
Ethyl Pyruvate Promotes Proliferation of Regulatory T Cells by Increasing Glycolysis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
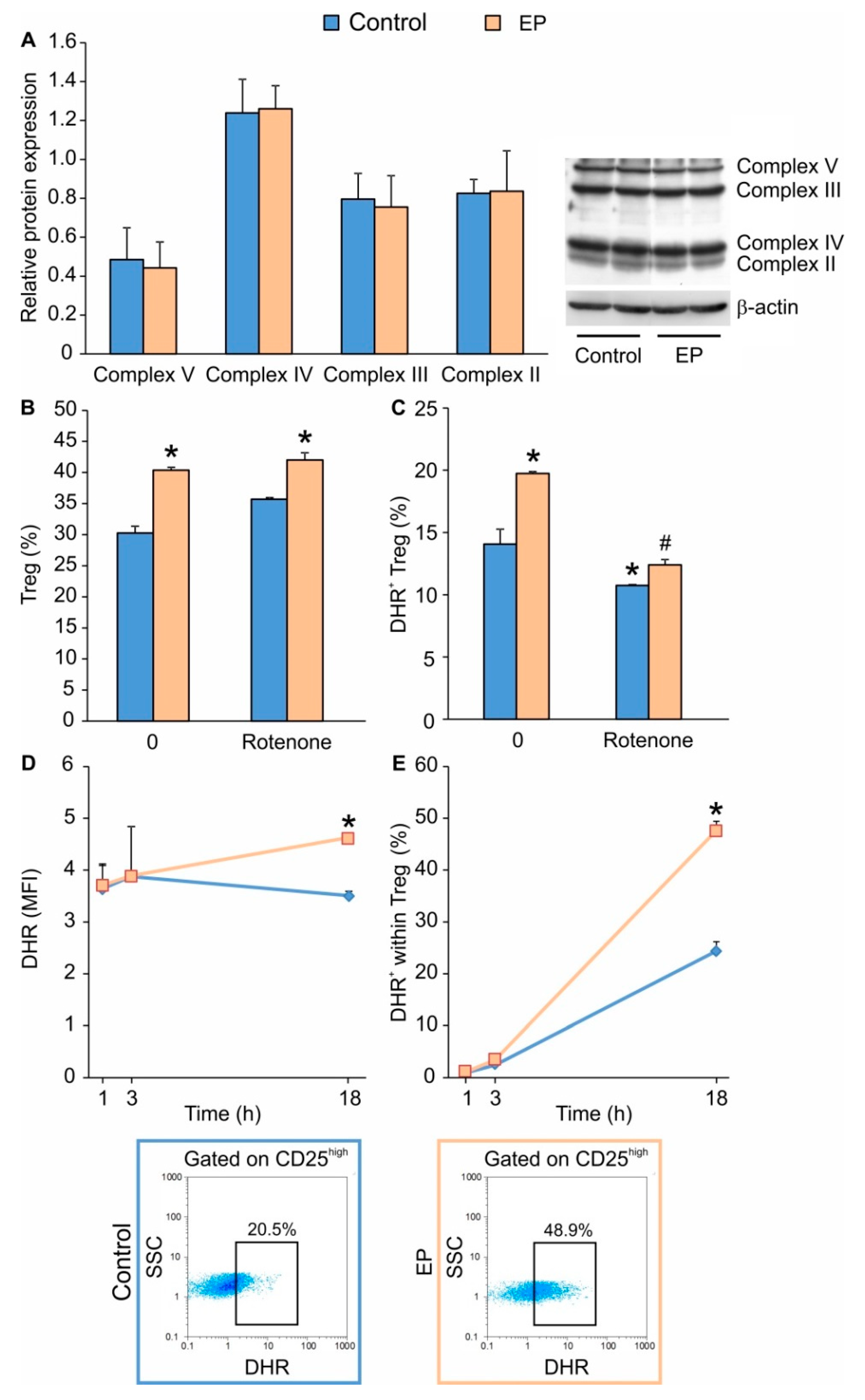
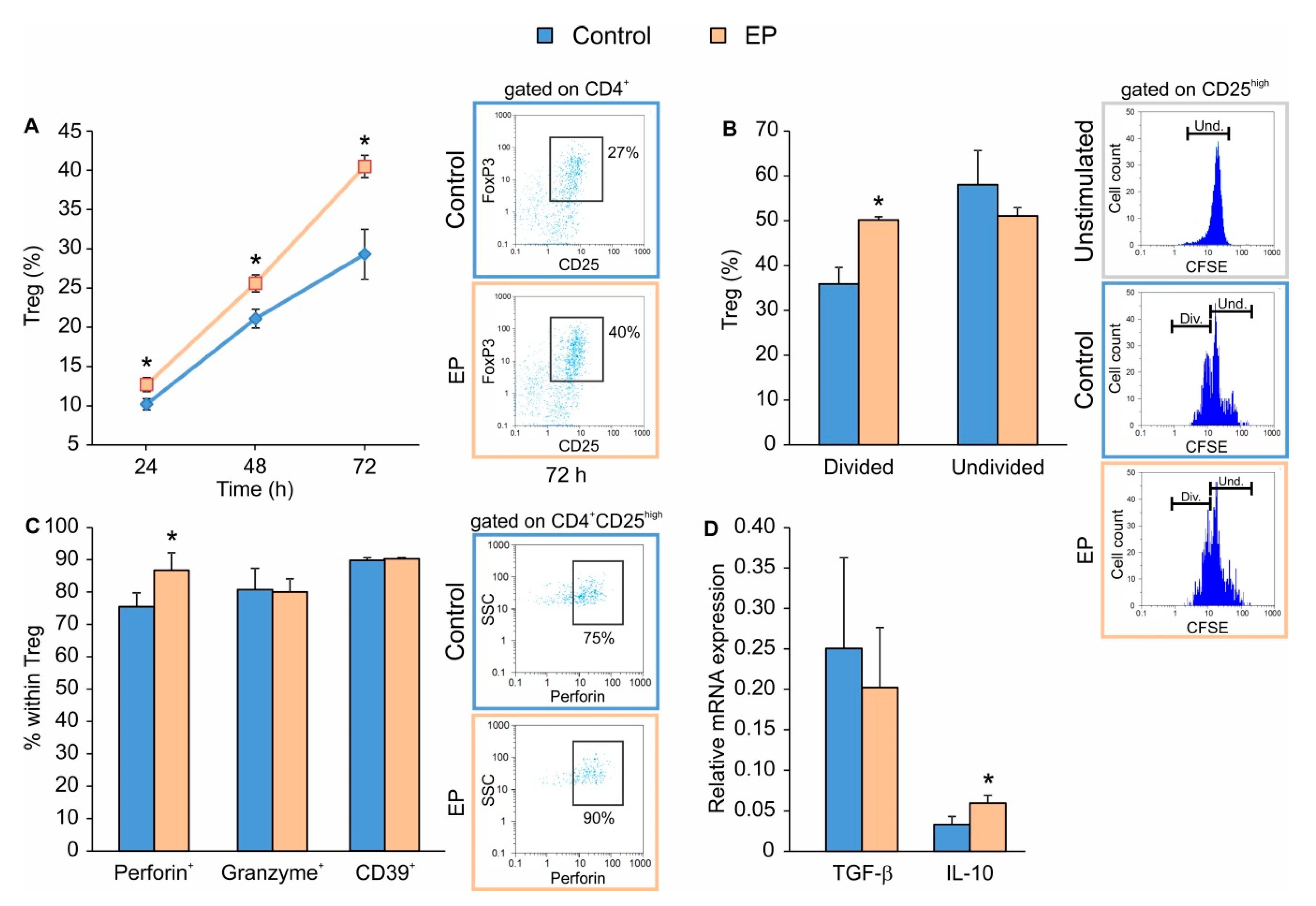
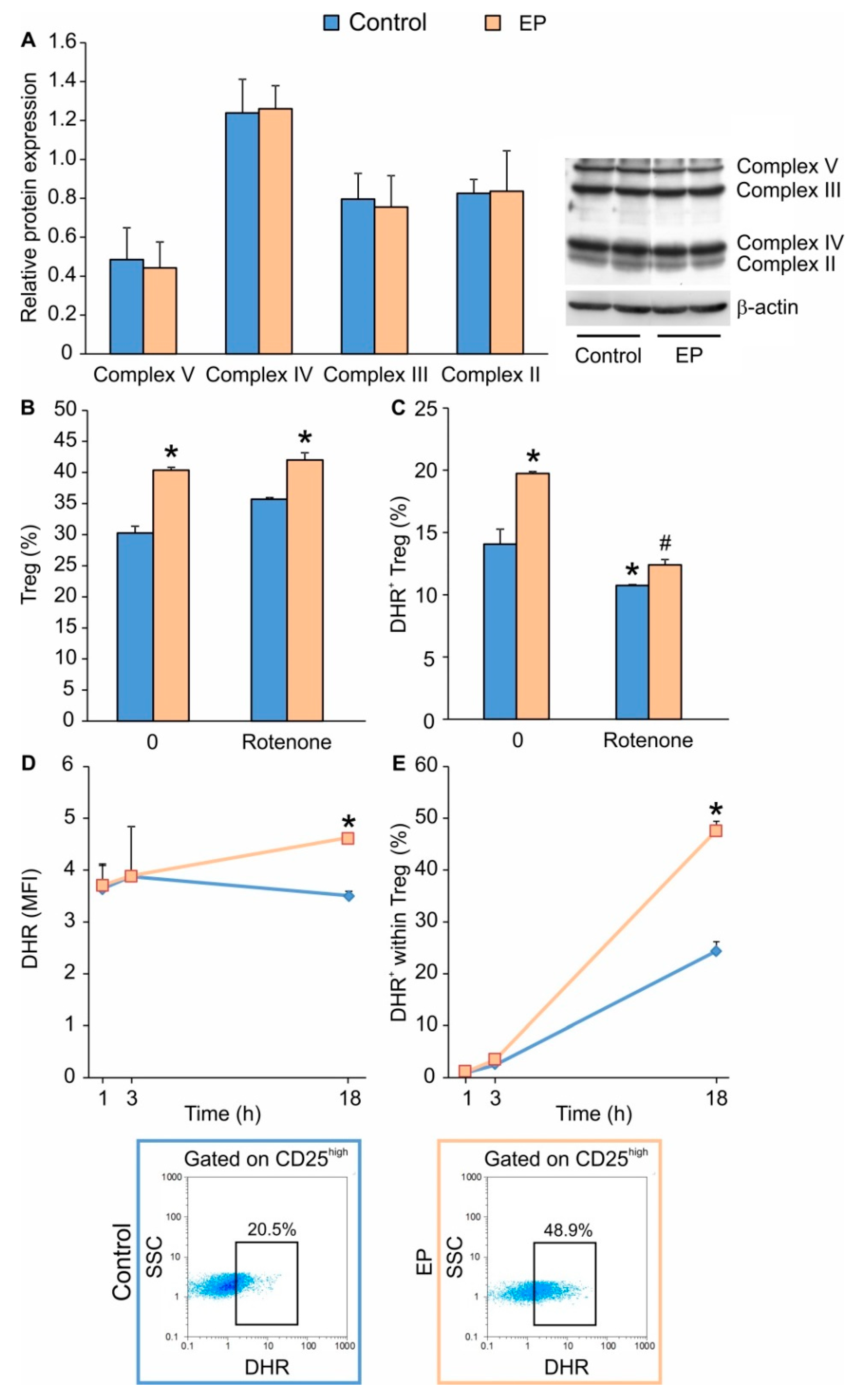
2.1. EP Stimulates the Proliferation of In Vitro Differentiated Treg
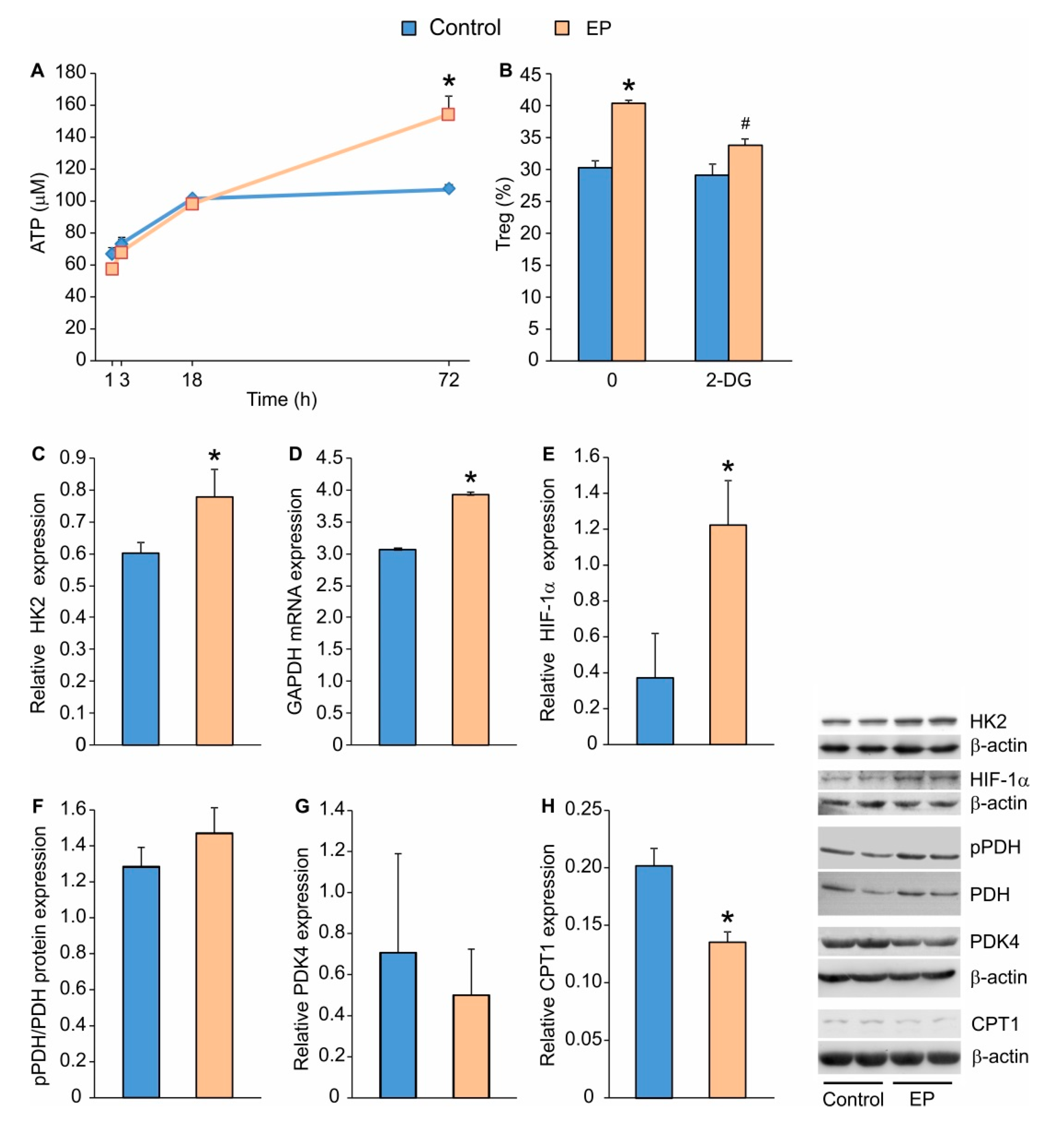
2.2. EP Increases Energy Production in Treg during Differentiation
2.3. The Effect of EP on the mTOR Pathway in Differentiating Treg
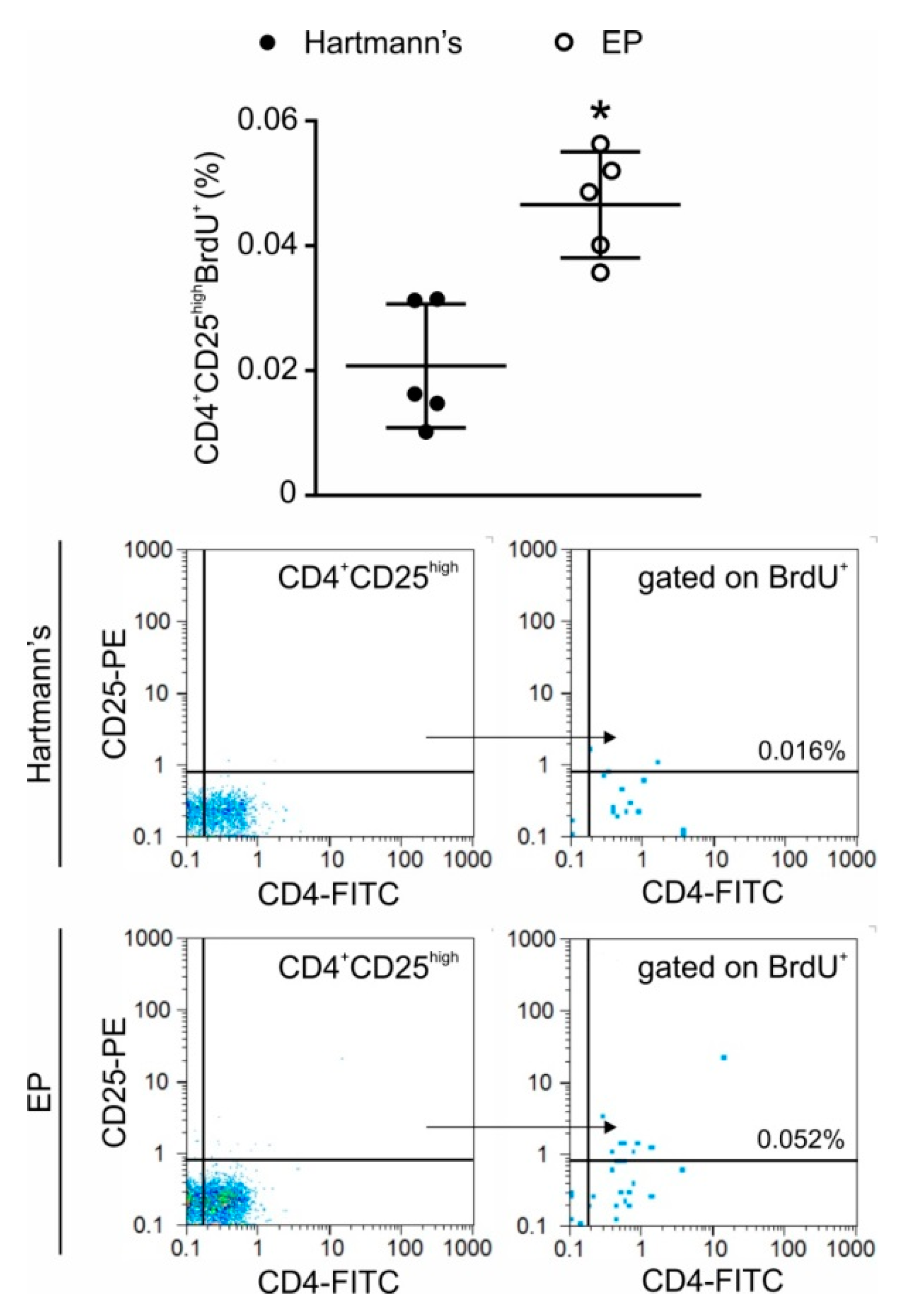
2.4. In Vivo Application of EP Increases Treg Numbers in the Peritoneum and the Gut
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. In Vivo EP Application
4.3. In Vivo Proliferation Assay
4.4. Treg Differentiation Protocol
4.5. Ex Vivo Cell Isolation
4.6. Flow Cytometry
4.7. Measurement of Intracellular Reactive Oxygen Species
4.8. ATP Bioluminescence Assay
4.9. Western Blot
4.10. Reverse Transcription and Real-Time PCR
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeng, J.; Liu, J.; Yang, G.Y.; Kelly, M.J.; James, T.L.; Litt, L. Exogenous ethyl pyruvate versus pyruvate during metabolic recovery after oxidative stress in neonatal rat cerebrocortical slices. Anesthesiology 2007, 107, 630–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.P. Ethyl pyruvate. Curr. Opin. Anaesthesiol. 2008, 21, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.K.; Clark, E.C.; Nath, K.A. Hydrogen peroxide-induced renal injury. A protective role for pyruvate in vitro and in vivo. J. Clin. Investig. 1991, 88, 1886–1893. [Google Scholar] [CrossRef]
- Kalariya, N.M.; Reddy, A.B.; Ansari, N.H.; VanKuijk, F.J.; Ramana, K.V. Preventive effects of ethyl pyruvate on endotoxin-induced uveitis in rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5144–5152. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Cheng, X.; Zhang, J.; Yuan, C.; Zhao, M.; Yang, X. Ethyl pyruvate confers protection against endotoxemia and sepsis by inhibiting caspase-11-dependent cell pyroptosis. Int. Immunopharmacol. 2020, 78, 106016. [Google Scholar] [CrossRef]
- Jang, I.S.; Park, M.Y.; Shin, I.W.; Sohn, J.T.; Lee, H.K.; Chung, Y.K. Ethyl pyruvate has anti-inflammatory and delayed myocardial protective effects after regional ischemia/reperfusion injury. Yonsei Med. J. 2010, 51, 838–844. [Google Scholar] [CrossRef]
- Kao, K.K.; Fink, M.P. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochem. Pharmacol. 2010, 80, 151–159. [Google Scholar] [CrossRef]
- Koprivica, I.; Vujičić, M.; Gajić, D.; Saksida, T.; Stojanović, I. Ethyl Pyruvate Stimulates Regulatory T Cells and Ameliorates Type 1 Diabetes Development in Mice. Front. Immunol. 2019, 9, 3130. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Galgani, M.; De Rosa, V.; La Cava, A.; Matarese, G. Role of Metabolism in the Immunobiology of Regulatory T Cells. J. Immunol. 2016, 197, 2567–2575. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M.; et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity 2016, 44, 406–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howie, D.; Cobbold, S.P.; Adams, E.; Ten Bokum, A.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2017, 2, e89160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anupam, K.; Kaushal, J.; Prabhakar, N.; Bhatnagar, A. Effect of redox status of peripheral blood on immune signature of circulating regulatory and cytotoxic T cells in streptozotocin induced rodent model of type I diabetes. Immunobiology 2018, 223, 586–597. [Google Scholar] [CrossRef]
- Kraaij, M.D.; van der Kooij, S.W.; Reinders, M.E.; Koekkoek, K.; Rabelink, T.J.; van Kooten, C.; Gelderman, K.A. Dexamethasone increases ROS production and T cell suppressive capacity by anti-inflammatory macrophages. Mol. Immunol. 2011, 49, 549–557. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef] [Green Version]
- Djedovic, N.; Mansilla, M.J.; Jevtić, B.; Navarro-Barriuso, J.; Saksida, T.; Martínez-Cáceres, E.M.; Miljković, Ð. Ethyl Pyruvate Induces Tolerogenic Dendritic Cells. Front. Immunol. 2019, 10, 157. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3⁺ T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, J.; Patel, N.; Wagmaister, J.; Choudhury, M.; Eshghi, M.; Konno, S. Prophylactic effect of ethyl pyruvate on renal ischemia/reperfusion injury mediated through oxidative stress. Int. Urol. Nephrol. 2019, 51, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Choi, J.S.; Park, C.; Jeong, J.W. Ethyl pyruvate stabilizes hypoxia-inducible factor 1 alpha via stimulation of the TCA cycle. Cancer Lett. 2010, 295, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Priyadharshini, B.; Loschi, M.; Newton, R.H.; Zhang, J.W.; Finn, K.K.; Gerriets, V.A.; Huynh, A.; Rathmell, J.C.; Blazar, B.R.; Turka, L.A. Cutting Edge: TGF-β and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J. Immunol. 2018, 201, 2215–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakhtoura, M.; Chain, R.W.; Sato, P.Y.; Qiu, C.C.; Lee, M.H.; Meissler, J.J.; Eisenstein, T.K.; Koch, W.J.; Caricchio, R.; Gallucci, S. Ethyl Pyruvate Modulates Murine Dendritic Cell Activation and Survival Through Their Immunometabolism. Front. Immunol. 2019, 10, 30. [Google Scholar] [CrossRef]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Yarosz, E.L.; Chang, C.H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef]
- Newton, R.; Priyadharshini, B.; Turka, L.A. Immunometabolism of regulatory T cells. Nat. Immunol. 2016, 17, 618–625. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sojka, D.K.; Huang, Y.H.; Fowell, D.J. Mechanisms of regulatory T-cell suppression—a diverse arsenal for a moving target. Immunology 2008, 124, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Frossi, B.; De Carli, M.; Piemonte, M.; Pucillo, C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol. Immunol. 2008, 45, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Xu, Q.; Qiu, Y.; Jin, X.; Xu, T.; Dong, S.; Wang, J.; Ke, Y.; Hu, H.; Cao, X.; et al. Suppression of Th17 cell differentiation by misshapen/NIK-related kinase MINK1. J. Exp. Med. 2017, 214, 1453–1469. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Chi, H. mTOR signaling, Tregs and immune modulation. Immunotherapy 2014, 6, 1295–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hukelmann, J.L.; Anderson, K.E.; Sinclair, L.V.; Grzes, K.M.; Murillo, A.B.; Hawkins, P.T.; Stephens, L.R.; Lamond, A.I.; Cantrell, D.A. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat. Immunol. 2016, 17, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Zhu, S.; Tonnessen, T.I. Ethyl pyruvate is a novel anti-inflammatory agent to treat multiple inflammatory organ injuries. J. Inflamm. 2016, 13, 37. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples are not available. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koprivica, I.; Gajić, D.; Pejnović, N.; Paunović, V.; Saksida, T.; Stojanović, I. Ethyl Pyruvate Promotes Proliferation of Regulatory T Cells by Increasing Glycolysis. Molecules 2020, 25, 4112. https://doi.org/10.3390/molecules25184112
Koprivica I, Gajić D, Pejnović N, Paunović V, Saksida T, Stojanović I. Ethyl Pyruvate Promotes Proliferation of Regulatory T Cells by Increasing Glycolysis. Molecules. 2020; 25(18):4112. https://doi.org/10.3390/molecules25184112
Chicago/Turabian StyleKoprivica, Ivan, Dragica Gajić, Nada Pejnović, Verica Paunović, Tamara Saksida, and Ivana Stojanović. 2020. "Ethyl Pyruvate Promotes Proliferation of Regulatory T Cells by Increasing Glycolysis" Molecules 25, no. 18: 4112. https://doi.org/10.3390/molecules25184112
APA StyleKoprivica, I., Gajić, D., Pejnović, N., Paunović, V., Saksida, T., & Stojanović, I. (2020). Ethyl Pyruvate Promotes Proliferation of Regulatory T Cells by Increasing Glycolysis. Molecules, 25(18), 4112. https://doi.org/10.3390/molecules25184112