
Strategies and Efforts towards the Total Synthesis of Palhinine Alkaloids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
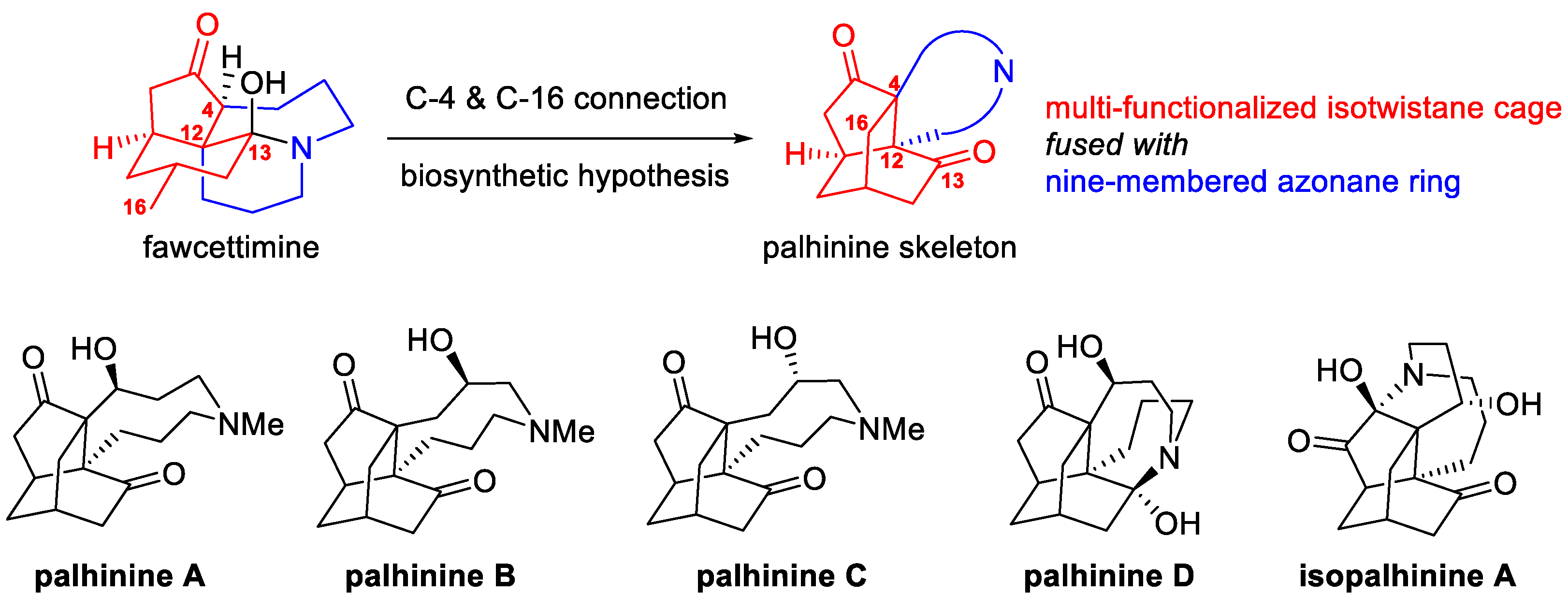
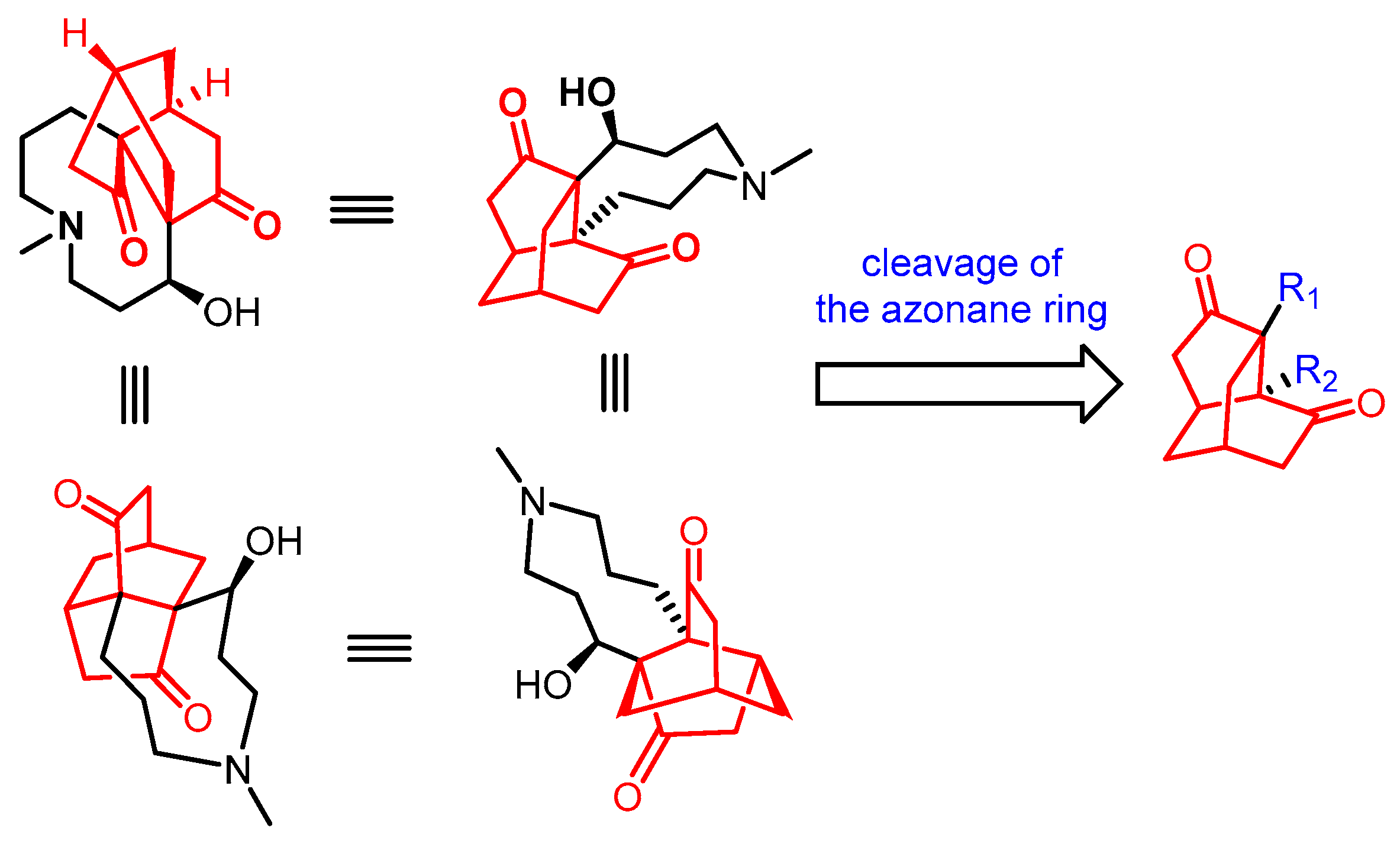
1. Introduction
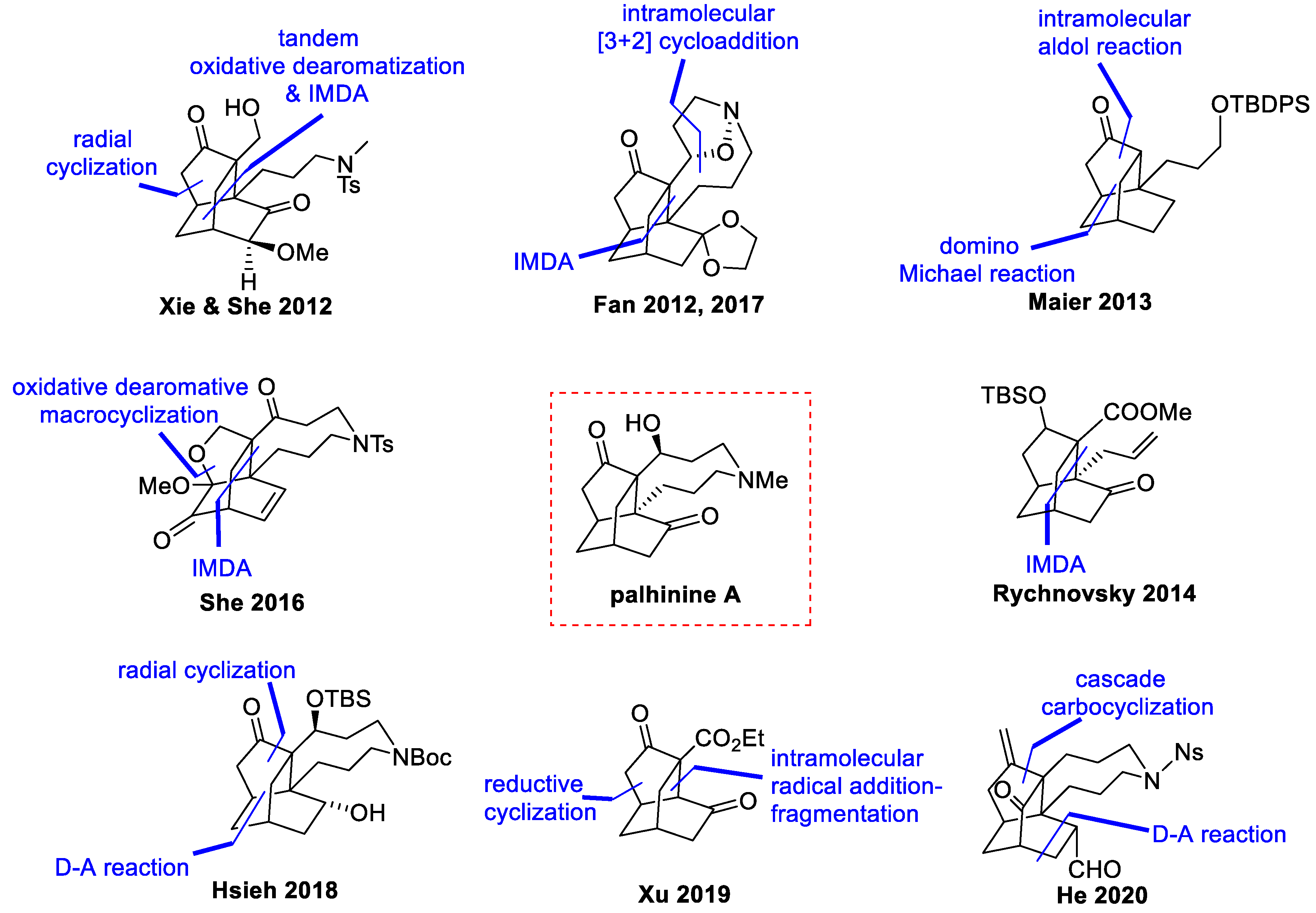
2. Synthetic Efforts Reported during 2010–2015
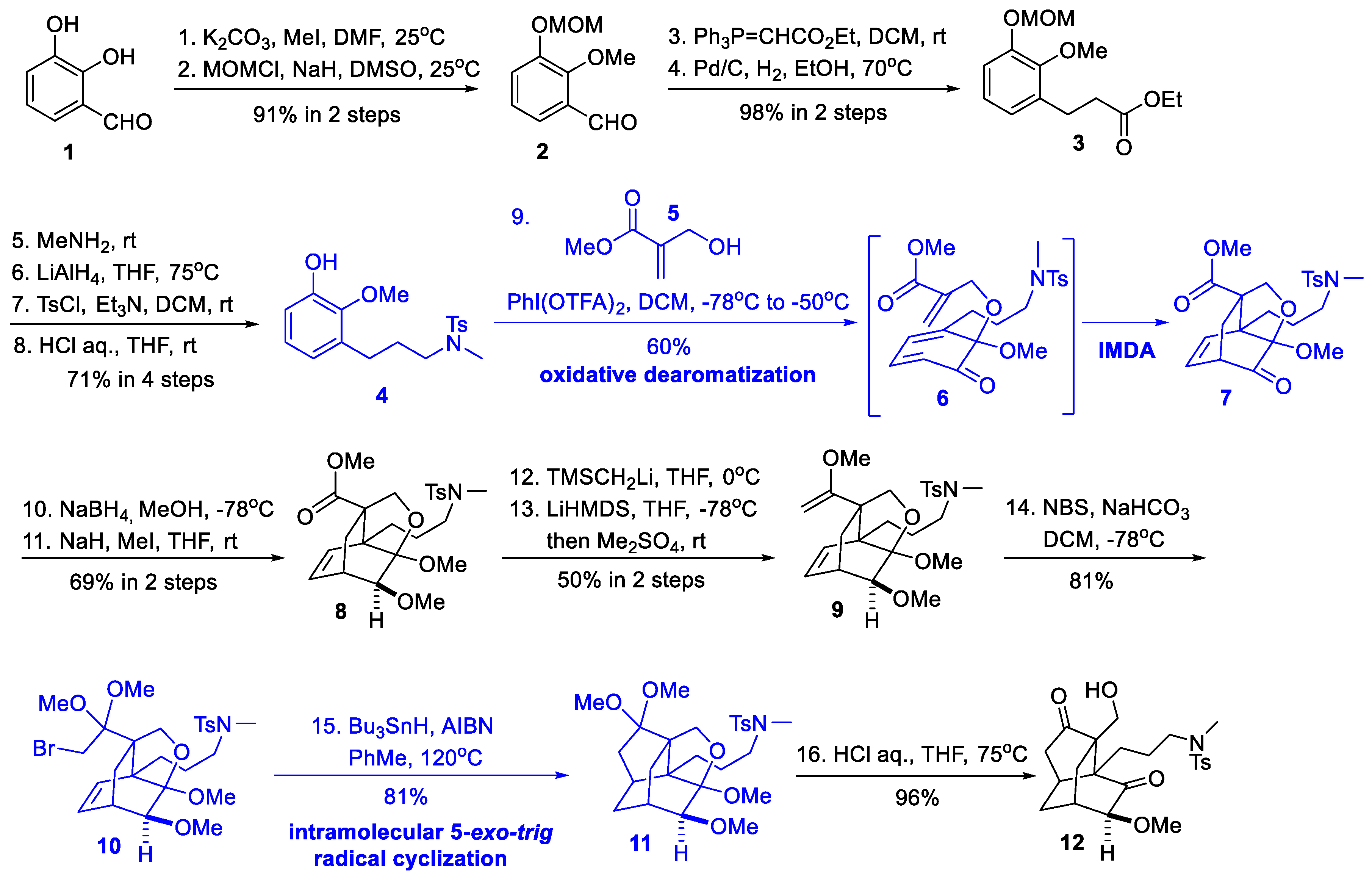
2.1. Synthetic Studies from Xie and She’s Group (2012)
2.2. Synthetic Studies from Fan’s Group (2012)
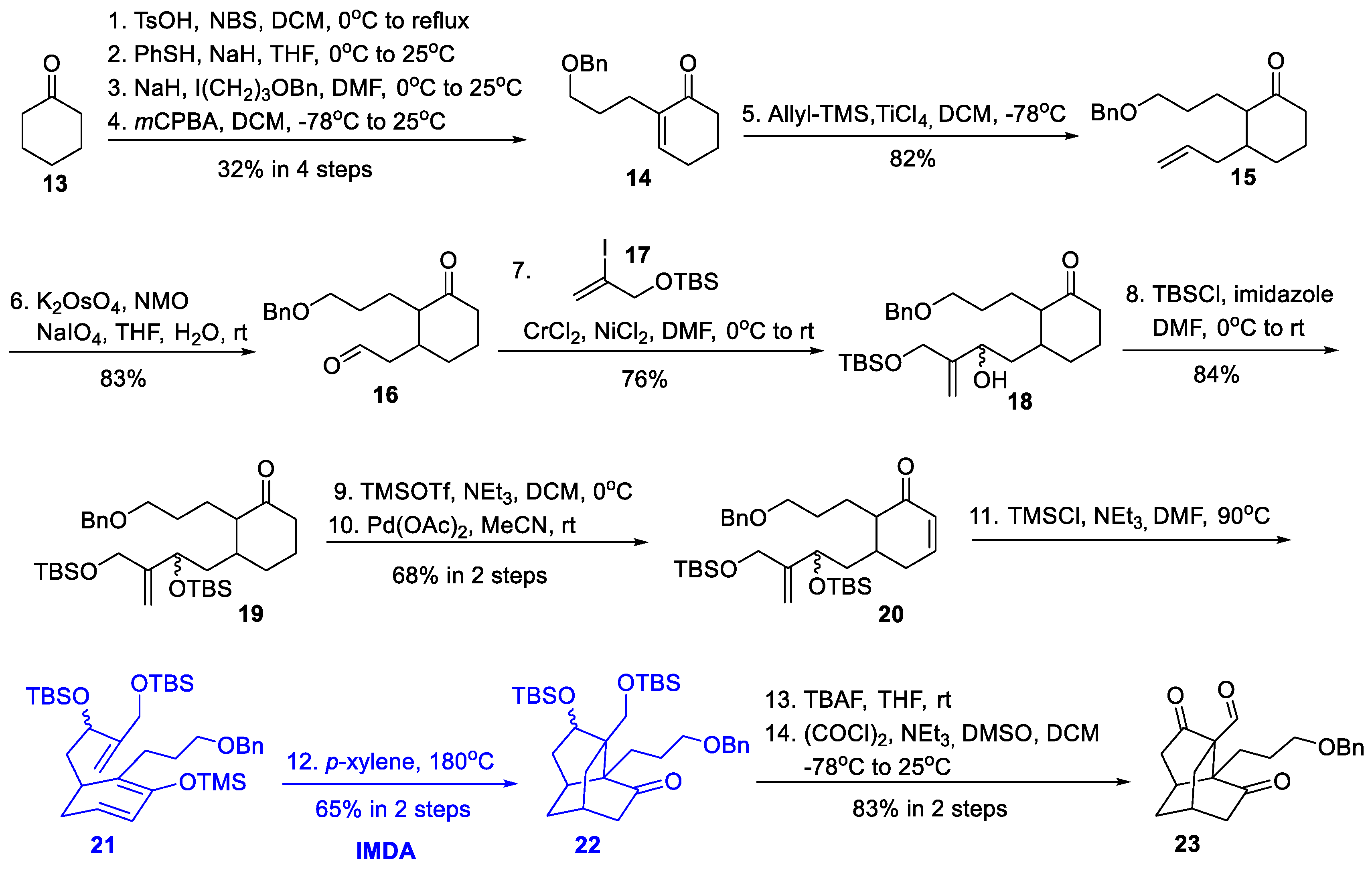
2.3. Synthetic Studies from Maier’s Group (2013)
2.4. Synthetic Studies from Rychnovsky’s Group (2014)
3. Synthetic Efforts Reported during 2016–2020
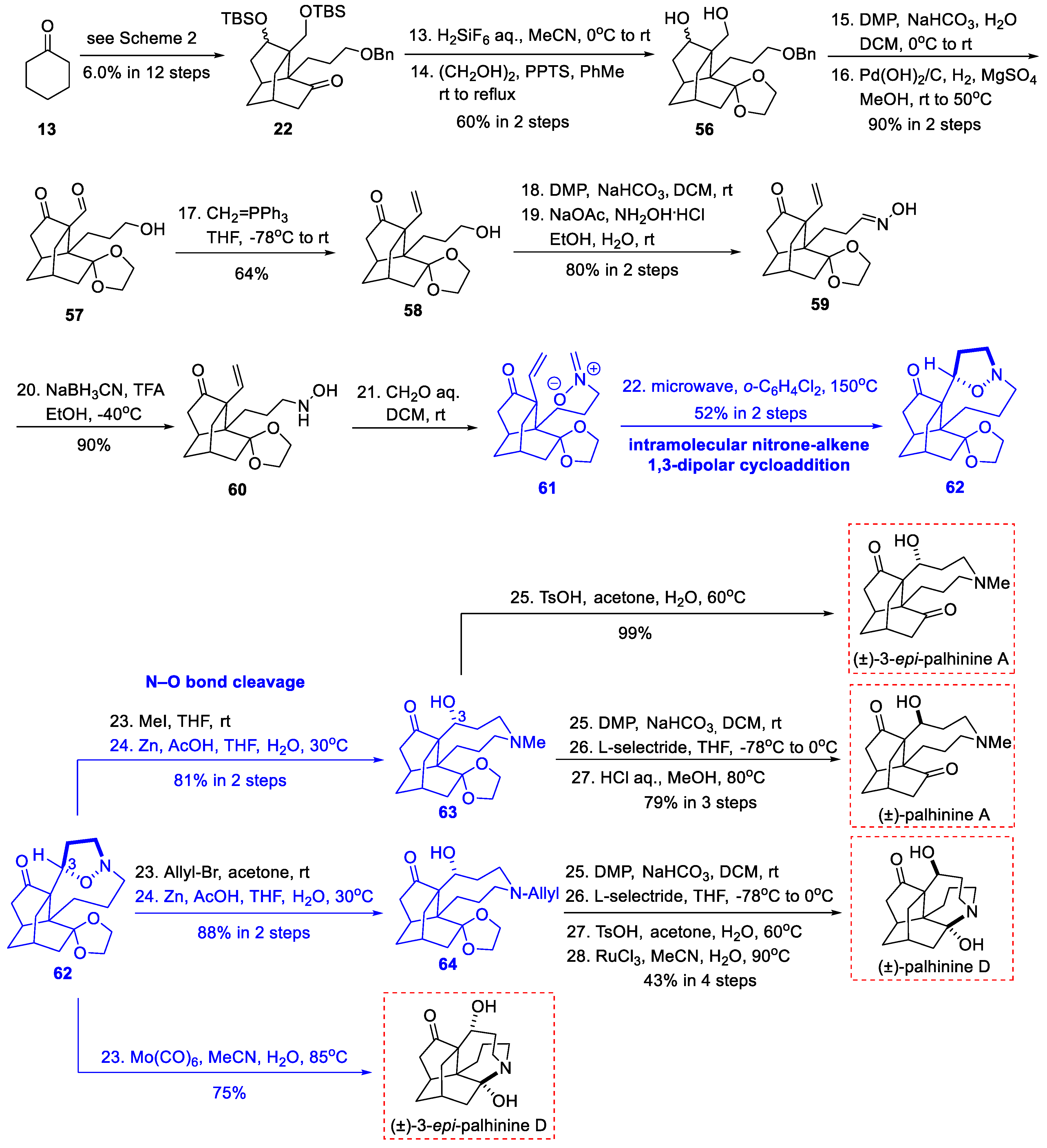
3.1. Synthetic Studies and Total Synthesis from Fan’s Group (2016, 2017)
3.2. Synthetic Studies from She’s Group (2016)
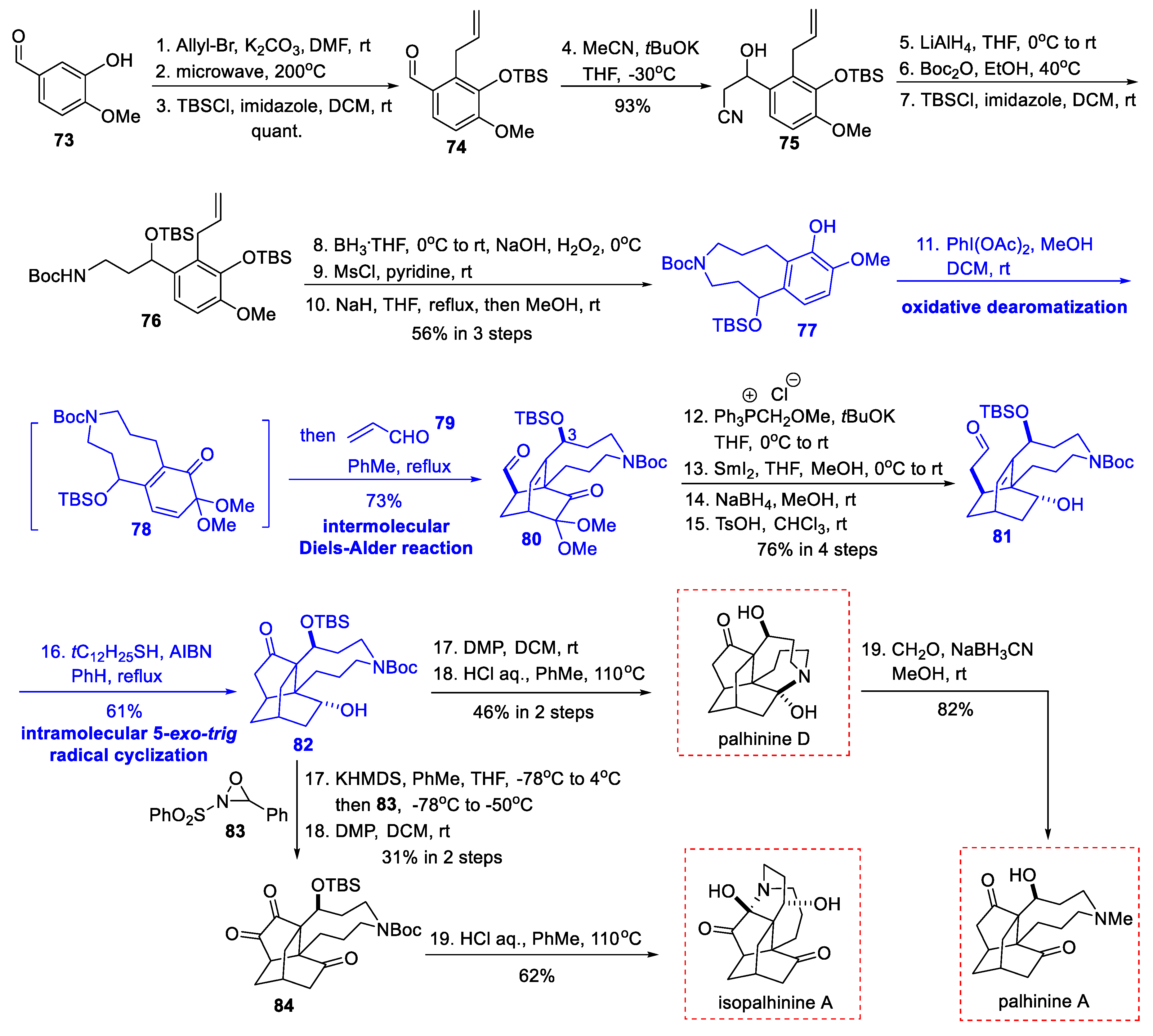
3.3. Total Synthesis from Hsieh’s Group (2018)
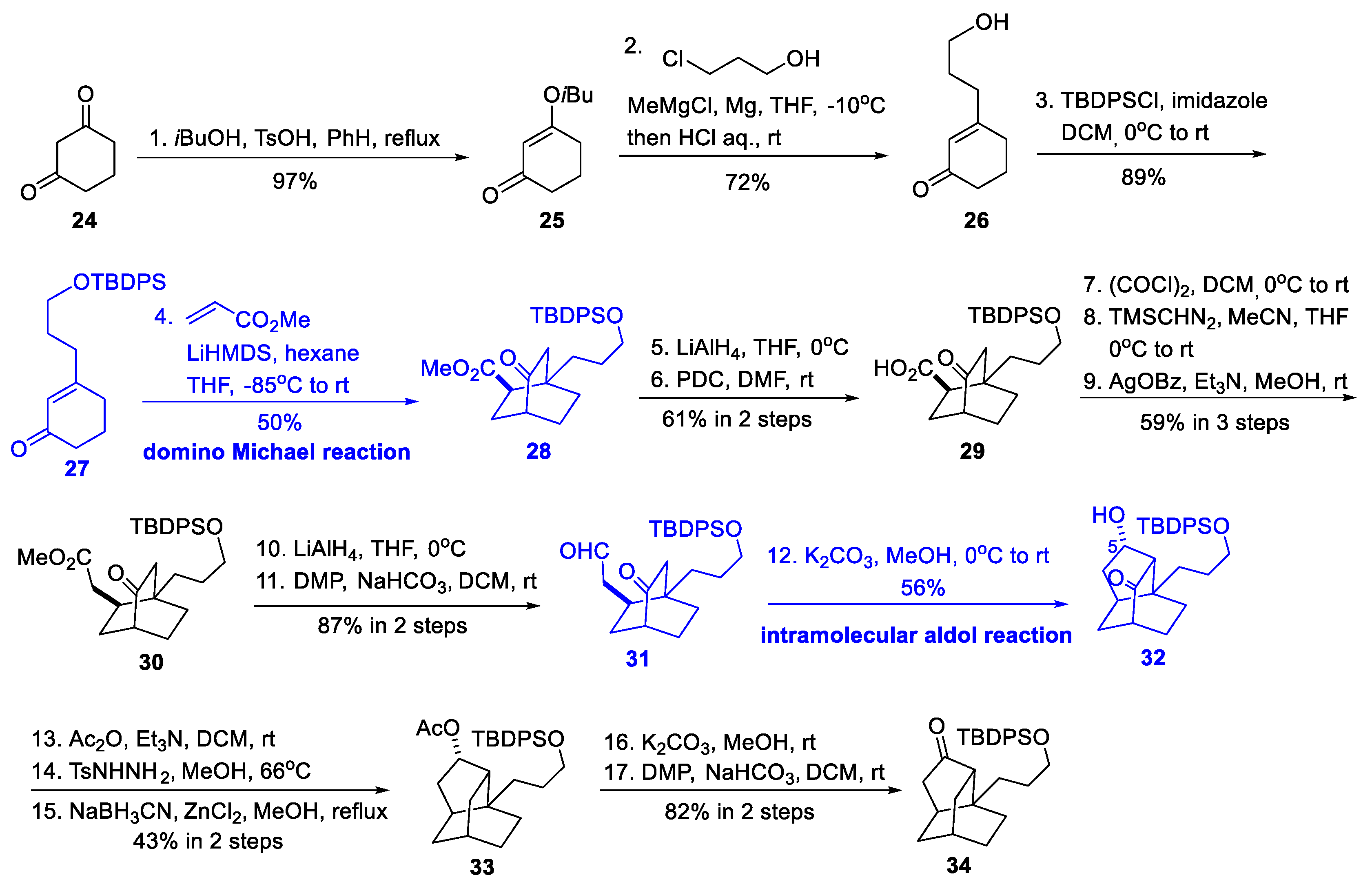
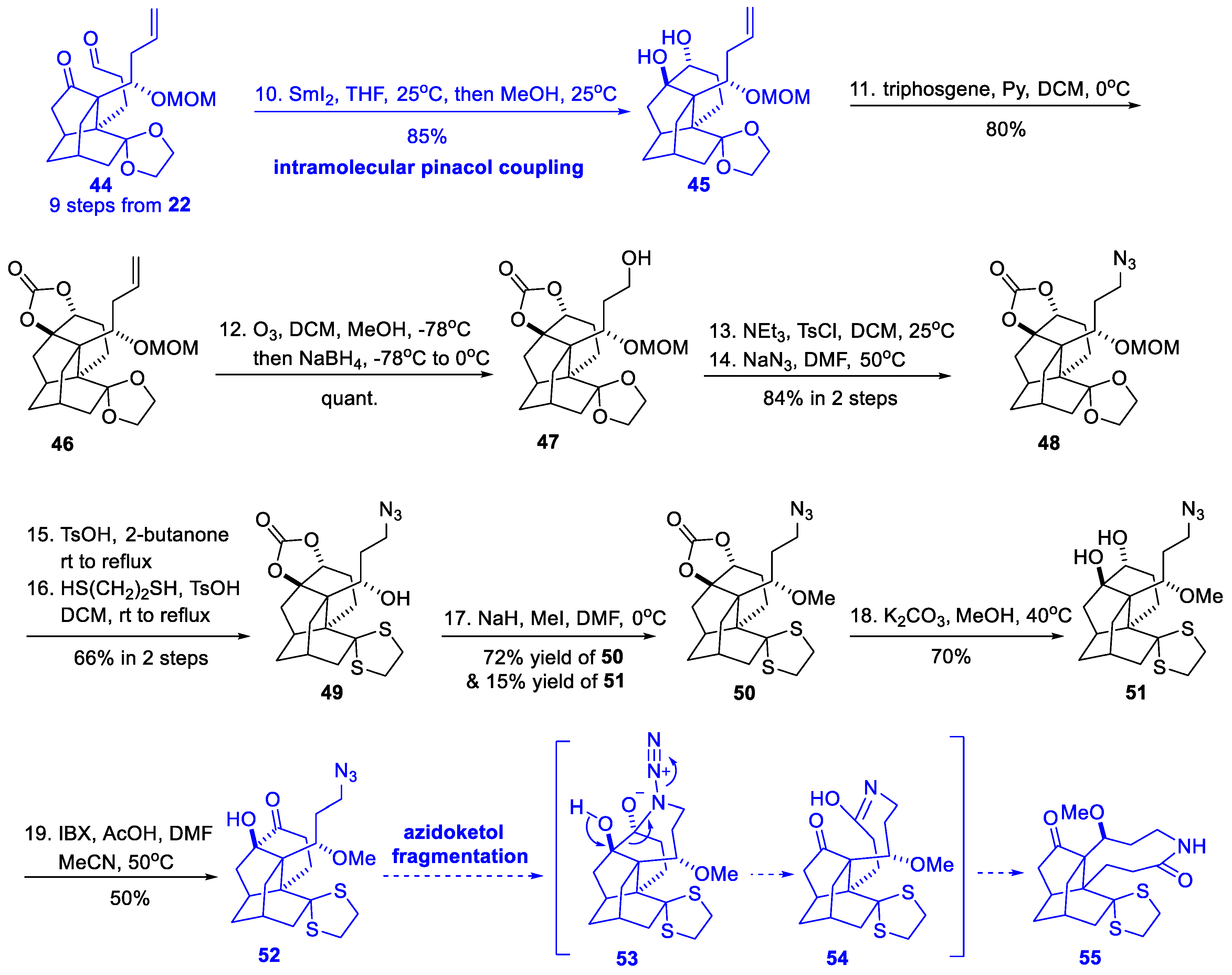
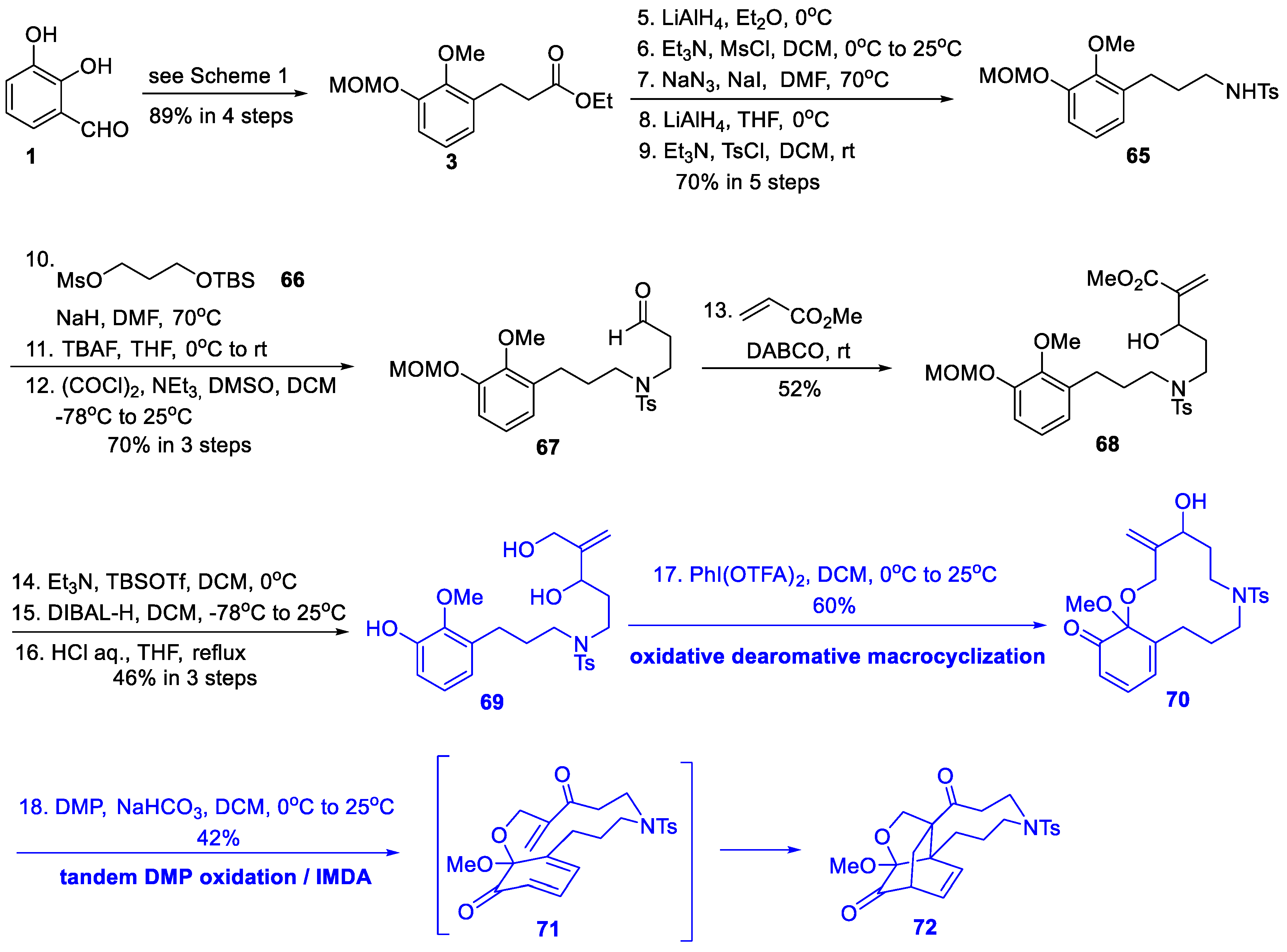
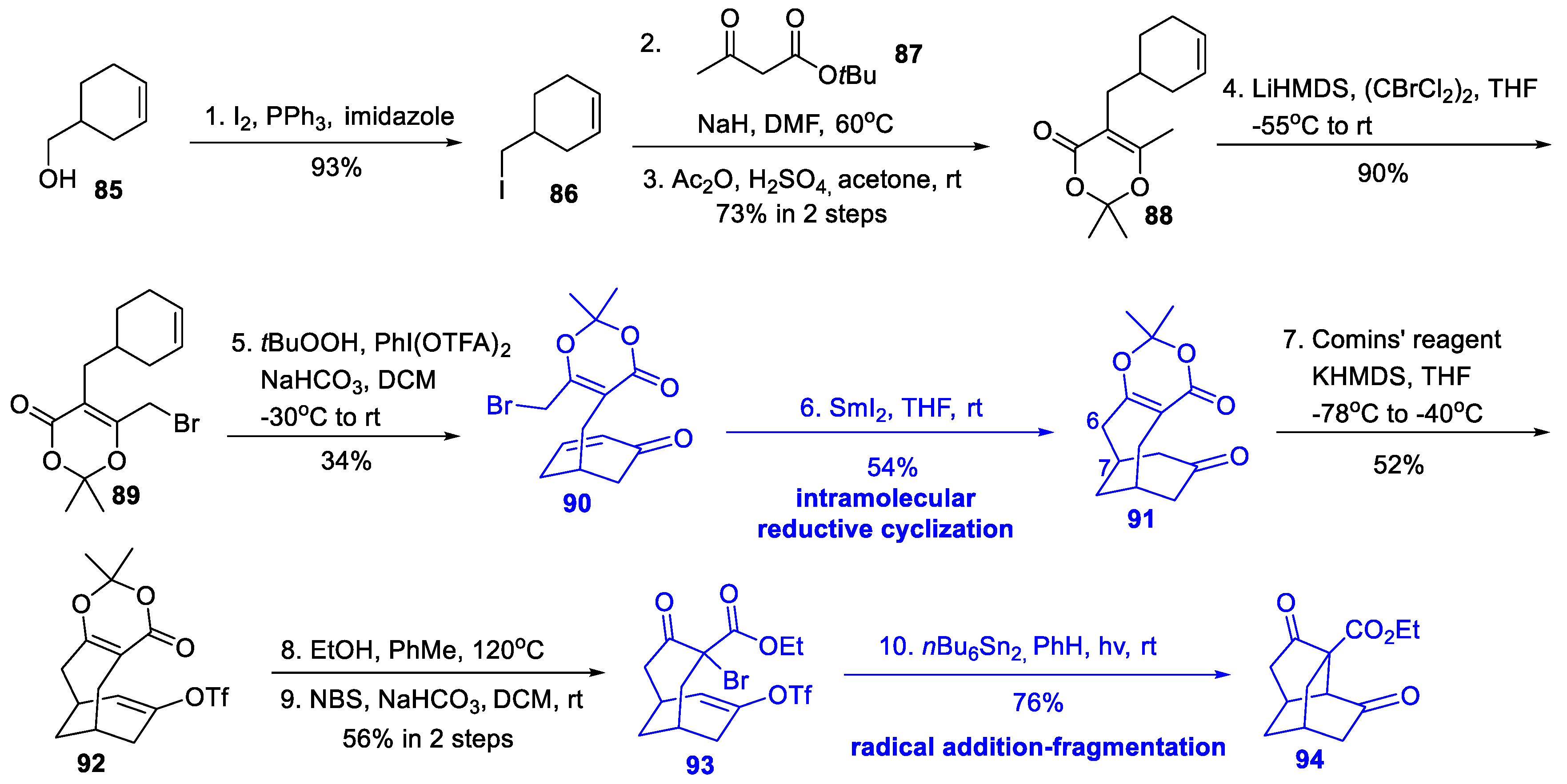
3.4. Synthetic Studies from Xu’s Group (2019)
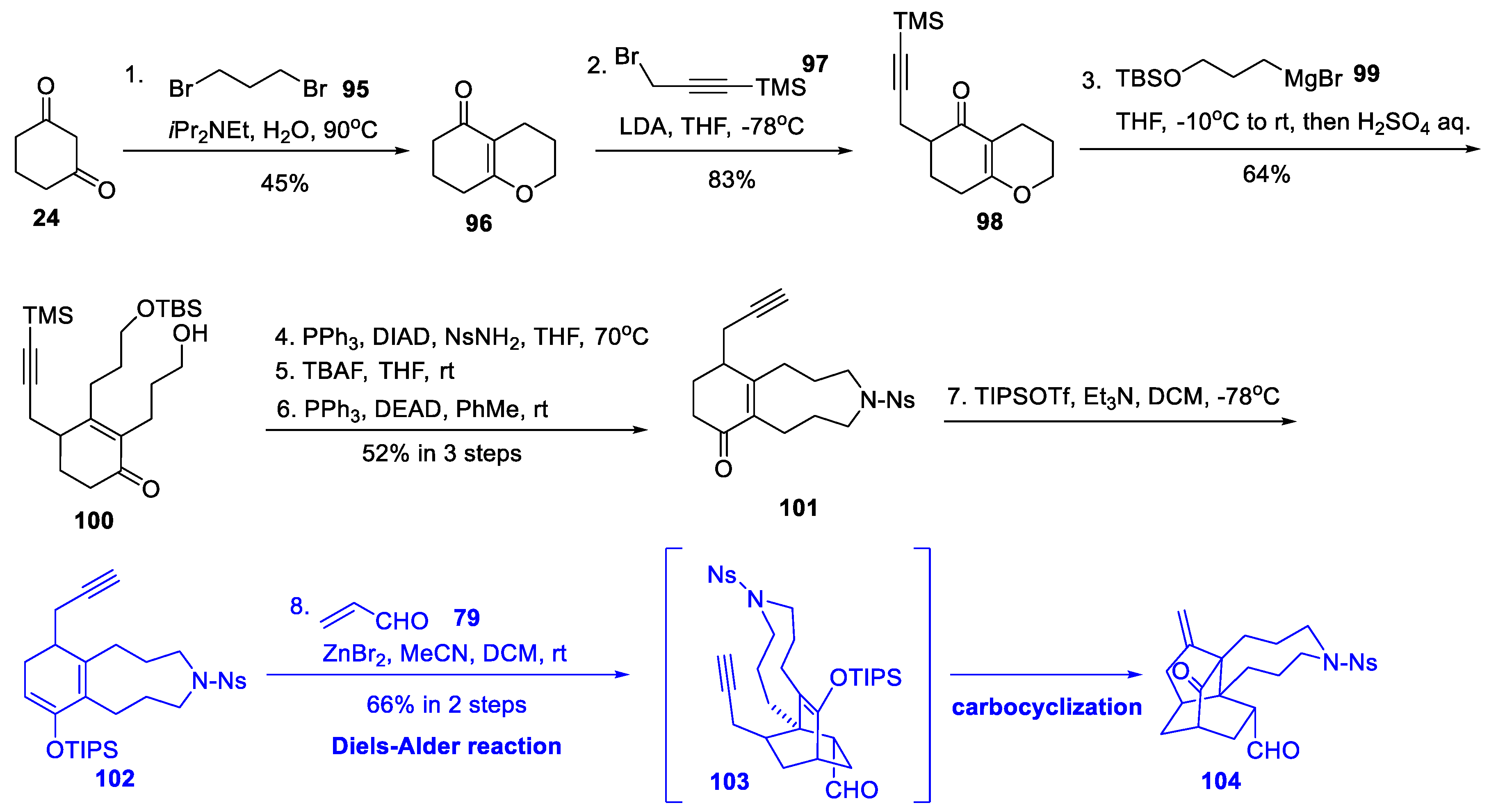
3.5. Synthetic Studies from He’s Group (2020)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| Ac | acetyl |
| AIBN | 2,2′-azobisisobutyronitrile |
| Bn | benzyl |
| Boc | tert-butoxycarbonyl |
| Bz | benzoyl |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| DCM | dichloromethane |
| DEAD | diethyl azodicarboxylate |
| DIAD | diisopropyl azodicarboxylate |
| DIBAL-H | diisobutylaluminium hydride |
| DMF | N,N-dimethylformamide |
| DMP | Dess–Martin periodinane |
| DMSO | dimethyl sulfoxide |
| IBX | o-iodoxybenzoic acid |
| IMDA | intramolecular Diels–Alder |
| KHMDS | potassium bis(trimethylsilyl)amide |
| LDA | lithium diisopropylamide |
| LiHMDS | lithium bis(trimethylsilyl)amide |
| mCPBA | meta-chloroperbenzoic acid |
| MOM | methoxymethyl |
| MPO | 4-methyoxypyridine 1-oxide |
| Ms | methanesulfonyl |
| NaHMDS | sodium bis(trimethylsilyl)amide |
| NBS | N-bromosuccinimide |
| NMO | N-methylmorpholine oxide |
| Ns | 2-nitrobenzenesulfonyl |
| PDC | pyridinium dichromate |
| Ph | phenyl |
| PPTS | pyridinium p-toluenesulfonate |
| Py | pyridine |
| quant. | quantitative yield |
| rt | room temperature |
| TASF | tris(diethylamino)sulfonium difluorotrimethylsilicate |
| TBAF | tetra-n-butylammonium fluoride |
| TBDPS | tert-butyldiphenylsilyl |
| TBS | tert-butyldimethylsilyl |
| TES | triethylsilyl |
| TFA | trifluoroacetyl |
| Tf | trifluoromethanesulfonyl |
| THF | tetrahydrofuran |
| TIPS | triisopropylsilyl |
| TMS | trimethylsilyl |
| Ts | p-toluenesulfonyl |
References
- Siengalewicz, P.; Mulzer, J.; Rinner, U. Lycopodium Alkaloids–Synthetic Highlights and Recent Developments. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Waltham, MA, USA, 2013; Volume 72, pp. 1–151. [Google Scholar] [CrossRef]
- Yang, H.L.; Ma, Y.S.; Wang, X.L.; Zhu, D. Huperzine A: A Mini-Review of Biological Characteristics, Natural Sources, Synthetic Origins, and Future Prospects. Russ. J. Org. Chem. 2020, 56, 148–157. [Google Scholar] [CrossRef]
- Zhao, F.-W.; Sun, Q.-Y.; Yang, F.-M.; Hu, G.-W.; Luo, J.-F.; Tang, G.-H.; Wang, Y.-H.; Long, C.-L. Palhinine A, a Novel Alkaloid from Palhinhaea cernua. Org. Lett. 2010, 12, 3922–3925. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.-B.; Gao, X.; Liu, F.; He, J.; Wu, X.-D.; Li, Y.; Zhao, Q.-S. Isopalhinine A, a Unique Pentacyclic Lycopodium Alkaloid from Palhinhaea cernua. Org. Lett. 2013, 15, 3570–3573. [Google Scholar] [CrossRef]
- Zhu, Y.; Dong, L.-B.; Zhang, Z.-J.; Fan, M.; Zhu, Q.-F.; Qi, Y.-Y.; Liu, Y.-C.; Peng, L.-Y.; Wu, X.-D.; Zhao, Q.-S. Three new Lycopodium alkaloids from Lycopodium japonicum. J. Asian Nat. Prod. Res. 2019, 21, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Li, L.; Yu, S.-S.; Ma, S.-G.; Qu, J.; Liu, Y.-B.; Li, Y.; Wang, Y.; Tang, W. Five New Fawcettimine-Related Alkaloids from Lycopodium japonicum Thunb. Fitoterapia 2013, 91, 74–81, Correction in 2016, 114, 194. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Y.; Zhan, R.; Chen, Y.-G. A New Fawcettimine-Related Alkaloid from Lycopodium japonicum. Chem. Nat. Compd. 2018, 54, 729–731. [Google Scholar] [CrossRef]
- Tang, Y.; Xiong, J.; Zou, Y.; Zhang, H.-Y.; Hu, J.-F. Palhicerines A–F, Lycopodium alkaloids from the club moss Palhinhaea cernua. Phytochemistry 2016, 131, 130–139. [Google Scholar] [CrossRef]
- Murphy, R.A.; Sarpong, R. Heathcock-Inspired Strategies for the Synthesis of Fawcettimine-Type Lycopodium Alkaloids. Chem. Eur. J. 2014, 20, 42–56. [Google Scholar] [CrossRef]
- Zhao, C.; Zheng, H.; Jing, P.; Fang, B.; Xie, X.; She, X. Tandem Oxidative Dearomatization/Intramolecular Diels−Alder Reaction for Construction of the Tricyclic Core of Palhinine A. Org. Lett. 2012, 14, 2293–2295. [Google Scholar] [CrossRef]
- Boger, D.L.; Hong, J.; Hikota, M.; Ishida, M.J. Total synthesis of phomazarin. Am. Chem. Soc. 1999, 121, 2471–2477. [Google Scholar] [CrossRef]
- Zhang, G.-B.; Wang, F.-X.; Du, J.-Y.; Qu, H.; Ma, X.-Y.; Wei, M.X.; Wang, C.-T.; Li, Q.; Fan, C.-A. Toward the Total Synthesis of Palhinine A: Expedient Assembly of Multifunctionalized Isotwistane Ring System with Contiguous Quaternary Stereocenters. Org. Lett. 2012, 14, 3696–3699. [Google Scholar] [CrossRef] [PubMed]
- Gaugele, D.; Maier, M.E. Approach to the Core Structure of the Polycyclic Alkaloid Palhinine A. Synlett 2013, 24, 955–958. [Google Scholar] [CrossRef][Green Version]
- Ryoma, H.; Takashi, F.; Hajime, K.; Hiroyuki, K.; Yoshiaki, H.; Isao, K. Enantioselective Total Synthesis of (+)-Taxusin. J. Am. Chem. Soc. 1999, 121, 3072–3082. [Google Scholar] [CrossRef]
- Sizemore, N.; Rychnovsky, S.D. Studies toward the Synthesis of Palhinine Lycopodium Alkaloids: A Morita−Baylis−Hillman/Intramolecular Diels−Alder Approach. Org. Lett. 2014, 16, 688–691. [Google Scholar] [CrossRef]
- Wang, F.-X.; Du, J.-Y.; Wang, H.-B.; Zhang, P.-L.; Zhang, G.-B.; Yu, K.-Y.; Zhang, X.-Z.; An, X.-T.; Cao, Y.-X.; Fan, C.-A. Total Synthesis of Lycopodium Alkaloids Palhinine A and Palhinine D. J. Am. Chem. Soc. 2017, 139, 4282–4285. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-X.; Zhang, P.-L.; Wang, H.-B.; Zhang, G.-B.; Fan, C.-A. A strategic study towards constructing the nine-membered azonane ring system of palhinine A via an azidoketol fragmentation reaction. Sci. China Chem. 2016, 59, 1188–1196. [Google Scholar] [CrossRef]
- Wang, F.-X. Studies on Total Synthesis of Lycopodium Alkaloids Palhinine A and Palhinine D. Ph.D. Thesis, Lanzhou University, Lanzhou, China, June 2017. [Google Scholar]
- Duan, S.-S.; Dan, L.; Zhao, C.-G.; Zhao, G.-Y.; Yuan, Z.-Y.; Xie, X.-G.; Fang, J.-G.; She, X.-G. Efficient construction of the A/C/D tricyclic skeleton of Palhinine A. Org. Chem. Front. 2016, 3, 1137–1143. [Google Scholar] [CrossRef]
- Chen, C.-M.; Shiao, H.-Y.; Uang, B.-J.; Hsieh, H.-P. Biomimetic Syntheses of (±)-Isopalhinine A, (±)-Palhinine A, and (±)-Palhinine D. Angew. Chem. Int. Ed. 2018, 57, 15572–15576. [Google Scholar] [CrossRef]
- Lu, S.-C.; Liang, Y.-Y.; Li, L.; Wang, X.-L.; Zhang, S.-P.; Gong, Y.-L.; Xu, S. Synthesis of the Tricyclic Caged Core of Palhinine Alkaloids Based on a Non-Diels-Alder-Type Strategy. Org. Lett. 2019, 21, 5567–5569. [Google Scholar] [CrossRef]
- Han, C.-C.; Chen, Y.; Ching, Y.-C.; Lee, C.-S.; He, S.-Z. An Approach towards the Tetracyclic Skeleton of Palhinine Alkaloids. Org. Chem. Front. 2020, 7, 2243–2246. [Google Scholar] [CrossRef]
- Taber, D.F.; Gerstenhaber, D.A.; Berry, J.F. Enantioselective Conjugate Allylation of Cyclic Enones. J. Org. Chem. 2011, 76, 7614–7617, Correction in 2012, 77, 1215. [Google Scholar] [CrossRef] [PubMed]
- Mash, E.A.; Gregg, T.M. Synthesis of the Enantiomers of 4-Vinylcyclohexene. J. Org. Chem. 1995, 60, 6180–6182. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.-Y.; Lu, S.-C.; Gong, Y.-L.; Xu, S. Strategies and Efforts towards the Total Synthesis of Palhinine Alkaloids. Molecules 2020, 25, 4211. https://doi.org/10.3390/molecules25184211
Liang Y-Y, Lu S-C, Gong Y-L, Xu S. Strategies and Efforts towards the Total Synthesis of Palhinine Alkaloids. Molecules. 2020; 25(18):4211. https://doi.org/10.3390/molecules25184211
Chicago/Turabian StyleLiang, Yu-Yan, Shi-Chao Lu, Ya-Ling Gong, and Shu Xu. 2020. "Strategies and Efforts towards the Total Synthesis of Palhinine Alkaloids" Molecules 25, no. 18: 4211. https://doi.org/10.3390/molecules25184211
APA StyleLiang, Y.-Y., Lu, S.-C., Gong, Y.-L., & Xu, S. (2020). Strategies and Efforts towards the Total Synthesis of Palhinine Alkaloids. Molecules, 25(18), 4211. https://doi.org/10.3390/molecules25184211