
Biological Activities and ADMET-Related Properties of Novel Set of Cinnamanilides †

 ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Biological Screening
2.1.1. In Vitro Antimicrobial Evaluation
2.1.2. In Vitro Cell Viability and Anti-Inflammatory Potential
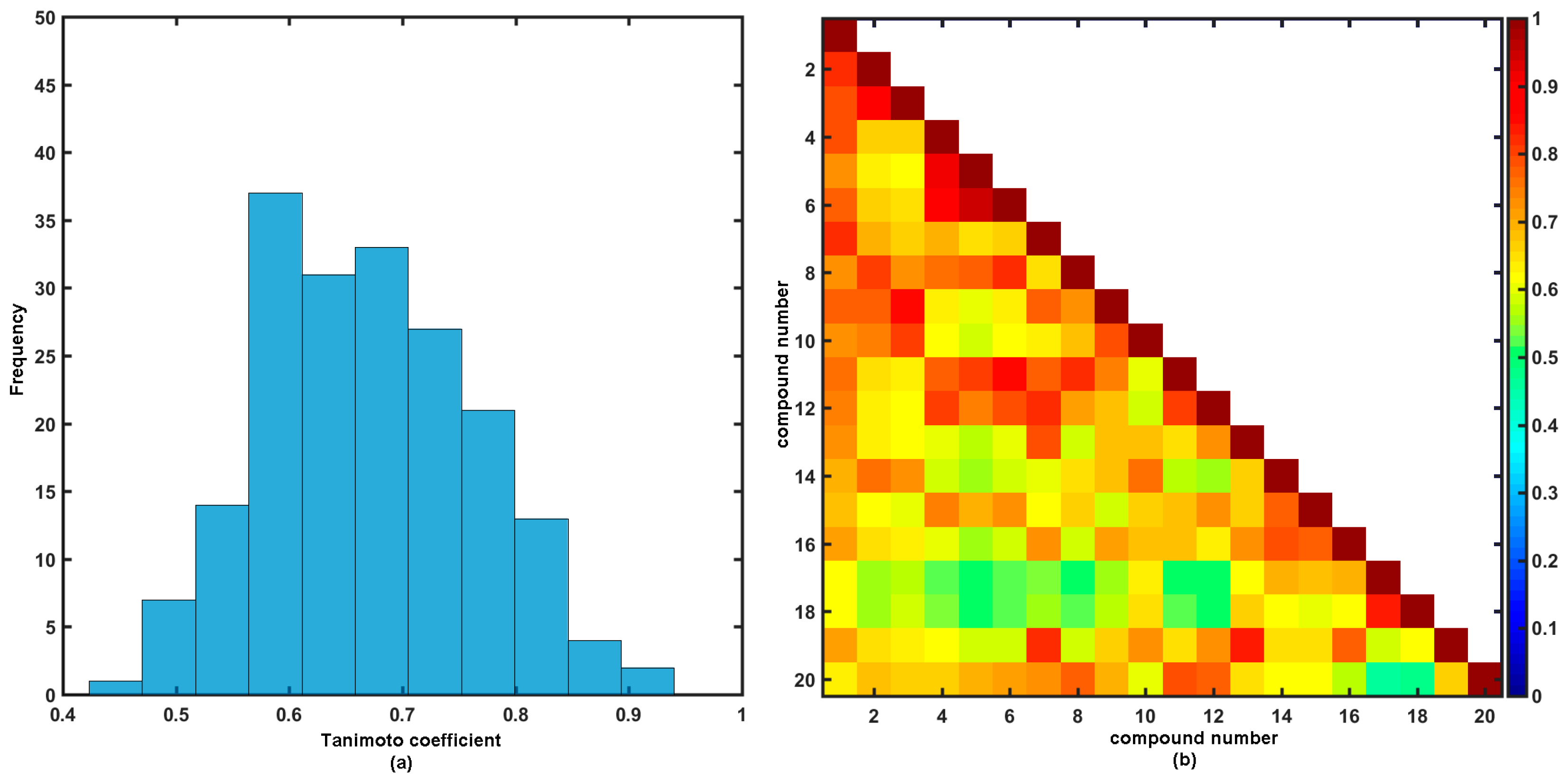
2.2. Similarity-Driven Property Evaluation
Comparative Molecular Shape Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis
3.2. Biological Testing
3.2.1. In Vitro Antibacterial Evaluation
3.2.2. In Vitro Antimycobacterial Evaluation
3.2.3. MTT Assay
3.2.4. In Vitro Cell Viability Assay
3.2.5. Determination of NF-κB Activity
3.3. Lipophilicity Determination by RP-HPLC (Capacity Factor k/Calculated log k)
3.4. Lipophilicity Determination by RP-TLC
3.5. Model Building and Experimental vs. Theoretical Lipophilicity Prediction
3.6. Similarity Assessment Using PCA and Tanimoto Coefficient
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Oehling, A.K. Bacterial infection as an important triggering factor in bronchial asthma. J. Investig. Allergol. Clin. Immunol. 1999, 9, 6–13. [Google Scholar]
- Sreenivasan, P.K.; Gaffar, A. Antibacterials as anti-inflammatory agents: Dual action agents for oral health. Antonie Van Leeuwenhoek 2008, 93, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.C.; Caricchio, R.; Gallucci, S. Triggers of autoimmunity: The role of bacterial infections in the extracellular exposure of lupus nuclear autoantigens. Front. Immunol. 2019, 10, 2608. [Google Scholar] [CrossRef] [PubMed]
- Van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Bhatia, P.; Bhatia, P.G.; Yaiw, K.C. Complementary approaches to existing target based drug discovery for identifying novel drug targets. Biomedicines 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Ul Islam, N.; Amin, R.; Shahid, M.; Amin, M.; Zaib, S.; Iqbal, J. A multi-target therapeutic potential of Prunus domestica gum stabilized nanoparticles exhibited prospective anticancer, antibacterial, urease-inhibition, anti-inflammatory and analgesic properties. BMC Complement. Altern. Med. 2017, 17, 276. [Google Scholar] [CrossRef]
- Brullo, C.; Massa, M.; Rapetti, F.; Alfei, S.; Bertolotto, M.B.; Montecucco, F.; Signorello, M.G.; Bruno, O. New hybrid pyrazole and imidazopyrazole antinflammatory agents able to reduce ROS production in different biological targets. Molecules 2020, 25, 899. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Bräse, S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar]
- Gaikwad, N.; Nanduri, S.; Madhavi, Y.V. Cinnamamide: An insight into the pharmacological advances and structure-activity relationships. Eur. J. Med. Chem. 2019, 181, 111561. [Google Scholar] [CrossRef] [PubMed]
- Das, A.B.; Goud, V.V.; Das, C. Phenolic compounds as functional ingredients in beverages. In Value-Added Ingredients and Enrichments of Beverages; Grumezescu, A.M., Holban, A.M., Eds.; Woodhead Publishing: Duxford, UK; Elsevier: Duxford, UK, 2019; pp. 285–323. [Google Scholar]
- Sharma, P. Cinnamic acid derivatives: A new chapter of various pharmacological activities. J. Chem. Pharm. Res. 2011, 3, 403–423. [Google Scholar]
- Peperidou, A.; Kapoukranidou, D.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Multitarget molecular hybrids of cinnamic acids. Molecules 2014, 19, 20197–20226. [Google Scholar] [CrossRef]
- Peperidou, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Voulgari, E.; Avgoustakis, K. Multifunctional cinnamic acid derivatives. Molecules 2017, 22, 1247. [Google Scholar] [CrossRef]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Lima, T.C.; Ferreira, A.R.; Silva, D.F.; Lima, E.O.; de Sousa, D.P. Antifungal activity of cinnamic acid and benzoic acid esters against Candida albicans strains. Nat. Prod. Res. 2018, 32, 572–575. [Google Scholar] [CrossRef]
- Dolab, J.G.; Lima, B.; Spaczynska, E.; Kos, J.; Cano, N.H.; Feresin, G.; Tapia, A.; Garibotto, F.; Petenatti, E.; Olivella, M.; et al. Antimicrobial activity of Annona emarginata (Schltdl.) H. Rainer and most active isolated compound against clinically important bacteria. Molecules 2018, 23, 1187. [Google Scholar] [CrossRef]
- Pontiki, E.; Peperidou, A.; Fotopoulos, I.; Hadjipavlou-Litina, D. Cinnamate hybrids: A unique family of compounds with multiple biological activities. Curr. Pharm. Biotechnol. 2018, 19, 1019–1048. [Google Scholar] [CrossRef]
- Silva, A.T.; Bento, C.M.; Pena, A.C.; Figueiredo, L.M.; Prudencio, C.; Aguiar, L.; Silva, T.; Ferraz, R.; Gomes, M.S.; Teixeira, C.; et al. Cinnamic acid conjugates in the rescuing and repurposing of classical antimalarial drugs. Molecules 2019, 25, 66. [Google Scholar] [CrossRef]
- Martinez, M.D.; Riva, D.A.; Garcia, C.; Duran, F.J.; Burton, G. Synthesis and antibacterial activity of difluoromethyl cinnamoyl amides. Molecules 2020, 25, 789. [Google Scholar] [CrossRef] [PubMed]
- Fungicide Resistance Action Committee. FRAC Code List© 2020: Fungal Control Agents Sorted by Cross Resistance Pattern and Mode of Action; Fungicide Resistance Action Committee, Croplife International: Brussels, Belgium, 2020. [Google Scholar]
- Pospisilova, S.; Kos, J.; Michnova, H.; Kapustikova, I.; Strharsky, T.; Oravec, M.; Moricz, A.M.; Bakonyi, J.; Kauerova, T.; Kollar, P.; et al. Synthesis and spectrum of biological activities of novel N-arylcinnamamides. Int. J. Mol. Sci. 2018, 19, 2318. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, S.; Kos, J.; Michnova, H.; Strharsky, T.; Cizek, A.; Jampilek, J. N-Arylcinnamamides as Antistaphylococcal Agents. In Proceedings of the 4th International Electronic Conference on Medicinal Chemistry, ECMC-4, 1–30 November 2018; p. 5576. Available online: https://sciforum.net/manuscripts/5576/slides.pdf (accessed on 4 August 2020).
- Hosek, J.; Kos, J.; Strharsky, T.; Cerna, L.; Starha, P.; Vanco, J.; Travnicek, Z.; Devinsky, F.; Jampilek, J. Investigation of anti-inflammatory potential of n-arylcinnamamide derivatives. Molecules 2019, 24, 4531. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modeling: Towads prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y.; Nakamura, H. Definition of drug-likeness for compound affinity. J. Chem. Inf. Model. 2011, 51, 1012–1016. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Efremov, R.G.; Chugunov, A.O.; Pyrkov, T.V.; Priestle, J.P.; Arseniev, A.S.; Jacoby, E. Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem. 2007, 14, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of logP methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 3, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.; Poda, G.I. Application of ALOGPS 2.1 to predict logD distribution coefficient for Pfizer proprietary compounds. J. Med. Chem. 2004, 47, 5601–5604. [Google Scholar] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Kah, M.; Brown, C.D. LogD: Lipophilicity for ionisable compounds. Chemosphere 2008, 72, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Bhal, S.K.; Kassam, K.; Peirson, I.G.; Pearl, G.M. The rule of five revisited: Applying logD in place of logP in drug-likeness filters. Mol. Pharm. 2007, 40, 556–560. [Google Scholar] [CrossRef]
- Xing, L.; Glen, C. Novel methods for the predicition of logP, pKa and logD. J. Chem. Inf. Comput. Sci. 2002, 42, 796–805. [Google Scholar] [CrossRef]
- Rupp, M.; Körner, R.; Tetko, I.V. Predicting the pKa of small molecules. Comb. Chem. High Throughput Screen. 2011, 14, 307–327. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Ferriz, J.M.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- International Organization for Standardization. ISO 10993-5:2009 Biological Evaluation of Medical Devices Part 5: Tests for In Vitro Cytotoxicity; International Organization for Standardization: Geneva, Switzerland, 2009; last revision 2017. [Google Scholar]
- Grela, E.; Kozłowska, J.; Grabowiecka, A. Current methodology of MTT assay in bacteria—A review. Acta Histochem. 2018, 120, 303–311. [Google Scholar] [CrossRef]
- Bender, A.; Scheiber, J.; Jenkins, J.L.; Sukuru, S.C. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef]
- Fialkowski, M.; Bishop, K.J.M.; Chubukov, V.A.; Campbell, C.J.; Grzybowski, B.A. Architecture and evolution of organic chemistry. Angew. Chem. Int. Ed. 2005, 44, 7263–7269. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated similarity assessment of the property profile for new, silicon-based AChE/BChE inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef]
- Martel, S.; Gillerat, F.; Carosati, E.; Maiarelli, D.; Tetko, I.V.; Mannhold, R.; Carrupt, P.A. Large, chemically diverse dataset of logP measurements for benchmarking studies. Eur. J. Pharm. Sci. 2013, 48, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V. Computing chemistry on the web. Drug Discov. Today 2005, 10, 1497–1500. [Google Scholar] [CrossRef]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: Concepts, challenges and recent advances. Future Med. Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. Methods Mol. Biol. 2011, 672, 39–100. [Google Scholar] [PubMed]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Du, Q.; Arteca, G.A. Interrelation between electrostatic and lipophilicity potentials on molecular surfaces. J. Mol. Graph. 1995, 13, 98–108. [Google Scholar] [CrossRef]
- Zupan, J.; Gasteiger, J. Neural Networks and Drug Design for Chemists, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Bak, A.; Wyszomirski, M.; Magdziarz, T.; Smolinski, A.; Polanski, J. Structure-based modeling of dye-fiber affinity with SOM-4D-QSAR paradigm: Application to set of anthraquinone derivatives. Comb. Chem. High Throughput Screen. 2014, 17, 485–502. [Google Scholar] [CrossRef]
- Michnova, H.; Pospisilova, S.; Gonec, T.; Kapustikova, I.; Kollar, P.; Kozik, V.; Musiol, R.; Jendrzejewska, I.; Vanco, J.; Travnicek, Z.; et al. Bioactivity of methoxylated and methylated 1-hydroxynaphthalene-2-carboxanilides: Comparative molecular surface analysis. Molecules 2019, 24, 2991. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel benzene-based carbamates for AChE/BChE inhibition: Synthesis and ligand/structure-oriented SAR study. Int. J. Mol. Sci. 2019, 20, 1524. [Google Scholar] [CrossRef]
- Xie, X.Q.; Chen, J.Z. Data mining a small molecule drug screening representative subset from NIH PubChem. J. Chem. Inf. Model. 2008, 48, 465–475. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Comp. | R | MIC (μM) | Tox IC50 (µM) ± SEM | |||
| SA | MRSA 63718 | MRSA SA 630 | Mtb | |||
| 1 * | H | >1146 | >1146 | >1146 | 286 | >20 |
| 2 | 2,4,6-F | >924 | >924 | >924 | >462 | >20 |
| 3 | 3,4,5-F | 462 | >924 | >924 | >462 | >20 |
| 4 | 2,4-Cl | >880 | >880 | >880 | >440 | >20 |
| 5 | 2,4,5-Cl | >790 | >790 | >790 | >395 | >20 |
| 6 | 3,4,5-Cl | >790 | >790 | >790 | >395 | >20 |
| 7 | 2,4-Br | >677 | >677 | >677 | >339 | >20 |
| 8 | 2-F-5-Cl | 931 | >931 | >931 | >465 | >20 |
| 9 | 3-F-4-Br | >805 | >805 | 805 | >403 | >20 |
| 10 | 3-F-4-CF3 | 25.9 | 25.9 | 12.9 | >414 | >20 |
| 11 | 3-Cl-4-Br | >766 | 192 | 192 | >383 | 6.5 ± 1.0 |
| 12 | 2-Br-4-Cl | >766 | >766 | >766 | >383 | >20 |
| 13 | 2-Br-5-CF3 | >696 | >696 | >696 | >348 | >20 |
| 14 | 2-CF3-4-F | >828 | >828 | >828 | >414 | >20 |
| 15 | 2-CF3-4-Cl | >788 | >788 | >788 | >394 | >20 |
| 16 | 2-CF3-4-Br | >696 | >696 | >696 | >348 | >20 |
| 17 | 2-CF3-4-NO2 | >762 | >762 | >762 | >381 | >20 |
| 18 | 3-CF3-4-NO2 | >762 | >762 | >762 | >381 | >20 |
| 19 | 2,6-Br-4-CF3 | >574 | >574 | >574 | >287 | >20 |
| 20 | 2,6-Br-3-Cl-4-F | >595 | >595 | >595 | >298 | >20 |
| AMP | – | 5.72 | 45.8 | 45.8 | – | – |
| INH | – | – | – | – | 36.6 | – |
| Comp. | R | RP-HPLC | RP-TLC | ||||
|---|---|---|---|---|---|---|---|
| Logk a | logD b | logD c | RM d | RM e | RM f | ||
| 1 | H | 0.0270 | 0.1251 | 0.1090 | −0.5087 | 0.2848 | 0.4038 |
| 2 | 2,4,6-F | −0.0831 | 0.0260 | 0.0107 | −0.6536 | 0.1218 | 0.1951 |
| 3 | 3,4,5-F | 0.4893 | 0.5495 | 0.5274 | −0.4206 | 0.6074 | 0.5495 |
| 4 | 2,4-Cl | 0.5278 | 0.5885 | 0.5685 | −0.3933 | 0.6546 | 0.5495 |
| 5 | 2,4,5-Cl | 0.8373 | 0.8879 | 0.8647 | −0.2497 | 0.9695 | 0.9832 |
| 6 | 3,4,5-Cl | 0.9671 | 1.0104 | 0.9897 | −0.1870 | 1.0852 | 1.1306 |
| 7 | 2,4-Br | 0.6152 | 0.6704 | 0.6518 | −0.3098 | 0.7331 | 0.7295 |
| 8 | 2-F-5-Cl | 0.3692 | 0.4334 | 0.4154 | −0.4711 | 0.5411 | 0.5495 |
| 9 | 3-F-4-Br | 0.5025 | 0.5597 | 0.5408 | −0.4081 | 0.6797 | 0.6895 |
| 10 | 3-F-4-CF3 | 0.5789 | 0.6327 | 0.6095 | −0.5032 | 0.7923 | 0.5960 |
| 11 | 3-Cl-4-Br | 0.6611 | 0.7126 | 0.6912 | −0.3376 | 0.8388 | 0.8181 |
| 12 | 2-Br-4-Cl | 0.5476 | 0.6064 | 0.5860 | −0.4072 | 0.6553 | 0.6484 |
| 13 | 2-Br-5-CF3 | 0.5561 | 0.6157 | 0.5936 | −0.4325 | 0.7194 | 0.5619 |
| 14 | 2-CF3-4-F | 0.1607 | 0.2426 | 0.2220 | −0.6797 | 0.3011 | 0.1330 |
| 15 | 2-CF3-4-Cl | 0.4113 | 0.4741 | 0.454 | −0.6102 | 0.5305 | 0.3219 |
| 16 | 2-CF3-4-Br | 0.4683 | 0.5316 | 0.5105 | −0.5624 | 0.5730 | 0.4273 |
| 17 | 2-CF3-4-NO2 | 0.3794 | 0.4431 | 0.3989 | −0.5633 | 0.4998 | 0.4311 |
| 18 | 3-CF3-4-NO2 | 0.5004 | 0.5571 | 0.5098 | −0.4804 | 0.6927 | 0.6160 |
| 19 | 2,6-Br-4-CF3 | 0.4321 | 0.4941 | 0.4731 | −0.5206 | 0.5301 | 0.3733 |
| 20 | 2,6-Br-3-Cl-4-F | 0.3139 | 0.3922 | 0.3699 | −0.5541 | 0.4320 | 0.4138 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kos, J.; Bak, A.; Kozik, V.; Jankech, T.; Strharsky, T.; Swietlicka, A.; Michnova, H.; Hosek, J.; Smolinski, A.; Oravec, M.; et al. Biological Activities and ADMET-Related Properties of Novel Set of Cinnamanilides. Molecules 2020, 25, 4121. https://doi.org/10.3390/molecules25184121
Kos J, Bak A, Kozik V, Jankech T, Strharsky T, Swietlicka A, Michnova H, Hosek J, Smolinski A, Oravec M, et al. Biological Activities and ADMET-Related Properties of Novel Set of Cinnamanilides. Molecules. 2020; 25(18):4121. https://doi.org/10.3390/molecules25184121
Chicago/Turabian StyleKos, Jiri, Andrzej Bak, Violetta Kozik, Timotej Jankech, Tomas Strharsky, Aleksandra Swietlicka, Hana Michnova, Jan Hosek, Adam Smolinski, Michal Oravec, and et al. 2020. "Biological Activities and ADMET-Related Properties of Novel Set of Cinnamanilides" Molecules 25, no. 18: 4121. https://doi.org/10.3390/molecules25184121
APA StyleKos, J., Bak, A., Kozik, V., Jankech, T., Strharsky, T., Swietlicka, A., Michnova, H., Hosek, J., Smolinski, A., Oravec, M., Devinsky, F., Hutta, M., & Jampilek, J. (2020). Biological Activities and ADMET-Related Properties of Novel Set of Cinnamanilides. Molecules, 25(18), 4121. https://doi.org/10.3390/molecules25184121