Synthesis, Characterization, Electrochemistry, Photoluminescence and Magnetic Properties of a Dinuclear Erbium(III)-Containing Monolacunary Dawson-Type Tungstophosphate: [{Er(H2O)(CH3COO)(P2W17O61)}2]16−

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
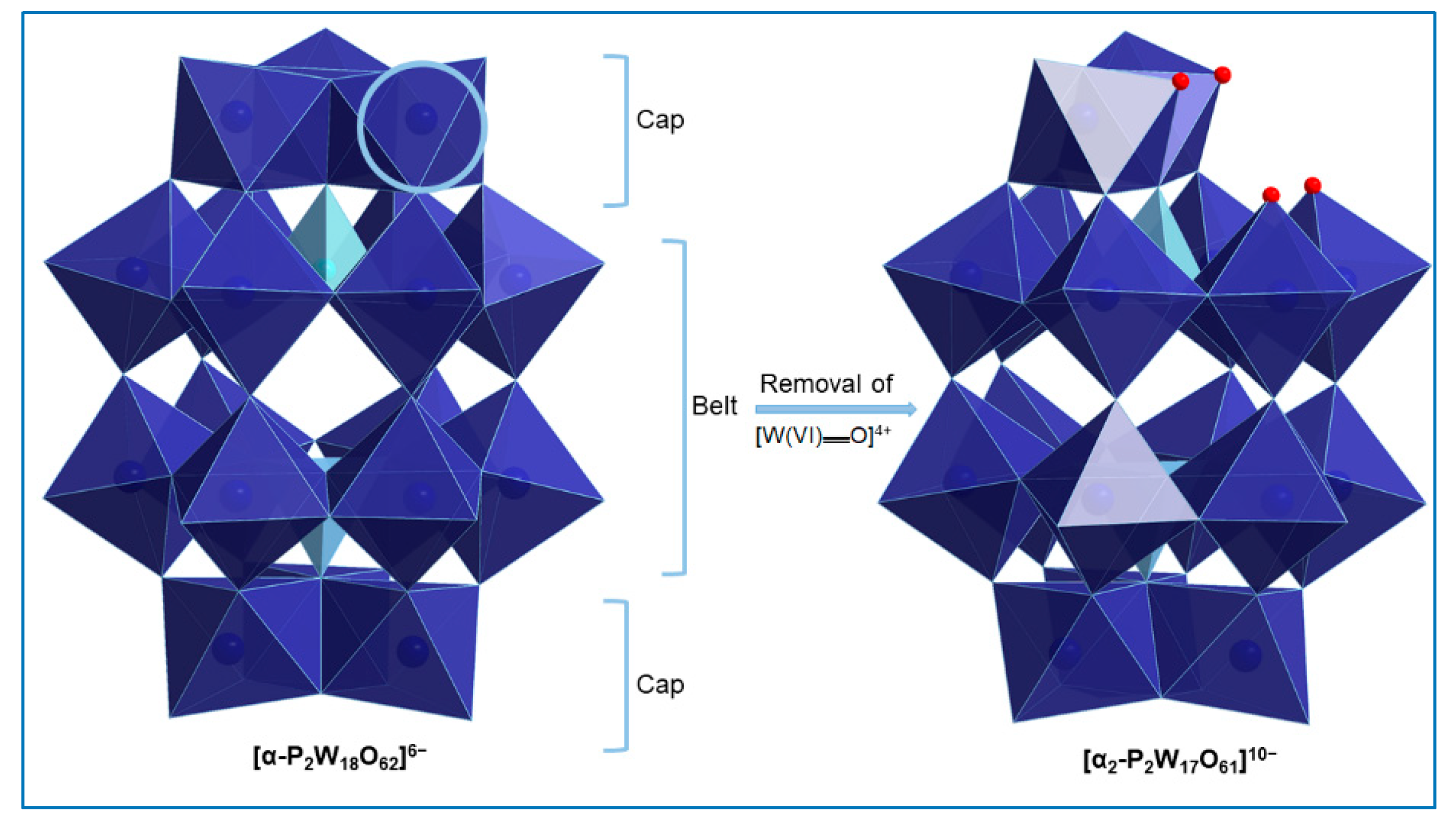
2.1. Synthesis
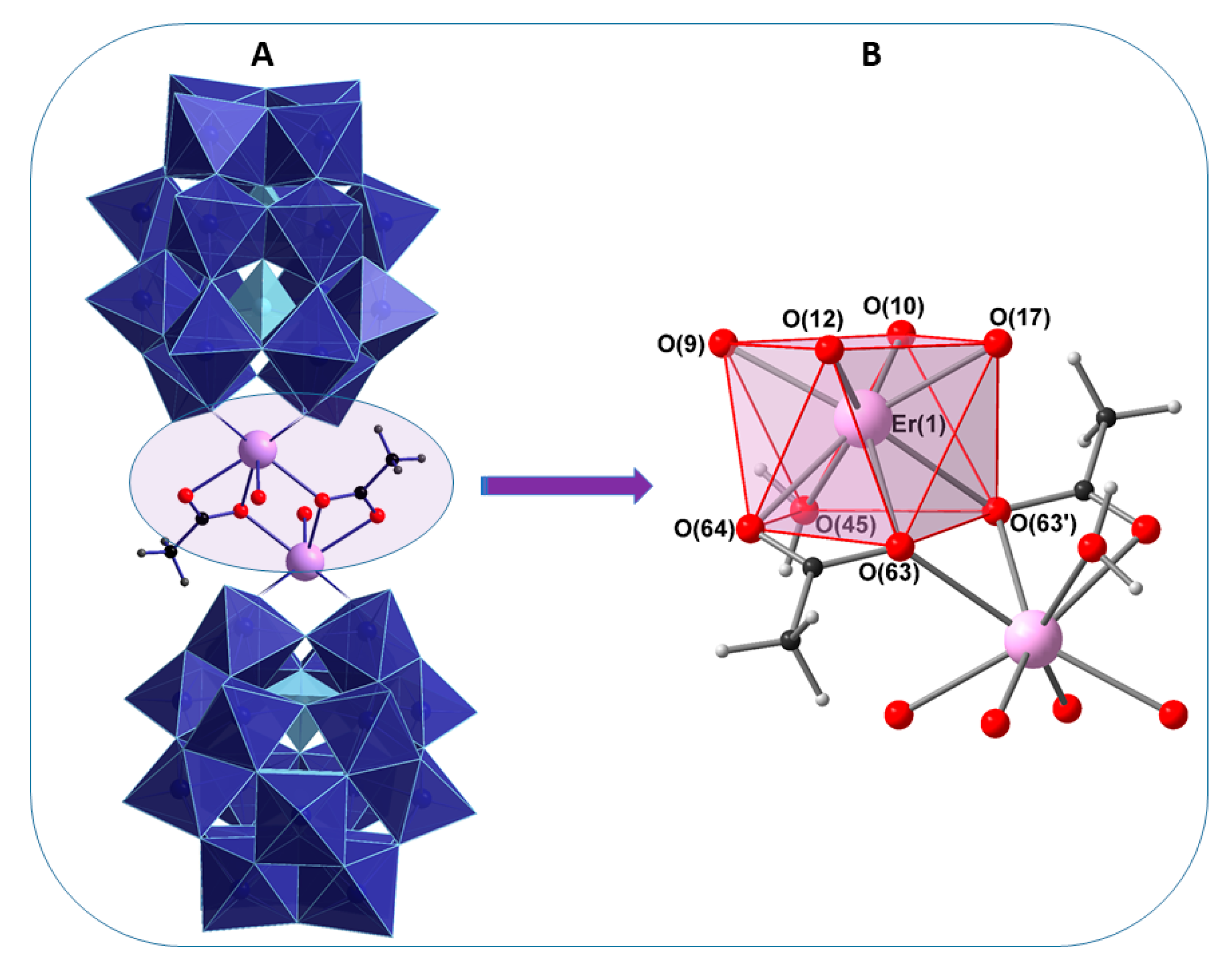
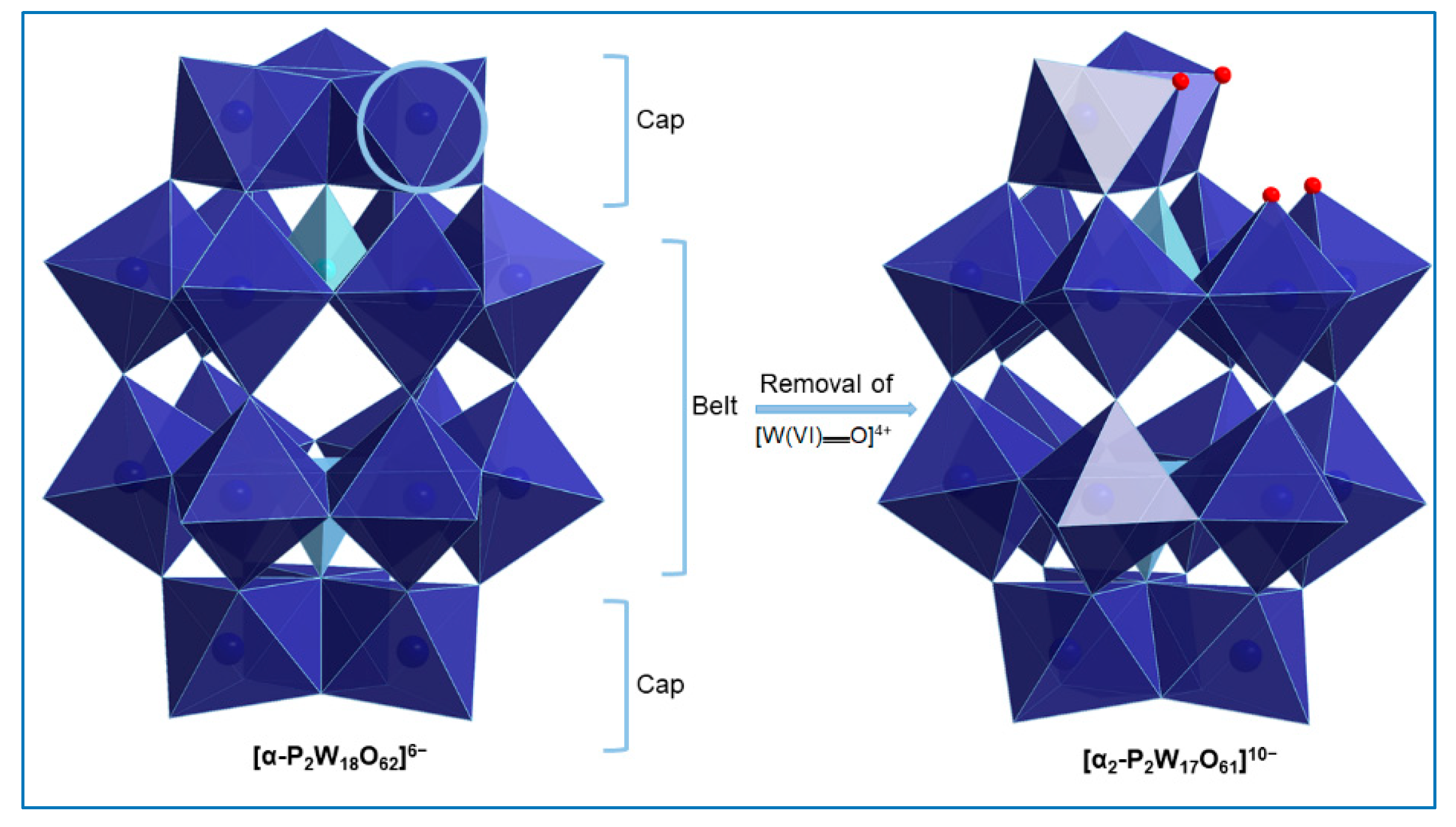
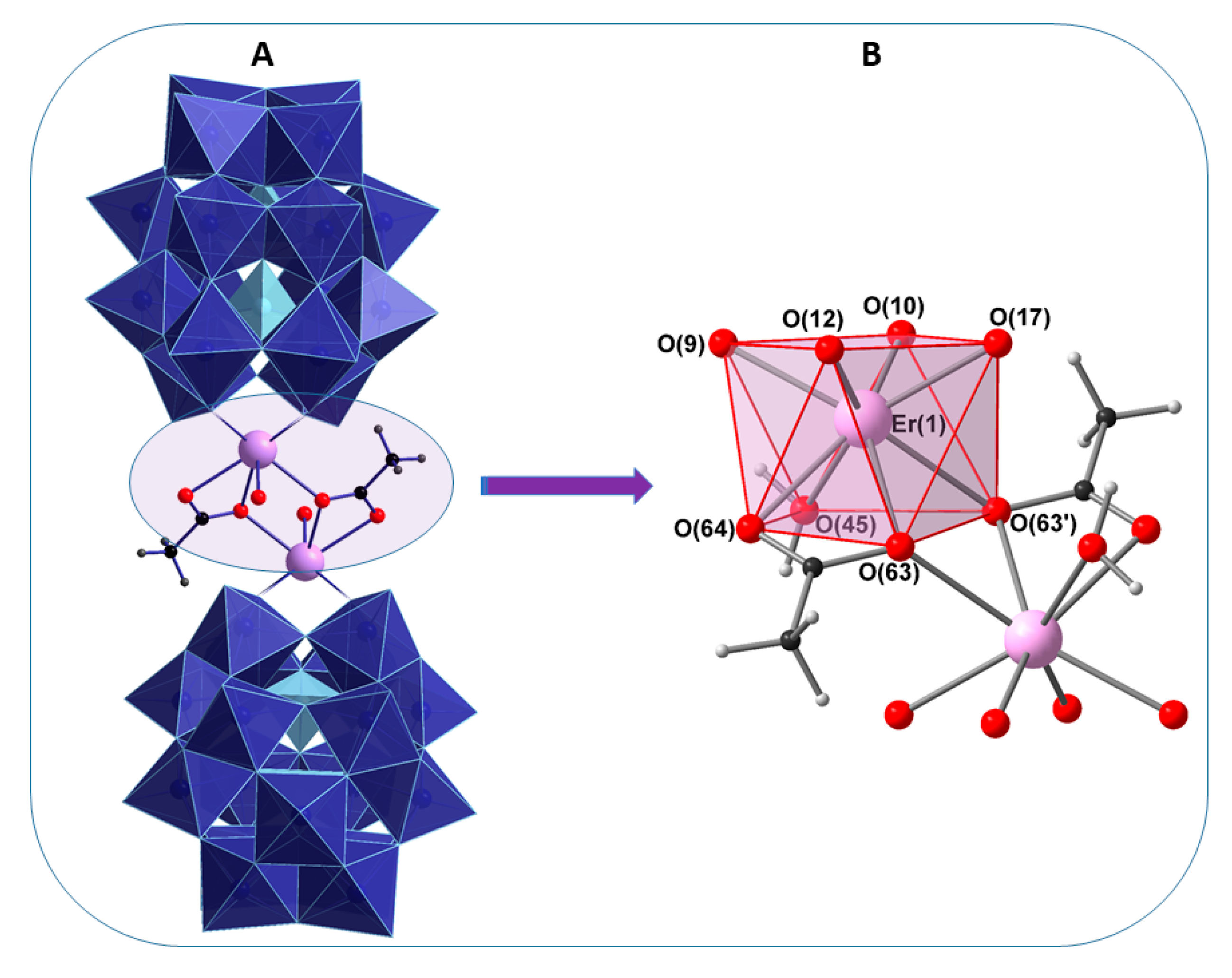
2.2. Single-Crystal X-Ray Structure Determination
2.3. PXRD Analysis
2.4. Vibrational Spectroscopy
2.5. Thermogravimetric Analysis
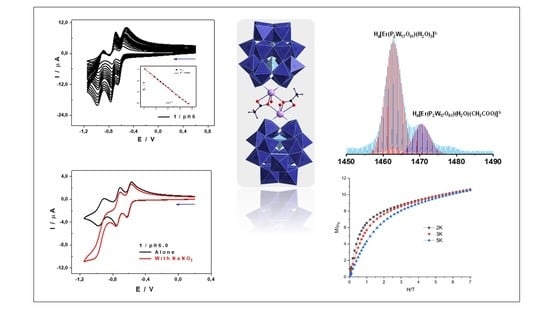
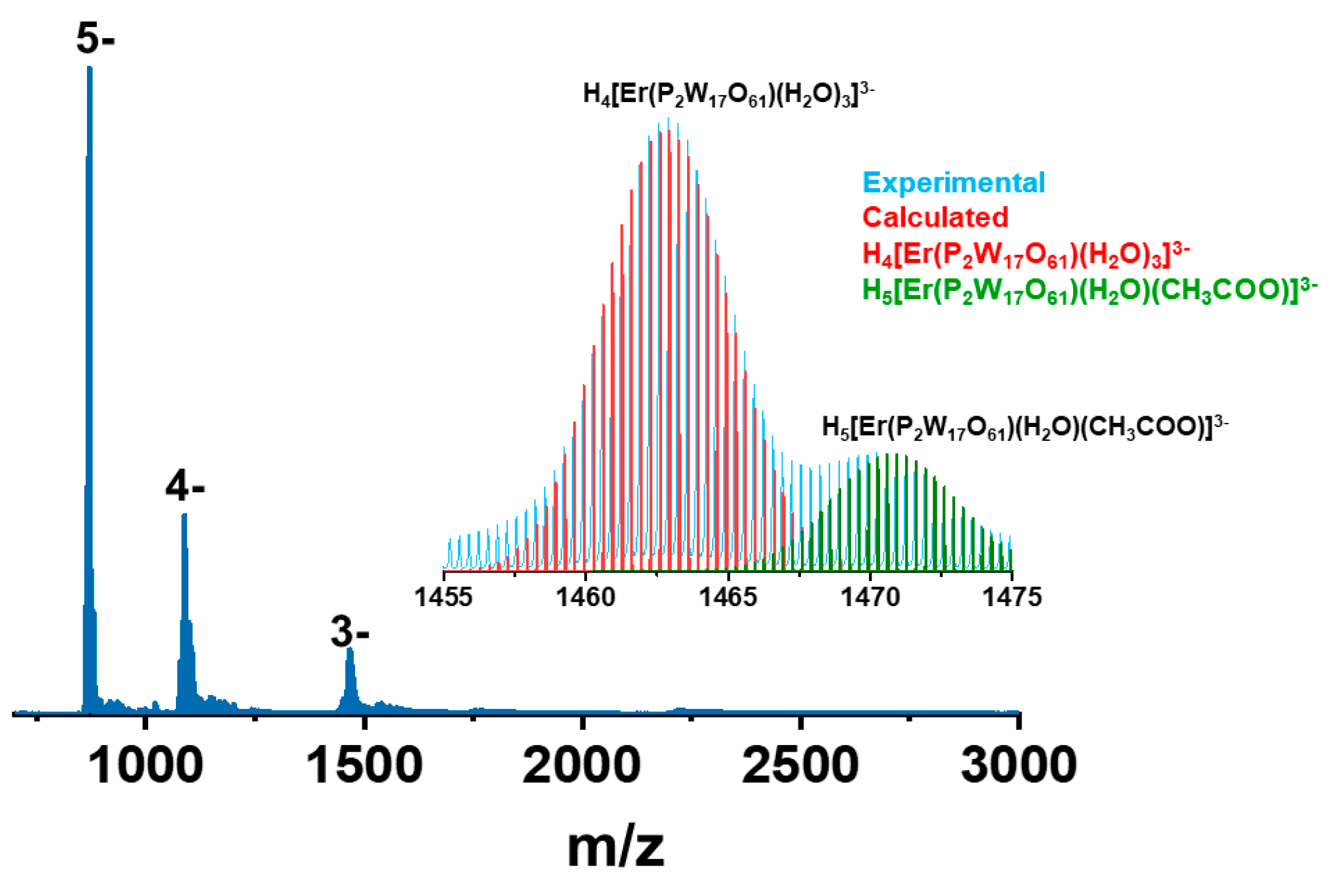
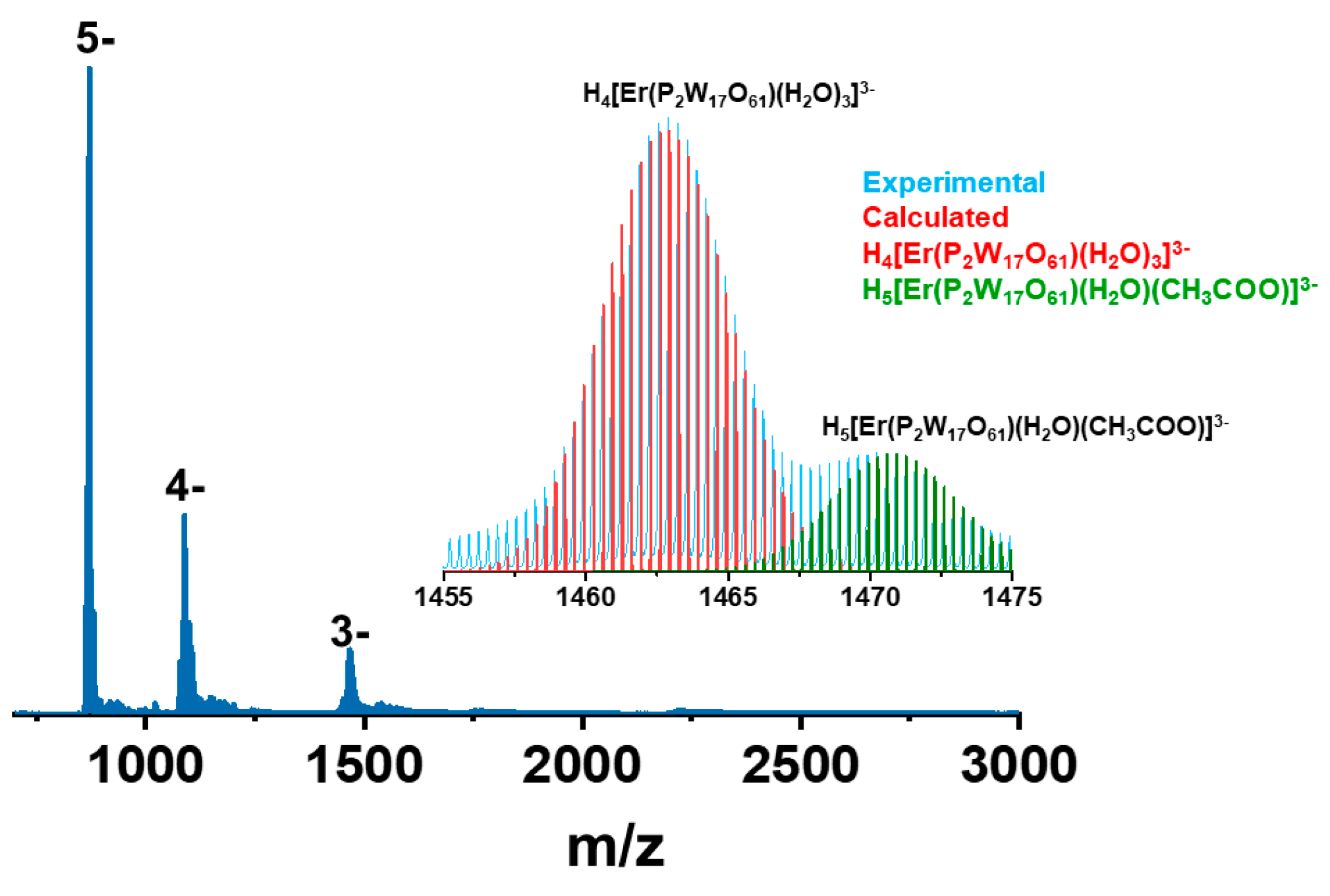
2.6. Mass Spectrometry
2.7. UV-Vis Spectroscopy
2.8. Emission Spectroscopy
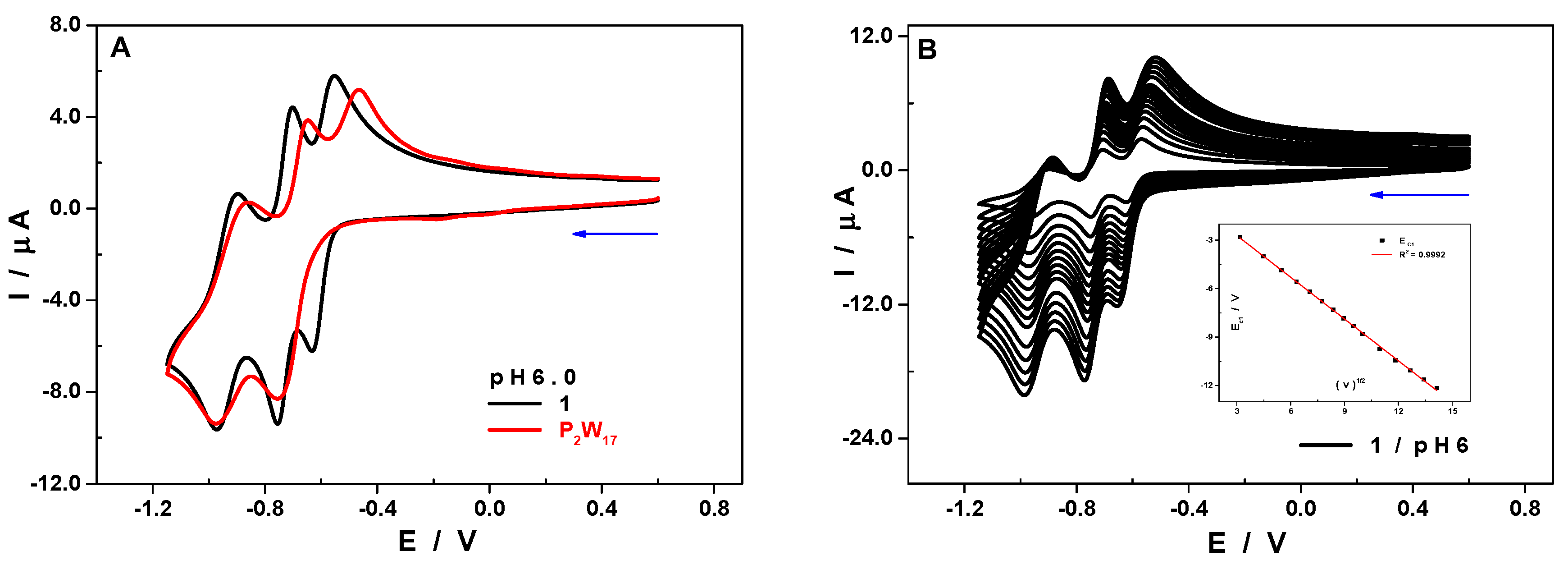
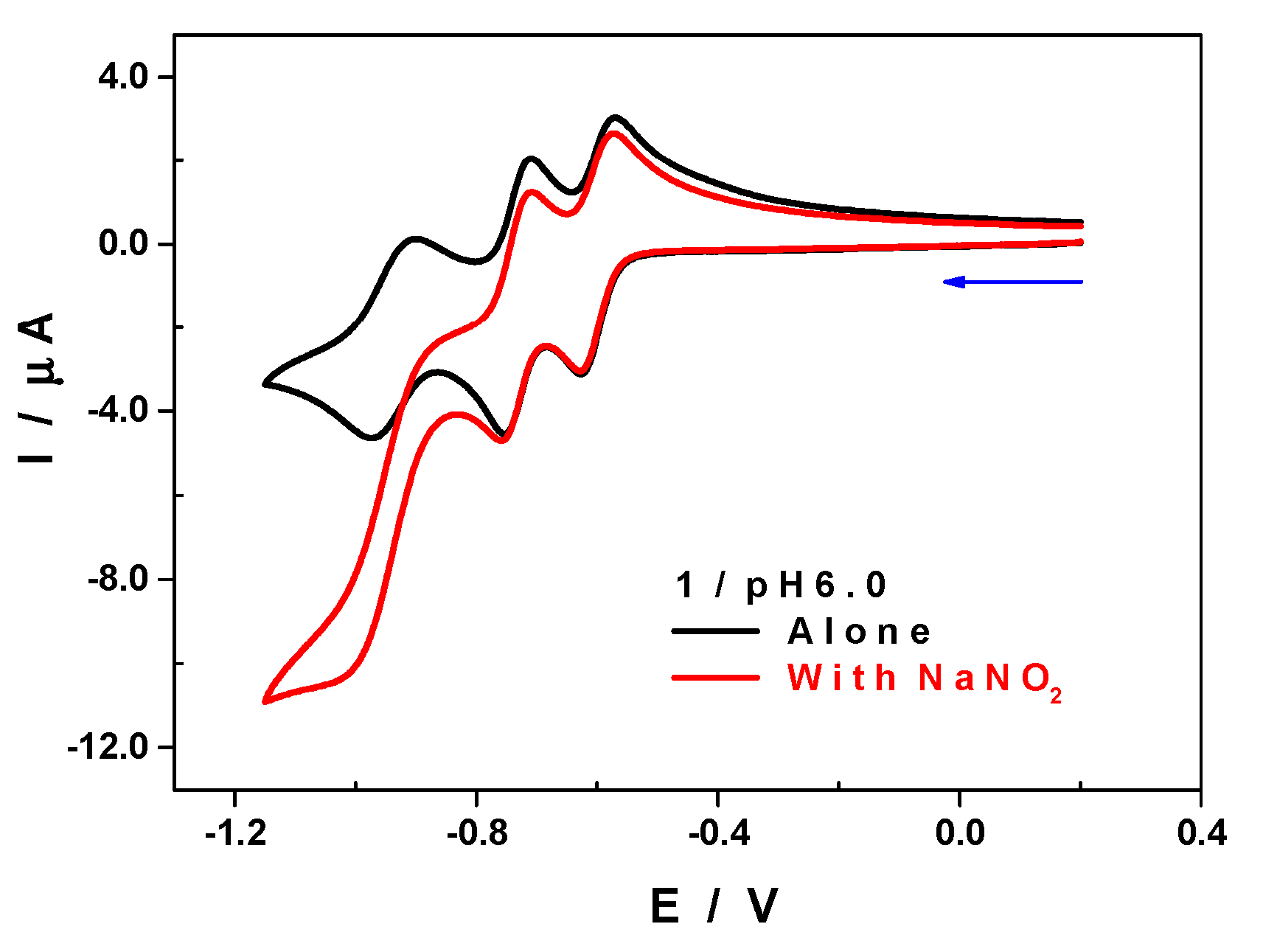
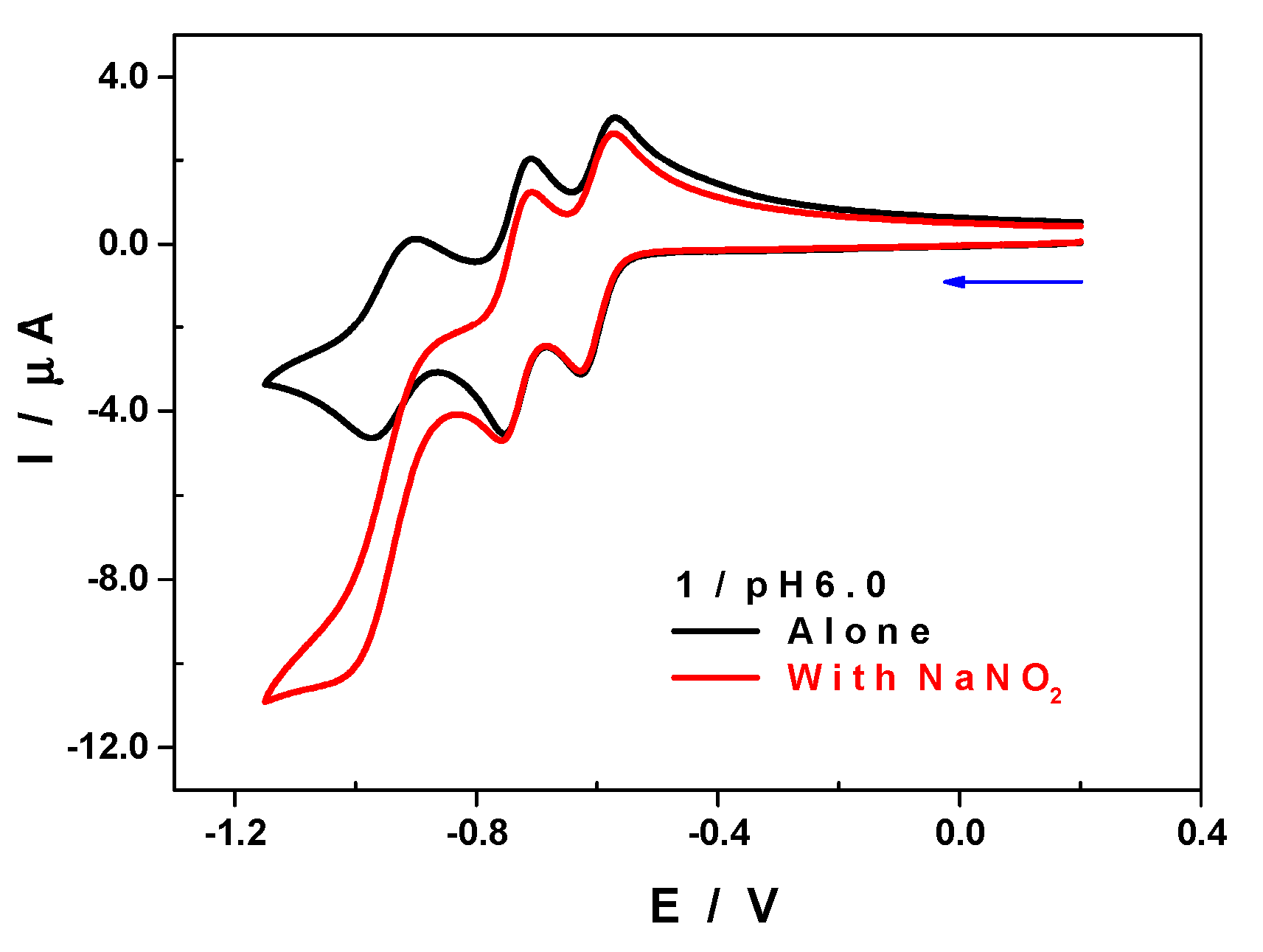
2.9. Electrochemical Characterization
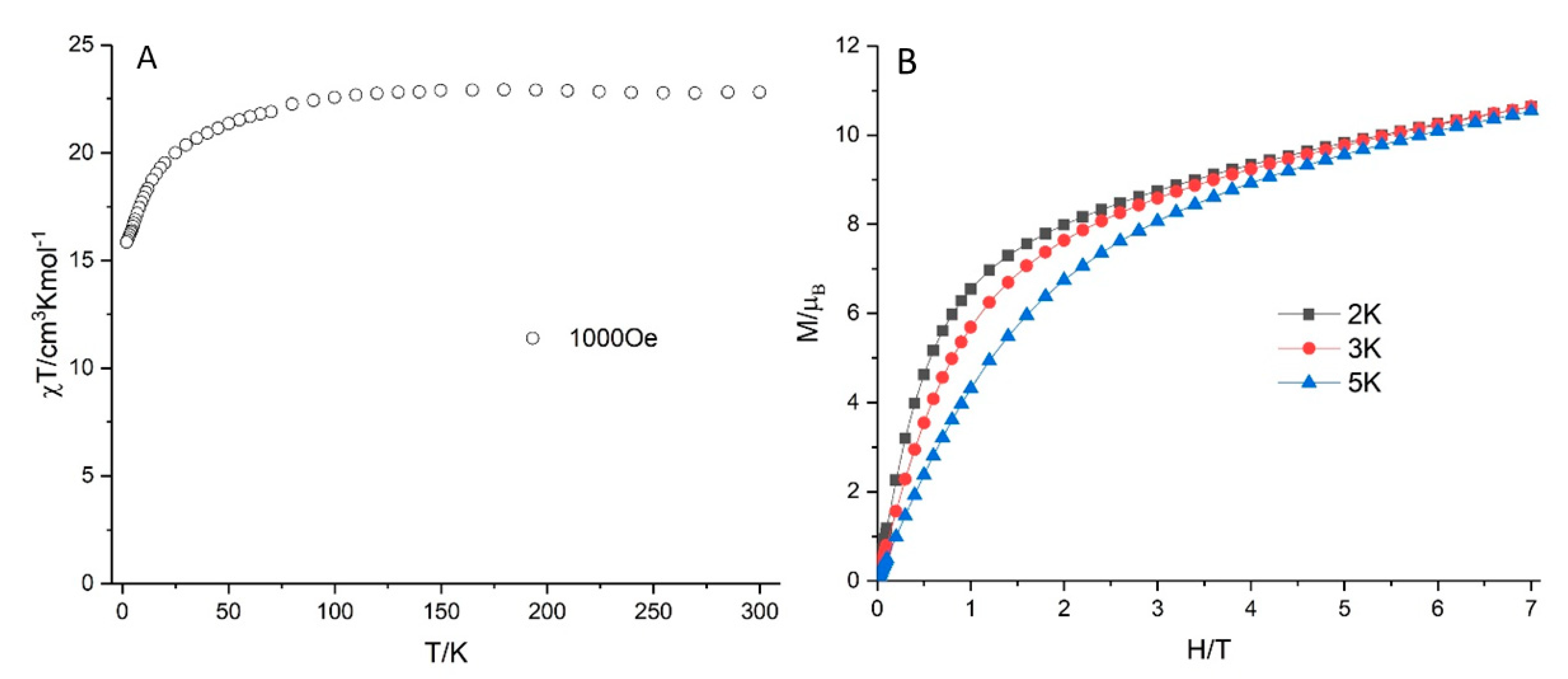
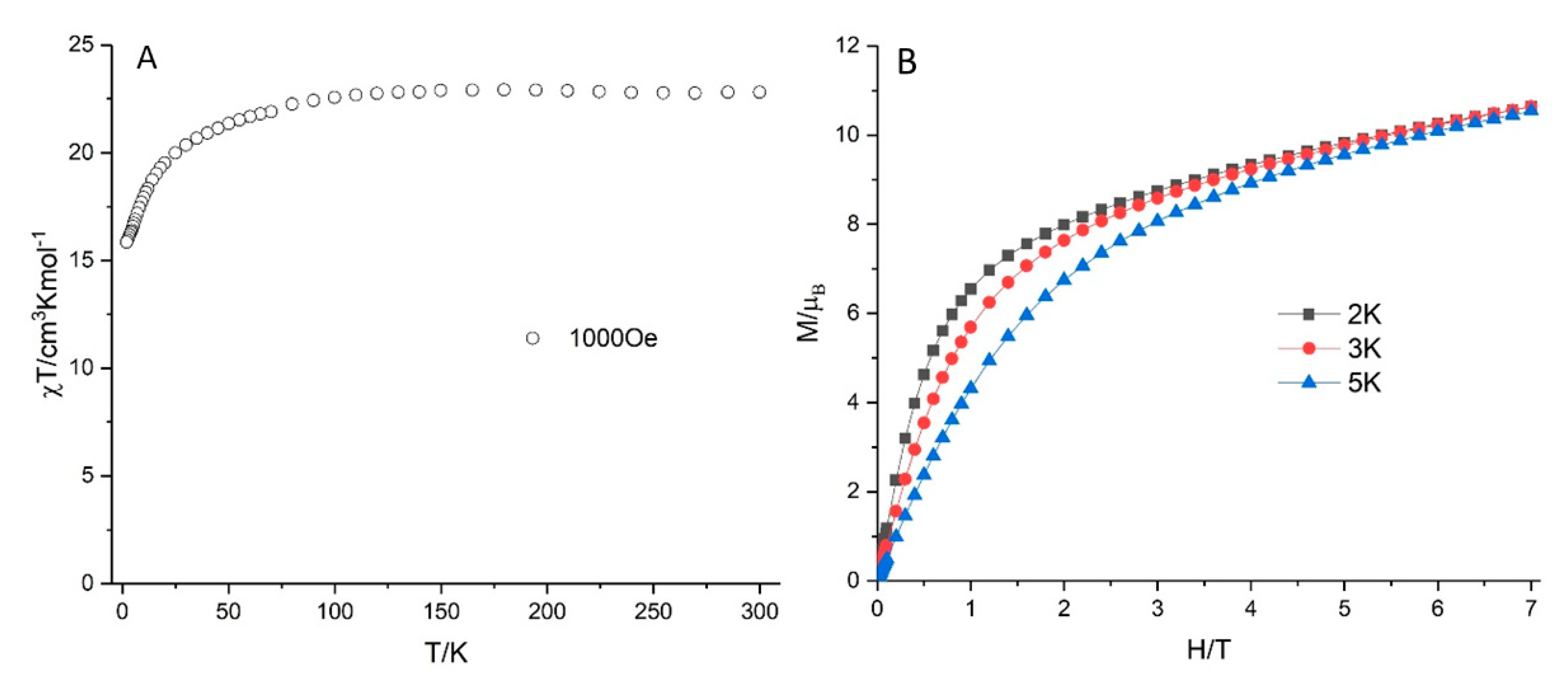
2.10. Magnetic Properties
3. Experimental Section
Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Han, Q.; Ding, Y. Recent advances in the field of light-driven water oxidation catalyzed by transition-metal substituted polyoxometalates. Dalton. Trans. 2018, 47, 8180–8188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.W.; Li, Y.Z.; Chen, L.J.; Yang, G.Y. Research progress on polyoxometalate-based transition-metal-rare-earth heterometallic derived materials: Synthetic strategies, structural overview and functional applications. Chem. Commun. 2016, 52, 4418–4445. [Google Scholar] [CrossRef] [PubMed]
- Bassil, B.S.; Kortz, U. Recent advances in lanthanide-containing polyoxotungstates. Zeitschrift Anorg. Allg. Chem. 2010, 636, 2222–2231. [Google Scholar] [CrossRef]
- Reinoso, S. Heterometallic 3d-4f polyoxometalates: Still an incipient field. Dalton. Trans. 2011, 40, 6610–6615. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Mereacre, V.; Leblanc, N.; Wernsdorfer, W.; Anson, C.E.; Powell, A.K. Self-assembly of a giant tetrahedral 3 d-4 f single-molecule magnet within a polyoxometalate system. Angew. Chem. Int. Ed. 2015, 54, 15574–15578. [Google Scholar] [CrossRef]
- Chen, W.C.; Jiao, C.Q.; Wang, X.L.; Shao, K.Z.; Su, Z.M. Self-assembly of nanoscale lanthanoid-containing selenotungstates: Synthesis, structures, and magnetic studies. Inorg. Chem. 2019, 58, 12895–12904. [Google Scholar] [CrossRef]
- Han, Q.; Li, Z.; Liang, X.; Ding, Y.; Zheng, S.T. Synthesis of a 6-nm-long transition-metal-rare-earth-containing polyoxometalate. Inorg. Chem. 2019, 58, 12534–12537. [Google Scholar] [CrossRef]
- Hussain, F.; Conrad, F.; Patzke, G.R. A gadolinium-bridged polytungstoarsenate(III) nanocluster: [Gd8As12W124O432(H2O)22]60−. Angew. Chem. Int. Ed. 2009, 48, 9088–9091. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.X.; Yang, T.; Cai, Z.W.; Zheng, S.T. Four-shell polyoxometalates featuring high-nuclearity Ln26 clusters: Structural transformations of nanoclusters into frameworks triggered by transition-metal ions. Angew. Chem. Int. Ed. 2017, 56, 2664–2669. [Google Scholar] [CrossRef]
- Ibrahim, M.; Moreno-pineda, E.; Bergfeldt, T.; Anson, C.E.; Powell, A.K. Synthesis and characterization of a heterometallic extended architecture based on a manganese (II) -substituted. Materials 2018, 11, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Chen, Y.C.; Wu, S.G.; Jia, J.H.; Chen, W.B.; Liu, J.L.; Tong, M.L. pH-controlled assembly of organophosphonate-bridged dysprosium(III) single-molecule magnets based on polyoxometalates. Inorg. Chem. 2018, 57, 6773–6777. [Google Scholar] [CrossRef]
- Wang, Y.J.; Wu, S.Y.; Sun, Y.Q.; Li, X.X.; Zheng, S.T. Octahedron-shaped three-shell Ln14 -substituted polyoxotungstogermanates encapsulating a W4O15 cluster: Luminescence and frequency dependent magnetic properties. Chem. Commun. 2019, 55, 2857–2860. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.H.; Eshtiagh-Hosseini, H.; Khoshnavazi, R. Synthesis, structure, and characterization of a new sandwich-type arsenotungstocerate, [As2W18Ce3O 71(H2O)3]12−. J. Mol. Struct. 2004, 688, 33–39. [Google Scholar] [CrossRef]
- Wang, W.; Izarova, N.V.; Van Leusen, J.; Kögerler, P. CeIII-functionalized polyoxotungstates: Discrete vs extended architectures. Cryst. Growth Des. 2019, 19, 4860–4870. [Google Scholar] [CrossRef]
- Hu, F.; Ma, P.; Han, M.; Wan, R.; Wang, J.; Niu, J. A new organic-inorganic hybrid tetrameric polyoxometalate assembled by monovacant Dawson [α2-P2W17O61]10- and trivalent cerium cations. Inorg. Chem. Commun. 2016, 67, 103–106. [Google Scholar] [CrossRef]
- Niu, J.; Wang, K.; Chen, H.; Zhao, J.; Pengtao, M.; Wang, J.; Li, M.; Bai, Y.; Dang, D. Assembly chemistry between lanthanide cations and monovacant keggin polyoxotungstates: Two types of lanthanide substituted phosphotungstates [(α-PW11O39H)Ln(H2O)39]6− and [(α-PW11O39)Ln(H2O)(η2-μ1,1)-CH3OO2]10−. Cryst. Growth Des. 2009, 9, 4362–4372. [Google Scholar] [CrossRef]
- Gong, P.; Li, Y.; Zhai, C.; Luo, J.; Tian, X.; Chen, L.; Zhao, J. Syntheses, structural characterization and photophysical properties of two series of rare-earth-isonicotinic-acid containing Waugh-type manganomolybdates. CrystEngComm 2017, 19, 834–852. [Google Scholar] [CrossRef]
- Khan, I.; Kaushik, R.; Tiwari, R.K.; Mbomekallé, I.; de Oliveira, P.; Matono, T.; Ooyama, H.E.; Sadakane, M.; Hussain, F. Self-assembled tetrameric lanthanide-containing germanotungstates [(Ln2GeW10O38)4(W3O8)(OH)4(H2O)2]26−: Syntheses, Crystal structure, photoluminescence and electrochemical properties. ChemistrySelect 2019, 4, 12668–12675. [Google Scholar] [CrossRef]
- Saini, M.K.; Gupta, R.; Parbhakar, S.; Kumar Mishra, A.; Mathur, R.; Hussain, F. Dimeric complexes of rare-earth substituted Keggin-type silicotungstates: Syntheses, crystal structure and solid state properties. RSC Adv. 2014, 4, 25357–25364. [Google Scholar] [CrossRef]
- Hussain, F.; Sandriesser, S.; Speldrich, M.; Patzke, G.R. A new series of lanthanoid containing Keggin-type germanotungstates with acetate chelators: [{Ln(CH3COO)GeW11O39(H2O)}2]12- {Ln = EuIII, GdIII, TbIII, DyIII, HoIII, ErIII, TmIII, and YbIII}. J. Solid State Chem. 2011, 184, 214–219. [Google Scholar] [CrossRef]
- Sato, R.; Suzuki, K.; Sugawa, M.; Mizuno, N. Heterodinuclear lanthanoid-containing polyoxometalates: Stepwise synthesis and single-molecule magnet behavior. Chem. Eur. J. 2013, 19, 12982–12990. [Google Scholar] [CrossRef]
- Fang, X.; Anderson, T.M.; Benelli, C.; Hill, C.L. Polyoxometalate-supported Y- and YbIII-hydroxo/oxo clusters from carbonate-assisted hydrolysis. Chem. Eur. J. 2005, 11, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Khoshnavazi, R.; Sadeghi, R.; Bahrami, L. High stable sandwich-type polyoxometallates based on A-β-SiW9O3410−. Synthesis, chemical properties and characterization of [(A-β-SiW9O34)2(MOH2)3CO3]13− (M = Y3+ and Yb3+). Polyhedron 2008, 27, 1855–1859. [Google Scholar] [CrossRef]
- Fang, X.; Anderson, T.M.; Neiwert, W.A.; Hill, C.L. Yttrium polyoxometalates. Synthesis and characterization of a carbonate-encapsulated sandwich-type complex. Inorg. Chem. 2003, 42, 8600–8602. [Google Scholar] [CrossRef]
- Knoth, W.H.; Domaille, P.J.; Harlow, R.L. Heteropolyanions of the types M3(W9PO34)212− and MM’M”(W9PO34)212−: Novel coordination of nitrate and nitrite. Inorg. Chem. 1986, 25, 1577–1584. [Google Scholar] [CrossRef]
- Ma, P.; Wan, R.; Si, Y.; Hu, F.; Wang, Y.; Niu, J.; Wang, J. Double-malate bridging tri-lanthanoid cluster encapsulated arsenotungstates: Syntheses, structures, luminescence and magnetic properties. Dalton. Trans. 2015, 44, 11514–11523. [Google Scholar] [CrossRef] [PubMed]
- Vonci, M.; Boskovic, C. Polyoxometalate-supported lanthanoid single-molecule magnets. Aust. J. Chem. 2014, 67, 1542–1552. [Google Scholar] [CrossRef]
- Ibrahim, M.; Krämer, S.; Schork, N.; Guthausen, G. Polyoxometalate-based high-spin cluster systems: A NMR relaxivity study up to 1.4 GHz/33 T. Dalton. Trans. 2019, 48, 15597–15604. [Google Scholar] [CrossRef]
- Yamase, T. Photo- and electrochromism of polyoxometalates and related materials. Chem. Rev. 2002, 98, 307–326. [Google Scholar] [CrossRef]
- Arab Fashapoyeh, M.; Mirzaei, M.; Eshtiagh-Hosseini, H.; Rajagopal, A.; Lechner, M.; Liu, R.; Streb, C. Photochemical and electrochemical hydrogen evolution reactivity of lanthanide-functionalized polyoxotungstates. Chem. Commun. 2018, 54, 10427–10430. [Google Scholar] [CrossRef]
- Suzuki, K.; Tang, F.; Kikukawa, Y.; Yamaguchi, K.; Mizuno, N. Visible-light-induced photoredox catalysis with a tetracerium-containing silicotungstate. Angew. Chem. Int. Ed. 2014, 53, 5356–5360. [Google Scholar] [CrossRef]
- Ibrahim, M.; Mbomekallé, I.M.; De Oliveira, P.; Baksi, A.; Carter, A.B.; Peng, Y.; Bergfeldt, T.; Malik, S.; Anson, C.E. Syntheses, crystal structure, electrocatalytic, and magnetic properties of the monolanthanide-containing germanotungstates [Ln(H2O) nGeW11O39]5− (Ln = Dy, Er, n = 4,3). ACS Omega 2019, 4, 21873–21882. [Google Scholar] [CrossRef] [Green Version]
- AlDamen, M.A.; Clemente-Juan, J.M.; Coronado, E.; Martii-Gastaldo, C.; Gaita-Arino, A. Mononuclear lanthanide single-molecule magnets based on polyoxometalates. J. Am. Chem. Soc. 2008, 130, 8874–8875. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Juan, J.M.; Coronado, E.; Gaita-Ariño, A. Magnetic polyoxometalates: From molecular magnetism to molecular spintronics and quantum computing. Chem. Soc. Rev. 2012, 41, 7464–7478. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Lin, Z.; Yin, A.; Yang, S.; Chi, Y.; Wang, Y.; Dong, J.; Liu, B.; Zhen, N.; Hill, C.L.; et al. Self-assembly of Ln(III)-containing tungstotellurates(VI): Correlation of structure and photoluminescence. Inorg. Chem. 2018, 57, 8831–8840. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Fournier, M.; Franck, R.; Thouvenot, R. Vibrational investigations of polyoxometalates. 2. Evidence for anion-anion interactions in molybdenum(VI) and tungsten(VI) compounds related to the Keggin structure. Inorg. Chem. 1983, 22, 207–216. [Google Scholar] [CrossRef]
- Ibrahim, M.; Mal, S.S.; Bassil, B.S.; Banerjee, A.; Kortz, U. Yttrium (III)-containing tungstoantimonate(III) stabilized by tetrahedral WO42− capping unit, [(Y(α-SbW9O31(OH)2)(CH3COO)(H2O)}3(WO4)]17−. Inorg. Chem. 2011, 50, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Mialane, P.; Dolbecq, A.; Sécheresse, F. Functionalization of polyoxometalates by carboxylato and azido ligands: Macromolecular complexes and extended compounds. Chem. Commun. 2006, 3477–3485. [Google Scholar] [CrossRef] [PubMed]
- Parent, L.; Aparicio, P.A.; De Oliveira, P.; Teillout, A.L.; Poblet, J.M.; López, X.; Mbomekallé, I.M. Effect of electron (De)localization and pairing in the electrochemistry of polyoxometalates: Study of Wells-Dawson molybdotungstophosphate derivatives. Inorg. Chem. 2014, 53, 5941–5949. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, M.; Steckhan, E. Electrochemical properties of polyoxometalates as electrocatalysts. Chem. Rev. 1998, 98, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Keita, B.; Nadjo, L. Electrochemistry of Isopoly and Heteropoly Oxometalates. In Encyclopedia of Electrochemistry; Bard, A.J., Ed.; Wiley VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keita, B.; Nadjo, L.; Contant, R. Electrocatalysis by polyoxometalate / polymer systems: Reduction of nitrite and nitric oxide. J. Electroanal. Chem. 1995, 381, 243–250. [Google Scholar] [CrossRef]
- Keita, B.; Nadjo, L. Polyoxometalate-based homogeneous catalysis of electrode reactions: Recent achievements. J. Mol. Catal. A Chem. 2007, 262, 190–215. [Google Scholar] [CrossRef]
- Bassil, B.S.; Bossoh, A.M.; Ibrahim, M.; Avo Bile, B.; Mougharbel, A.S.; Lin, Z.; de Oliveira, P.; Mbomekalle, I.-M.; Kortz, U. Synthesis, structure and electrochemistry of the dinickel(II)-containing 30-tungsto-4-phosphate [Ni2Na2(H2O)2(P2W15O56)2]18−. Curr. Inorg. Chem. 2017, 7, 21–27. [Google Scholar]
- Keita, B.; Mbomekalle, I.-M.; Nadjo, L.; Contant, R. [H4AsW18O62]7−, A novel Dawson heteropolyanion and two of its sandwich-type derivatives [Zn4(H2O)2(H4AsW15O56)2]18−, [Cu4(H2O)2(H4AsW15O56)2]18−: Cyclic voltammetry and electrocatalytic properties towards nitrite and nitrate. Electrochem. Commun. 2001, 3, 267–273. [Google Scholar] [CrossRef]
- Feng, M.; Lyu, B.H.; Wang, M.H.; Wu, W.W.; Chen, Y.C.; Huang, G.Z.; Lin, W.Q.; Wu, S.G.; Liu, J.L.; Tong, M.L. Chiral erbium(III) complexes: Single-molecule magnet behavior, chirality, and nuclearity control. Inorg. Chem. 2019, 58, 10694–10703. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Chen, Y.-C.; Tong, M.-L. Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 2018, 47, 2431–2453. [Google Scholar] [CrossRef] [PubMed]
- Finke, R.G.; Droege, M.W.; Domaille, P.J. Rational syntheses, characterization, two-dimensional 183W NMR, and properties of tungstometallophosphates P2W18M4(H2O)2O6810− and P4W30M4(H2O)2O11216− (M = cobalt, copper, zinc). Inorg. Chem. 1987, 26, 3886–3896. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound (NH2Me2)13Na3[{Er(H2O)(CH3COO)(α2P2W17O61)}2] 40 H2O is available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | C30H174Er2N13Na3O158P4W34 |
|---|---|
| Formula weight/g mol−1 | 10,024.08 |
| Crystal System | triclinic |
| Space Group | P |
| a/Å | 13.2960(2) |
| b/Å | 13.4153(2) |
| c/Å | 27.0804(4) |
| α/° | 91.764(1) |
| β/° | 92.500(1) |
| γ/° | 111.721(1) |
| U/Å3 | 4432.70(12) |
| Z | 1 |
| T/K | 180(2) |
| F(000) | 4454 |
| Dc/Mg m−3 | 3.755 |
| µ(Ga-Kα)/mm−1 | 32.074 |
| Data Measured | 77,253 |
| Unique Data | 18,640 |
| Rint | 0.0553 |
| Data with I ≥ 2σ(I) | 15,225 |
| wR2 (all data) | 0.1944 |
| S (all data) | 1.040 |
| R1 [II ≥ 2σ(I)] | 0.0641 |
| Parameters/Restraints | 1169/70 |
| Biggest diff. peak/hole/eÅ−3 | 3.66/−4.90 |
| CSD number | 2,021,556 |
| Ec1 | Ec2 | Ec3 | Ea1 | Ea2 | Ea3 | ||
|---|---|---|---|---|---|---|---|
| pH 6 | P2W17 | −0.76 | −0.98 | −0.47 | −0.65 | −0.86 | |
| 1 | −0.63 | −0.76 | −0.97 | −0.55 | −0.70 | −0.90 | |
| pH 5 | 1 | −0.60 | −0.70 | −0.94 | −0.45 | −0.63 | −0.83 |
| pH 4 | 1 | −0.50 | −0.65 | −0.90 | −0.30 | −0.43 | −0.78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.; Baksi, A.; Peng, Y.; Al-Zeidaneen, F.K.; Mbomekallé, I.M.; de Oliveira, P.; Anson, C.E. Synthesis, Characterization, Electrochemistry, Photoluminescence and Magnetic Properties of a Dinuclear Erbium(III)-Containing Monolacunary Dawson-Type Tungstophosphate: [{Er(H2O)(CH3COO)(P2W17O61)}2]16−. Molecules 2020, 25, 4229. https://doi.org/10.3390/molecules25184229
Ibrahim M, Baksi A, Peng Y, Al-Zeidaneen FK, Mbomekallé IM, de Oliveira P, Anson CE. Synthesis, Characterization, Electrochemistry, Photoluminescence and Magnetic Properties of a Dinuclear Erbium(III)-Containing Monolacunary Dawson-Type Tungstophosphate: [{Er(H2O)(CH3COO)(P2W17O61)}2]16−. Molecules. 2020; 25(18):4229. https://doi.org/10.3390/molecules25184229
Chicago/Turabian StyleIbrahim, Masooma, Ananya Baksi, Yan Peng, Firas Khalil Al-Zeidaneen, Israël M. Mbomekallé, Pedro de Oliveira, and Christopher E. Anson. 2020. "Synthesis, Characterization, Electrochemistry, Photoluminescence and Magnetic Properties of a Dinuclear Erbium(III)-Containing Monolacunary Dawson-Type Tungstophosphate: [{Er(H2O)(CH3COO)(P2W17O61)}2]16−" Molecules 25, no. 18: 4229. https://doi.org/10.3390/molecules25184229