | 0 |
| 3 | DMSO | DBU | 80 | 8 | 25 1 | 0 |
| 4 | acetone | K2CO3 | 60 | 8 | 55 1 | 5 1 |
| 5 | DMF | NaH | 60 | 2 | 87 | 13 |
| 6 | DMF | NaH | 80 | 1 | 80 | 20 |
| 7 | DMF | K2CO3 | 80 | 4 | 99 | 1 |
| 8 | DMSO | K2CO3 | 80 | 2 | 99 | 0.8 |
| 9 | DMF | NaH | 80 | 8 | 99 | 0.4 |
| 10 | DMF | K2CO3 | 80 | 8 | 99 | 0.6 |
| Mol | BAS | Final Intermolecular Energy (kcal/mol) | Est. Binding Energy (kcal/mol) | cLogP | TPSA [26] |
|---|---|---|---|---|---|
| reference | −28.2 | −16.55 | −14.76 | 2.61 | 76.05 |
| 4 | −25 | −13.0 | −12.0 | 2.52 | 48.42 |
| 6(a) | −23.15 | −13.0 | −12.39 | 2.13 | 70.4 |
| 5 | −22.07 | −12.14 | −11.24 | 1.82 | 48.3 |
| 3(a) | −21.81 | −12.92 | −13.03 | 2.22 | 59.42 |
| 2(c) | −20.71 | −11.52 | −10.92 | 2.16 | 59.42 |
| Mol | HBeAg 1 Inhibition, % | Cell Survival, % |
|---|---|---|
| 4 | 83 ± 7% | 91 ± 6% |
| 6 | 79 ± 4% | 78 ± 7% |
| 3 | 68 ± 6% | 87 ± 5% |
| zafirlukast (positive control) | 82 ± 9% | 89 ± 7% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovalenko, S.M.; Drushlyak, O.G.; Shishkina, S.V.; Konovalova, I.S.; Mariutsa, I.O.; Bunyatyan, N.D.; Kravchenko, D.V.; Ivanov, V.V.; Ivachtchenko, A.V.; Langer, T. Methylation of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic, Crystallographic, and Molecular Docking Studies. Molecules 2020, 25, 4238. https://doi.org/10.3390/molecules25184238
Kovalenko SM, Drushlyak OG, Shishkina SV, Konovalova IS, Mariutsa IO, Bunyatyan ND, Kravchenko DV, Ivanov VV, Ivachtchenko AV, Langer T. Methylation of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic, Crystallographic, and Molecular Docking Studies. Molecules. 2020; 25(18):4238. https://doi.org/10.3390/molecules25184238
Chicago/Turabian StyleKovalenko, Sergiy M., Oleksandr G. Drushlyak, Svitlana V. Shishkina, Irina S. Konovalova, Illia O. Mariutsa, Natalya D. Bunyatyan, Dmitry V. Kravchenko, Vladimir V. Ivanov, Alexandre V. Ivachtchenko, and Thierry Langer. 2020. "Methylation of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic, Crystallographic, and Molecular Docking Studies" Molecules 25, no. 18: 4238. https://doi.org/10.3390/molecules25184238
APA StyleKovalenko, S. M., Drushlyak, O. G., Shishkina, S. V., Konovalova, I. S., Mariutsa, I. O., Bunyatyan, N. D., Kravchenko, D. V., Ivanov, V. V., Ivachtchenko, A. V., & Langer, T. (2020). Methylation of Methyl 4-Hydroxy-2-thioxo-1,2-dihydroquinoline-3-carboxylate: Synthetic, Crystallographic, and Molecular Docking Studies. Molecules, 25(18), 4238. https://doi.org/10.3390/molecules25184238