Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy

 , ,
, , 
Abstract
1. Introduction
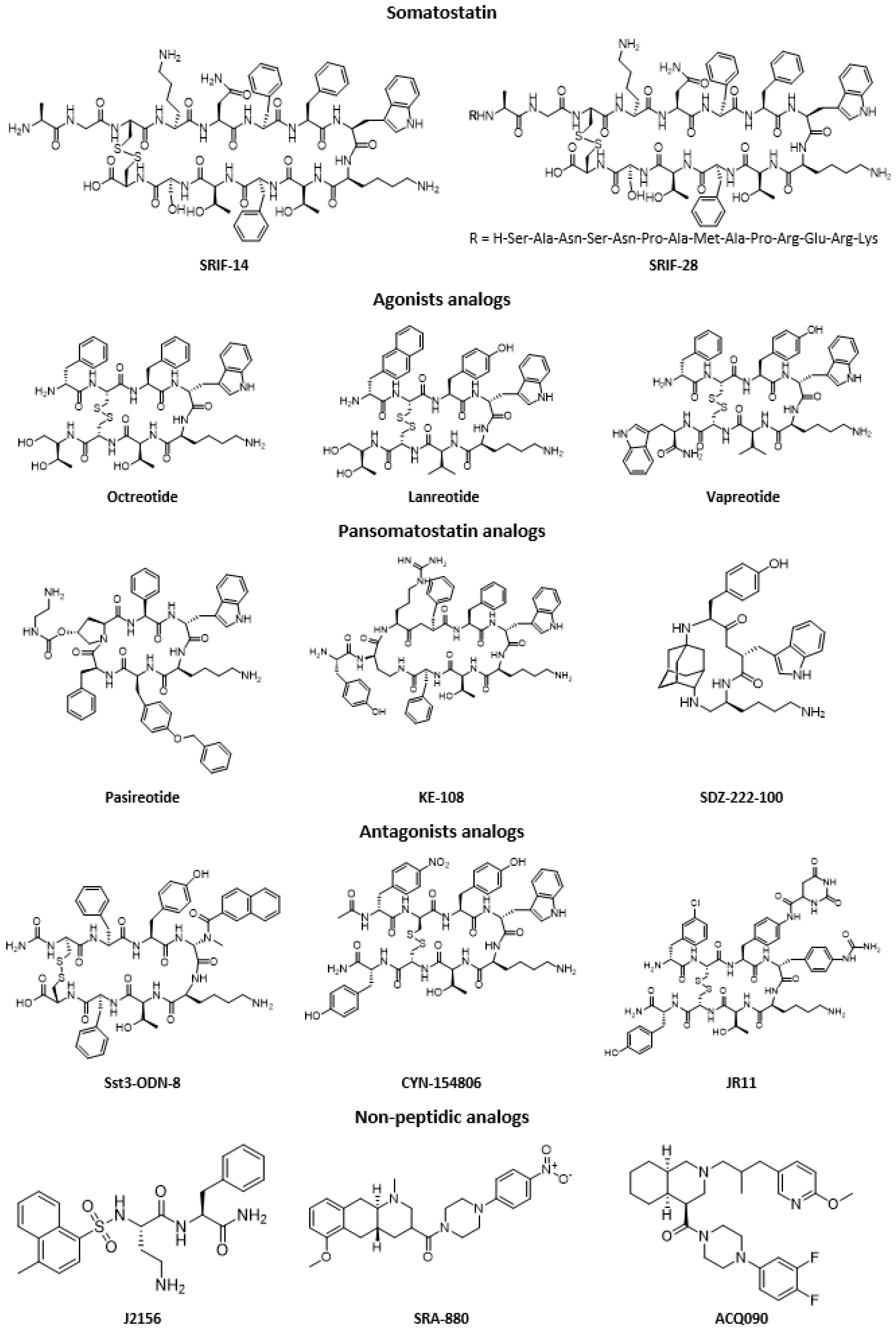
2. Somatostatin Analogs
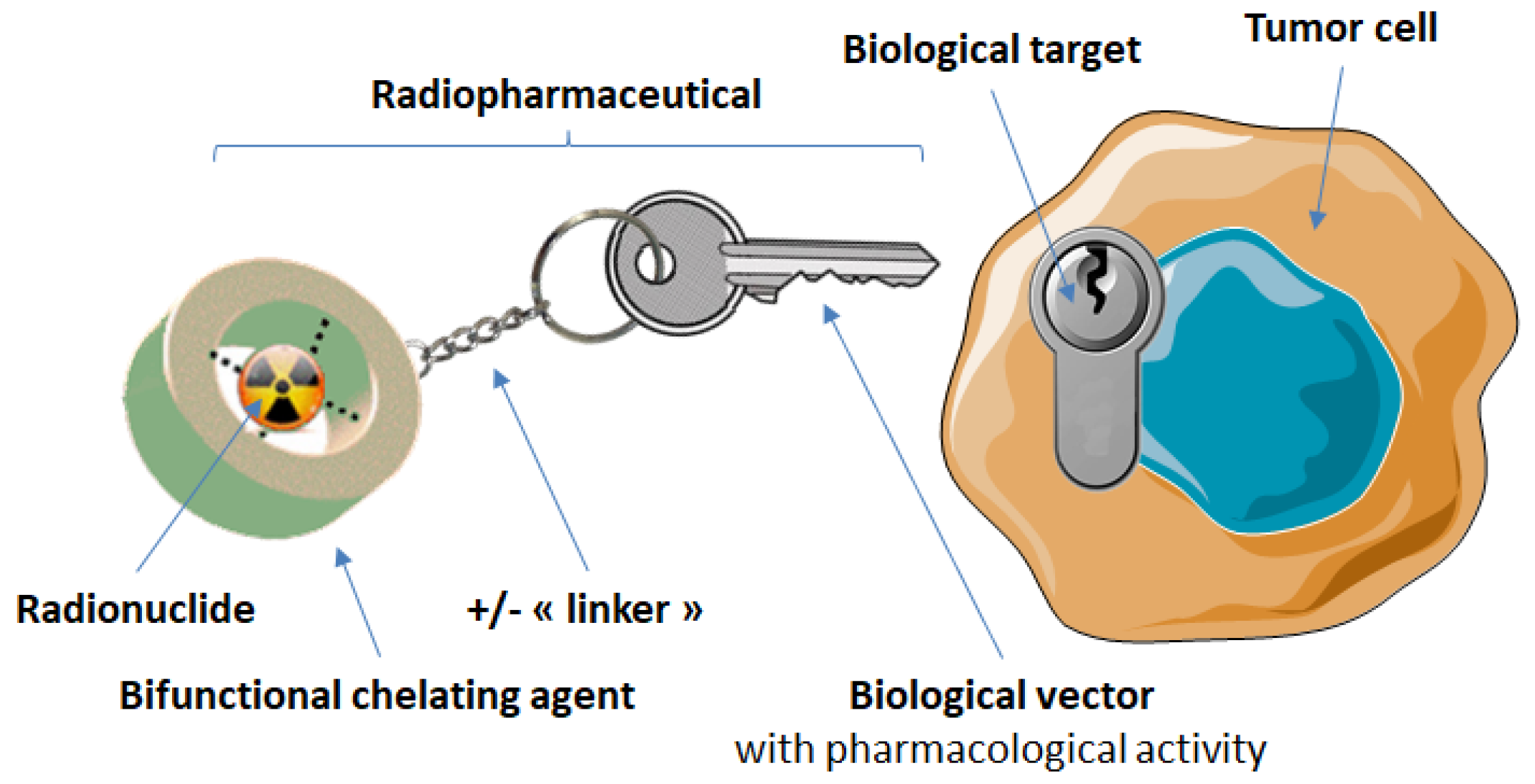
3. Targeting of Somatostatin Receptors with Radiopharmaceuticals
3.1. Radiolabeled Somatostatin Analogs for Imaging
3.1.1. Gallium-68 and Indium-111
3.1.2. Technetium-99m
3.1.3. Copper-64
3.1.4. Other Radiometals
3.1.5. Fluorine-18
3.2. Radiolabeled Somatostatin Analogs for Therapy
3.2.1. Yttrium-90 and Lutetium-177
3.2.2. Rhenium-188 and Other β-Emitting Radionuclides
3.2.3. Alpha and Auger Emitters
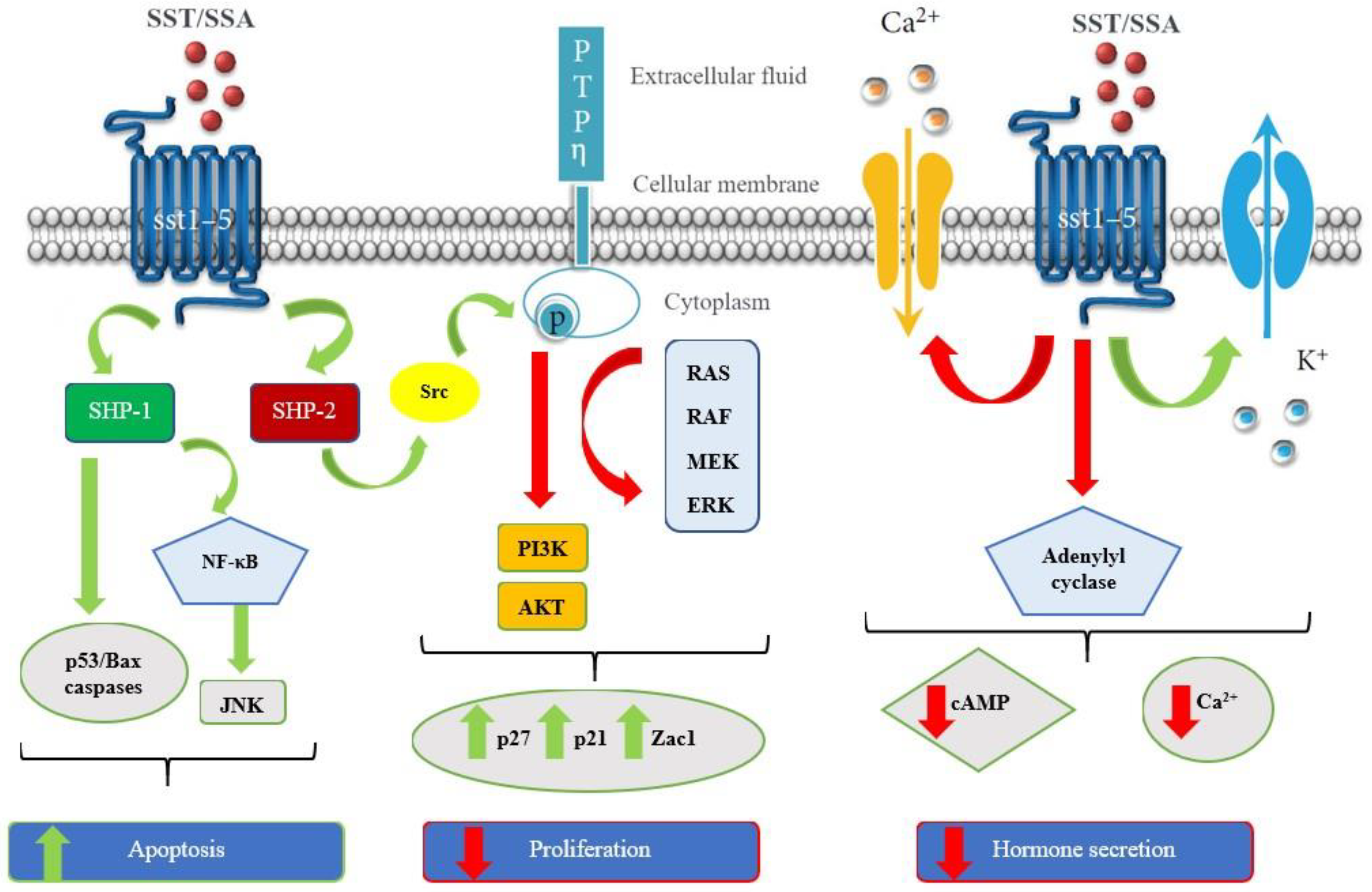
4. Antagonists vs. Agonists
5. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973, 179, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C.; Greenwood, M.T.; Panetta, R.; Demchyshyn, L.; Niznik, H.; Srikant, C.B. The somatostatin receptor family. Life Sci. 1995, 57, 1249–1265. [Google Scholar] [CrossRef]
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocrinol. 1999, 20, 157–198. [Google Scholar] [CrossRef] [PubMed]
- Weckbecker, G.; Lewis, I.; Albert, R.; Schmid, H.A.; Hoyer, D.; Bruns, C. Opportunities in somatostatin research: Biological, chemical and therapeutic aspects. Nat. Rev. Drug Discov. 2003, 2, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O.; Lamarca, A.; Valle, J.W.; Hubner, R.A. Somatostatin receptor expression in hepatocellular carcinoma: Prognostic and therapeutic considerations. Endocr. Relat. Cancer 2014, 21, R485–R493. [Google Scholar] [CrossRef][Green Version]
- Pyronnet, S.; Bousquet, C.; Najib, S.; Azar, R.; Laklai, H.; Susini, C. Antitumor effects of somatostatin. Mol. Cell. Endocrinol. 2008, 286, 230–237. [Google Scholar] [CrossRef]
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Würth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide receptor targeting in cancer: The somatostatin paradigm. Int. J. Pept. 2013, 2013, 926295. [Google Scholar] [CrossRef]
- Rai, U.; Thrimawithana, T.R.; Valery, C.; Young, S.A. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol. Ther. 2015, 152, 98–110. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.C.; Laissue, J.A.; Waser, B. Somatostatin receptors and their subtypes in human tumors and in peritumoral vessels. Metabolism 1996, 45, 39–41. [Google Scholar] [CrossRef]
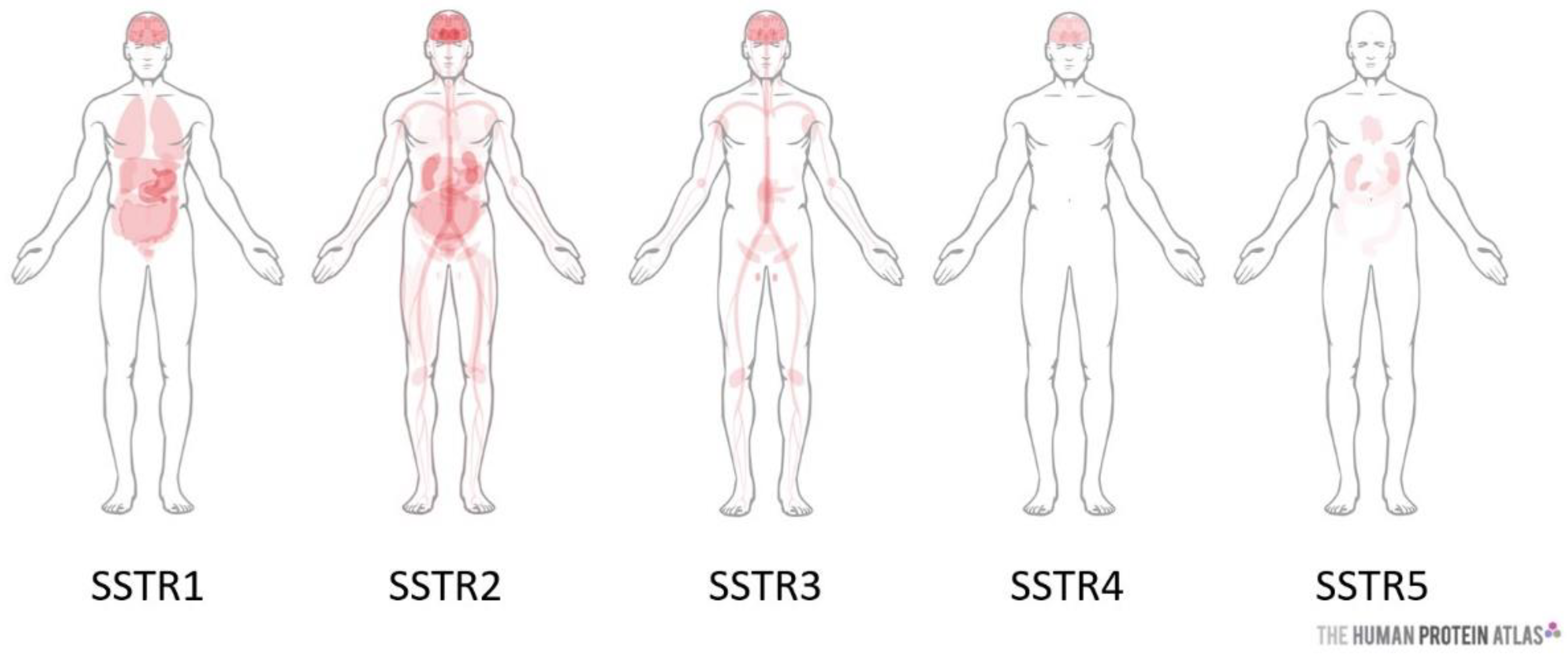
- Reubi, J.C.; Waser, B.; Schaer, J.C.; Laissue, J.A. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl. Med. 2001, 28, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Lahlou, H.; Pyronnet, S.; Bousquet, C.; Susini, C. Somatostatin receptors as tools for diagnosis and therapy: Molecular aspects. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 535551. [Google Scholar] [CrossRef]
- Hasskarl, J.; Kaufmann, M.; Schmid, H.A. Somatostatin receptors in non-neuroendocrine malignancies: The potential role of somatostatin analogs in solid tumors. Future Oncol. 2011, 7, 895–913. [Google Scholar] [CrossRef]
- Gomes-Porras, M.; Cárdenas-Salas, J.; Álvarez-Escolá, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682. [Google Scholar] [CrossRef]
- Hejna, M.; Schmidinger, M.; Raderer, M. The clinical role of somatostatin analogues as antineoplastic agents: Much ado about nothing? Ann. Oncol. 2002, 13, 653–668. [Google Scholar] [CrossRef]
- Keskin, O.; Yalcin, S. A review of the use of somatostatin analogs in oncology. OncoTargets Ther. 2013, 6, 471–483. [Google Scholar] [CrossRef][Green Version]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef]
- Zhao, B.; Zhao, H.; Zhao, N.; Zhu, X.G. Cholangiocarcinoma cells express somatostatin receptor subtype 2 and respond to octreotide treatment. J. Hepatobiliary Pancreat. Surg. 2002, 9, 497–502. [Google Scholar] [CrossRef]
- Bläker, M.; Schmitz, M.; Gocht, A.; Burghardt, S.; Schulz, M.; Bröring, D.C.; Pace, A.; Greten, H.; De Weerth, A. Differential expression of somatostatin receptor subtypes in hepatocellular carcinomas. J. Hepatol. 2004, 41, 112–118. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 781–793. [Google Scholar] [CrossRef]
- Reubi, J.C.; Zimmermann, A.; Jonas, S.; Waser, B.; Neuhaus, P.; Läderach, U.; Wiedenmann, B. Regulatory peptide receptors in human hepatocellular carcinomas. Gut 1999, 45, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, H.; Rombouts, K.; Vandermonde, A.; Urbain, D.; Kumar, U.; Bioulac-Sage, P.; Pinzani, M.; Rosenbaum, J.; Geerts, A. Expression of somatostatin receptors in normal and cirrhotic human liver and in hepatocellular carcinoma. Gut 2004, 53, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Khac, E.; Ollivier, I.; Aparicio, T.; Moullart, V.; Hugentobler, A.; Lebtahi, R.; Lobry, C.; Susini, C.; Duhamel, C.; Hommel, S.; et al. Somatostatin receptor scintigraphy screening in advanced hepatocarcinoma: A multi-center French study. Cancer Biol. Ther. 2009, 8, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, C.; van Dekken, H.; Hofland, L.J.; Zondervan, P.E.; de Wilt, J.H.; van Marion, R.; de Man, R.A.; IJzermans, J.N.; van Eijck, C.H. Somatostatin receptors in human hepatocellular carcinomas: Biological, patient and tumor characteristics. Dig. Surg. 2008, 25, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Huo, L.; Wang, L. Octreotide inhibits proliferation and induces apoptosis of hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2004, 25, 1380–1386. [Google Scholar]
- Li, S.; Liu, Y.; Shen, Z. Characterization of somatostatin receptor 2 and 5 expression in operable hepatocellular carcinomas. Hepatogastroenterology 2012, 59, 2054–2058. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Mu, Y. Somatostatin receptor subtypes 2 and 5 are associated with better survival in operable hepatitis B-related hepatocellular carcinoma following octreotide long-acting release treatment. Oncol. Lett. 2013, 6, 821–828. [Google Scholar] [CrossRef][Green Version]
- Huang, C.Z.; Huang, A.M.; Liu, J.F.; Wang, B.; Lin, K.C.; Ye, Y.B. Somatostatin Octapeptide Inhibits Cell Invasion and Metastasis in Hepatocellular Carcinoma Through PEBP1. Cell Physiol. Biochem. 2018, 47, 2340–2349. [Google Scholar] [CrossRef]
- Hua, Y.P.; Huang, J.F.; Liang, L.J.; Li, S.Q.; Lai, J.M.; Liang, H.Z. The study of inhibition effect of octreotide on the growth of hepatocellular carcinoma xenografts in situ in nude mice. Chin. J. Surg. 2005, 43, 721–725. [Google Scholar]
- Jia, W.D.; Xu, G.L.; Wang, W.; Wang, Z.H.; Li, J.S.; Ma, J.L.; Ren, W.H.; Ge, Y.S.; Yu, J.H.; Liu, W.B. A somatostatin analogue, octreotide, inhibits the occurrence of second primary tumors and lung metastasis after resection of hepatocellular carcinoma in mice. Tohoku J. Exp. Med. 2009, 218, 155160. [Google Scholar] [CrossRef][Green Version]
- Reynaert, H.; Colle, I. Treatment of Advanced Hepatocellular Carcinoma with Somatostatin Analogues: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 4811. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E.; Skordilis, P.; Thermos, K.; Vasilaki, A.; Moschandrea, J.; Manousos, O.N. Treatment of hepatocellular carcinoma with octreotide: A randomised controlled study. Gut 1998, 42, 442–447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dimitroulopoulos, D.; Xinopoulos, D.; Tsamakidis, K.; Zisimopoulos, A.; Andriotis, E.; Panagiotakos, D.; Fotopoulou, A.; Chrysohoou, C.; Bazinis, A.; Daskalopoulou, D.; et al. Long acting octreotide in the treatment of advanced hepatocellular cancer and overexpression of somatostatin receptors: Randomized placebo-controlled trial. World J. Gastroenterol. 2007, 13, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.; Allgaier, H.P.; Olschewski, M.; Zähringer, A.; Blum, H.E.; HECTOR Study Group. Long-acting octreotide versus placebo for treatment of advanced HCC: A randomized controlled double-blind study. Hepatology 2007, 45, 9–15. [Google Scholar] [CrossRef]
- Barbare, J.C.; Bouché, O.; Bonnetain, F.; Dahan, L.; Lombard-Bohas, C.; Faroux, R.; Raoul, J.L.; Cattan, S.; Lemoine, A.; Blanc, J.F.; et al. Treatment of advanced hepatocellular carcinoma with long-acting octreotide: A phase III multicenter, randomised, double blind placebo-controlled study. Eur. J. Cancer 2009, 45, 1788–1797. [Google Scholar] [CrossRef]
- Samonakis, D.N.; Notas, G.; Christodoulakis, N.; Kouroumalis, E.A. Mechanisms of action and resistance of somatostatin analogues for the treatment of hepatocellular carcinoma: A message not well taken. Dig. Dis. Sci. 2008, 53, 2359–2365. [Google Scholar] [CrossRef]
- Kaemmerer, D.; Schindler, R.; Mußbach, F.; Dahmen, U.; Altendorf-Hofmann, A.; Dirsch, O.; Sänger, J.; Schulz, S.; Lupp, A. Somatostatin and CXCR4 chemokine receptor expression in hepatocellular and cholangiocellular carcinomas: Tumor capillaries as promising targets. BMC Cancer 2017, 17, 896. [Google Scholar] [CrossRef]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID study group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Anti-tumour effects of lanreotide for pancreatic and intestinal neuroendocrine tumours: The CLARINET open-label extension study. Endocr. Relat. Cancer 2016, 23, 191–199. [Google Scholar] [CrossRef]
- Sheppard, M.; Shapiro, B.; Pimstone, B.; Kronheim, S.; Berelowitz, M.; Gregory, M. Metabolic clearance and plasma half-disappearance time of exogenous somatostatin in man. J. Clin. Endocrinol. Metab. 1979, 48, 50–53. [Google Scholar] [CrossRef]
- Lowell, A.; Freda, P.U. From somatostatin to octreotide LAR: Evolution of a somatostatin analogue. Curr. Med. Res. Opin. 2009, 25, 2989–2999. [Google Scholar] [CrossRef]
- Öberg, K.; Lamberts, S.W.J. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: Past, present and future. Endocr. Relat. Cancer 2016, 23, R551–R566. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; McBride, A.; Ray, D.; Pulgar, S.; Ramirez, R.A.; Elquza, E.; Favaro, J.P.; Dranitsaris, G. Lanreotide vs. octreotide LAR for patients with advanced gastroenteropancreatic neuroendocrine tumors: An observational time and motion analysis. J. Oncol. Pharm. Pract. 2019, 25, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Feelders, R.A.; Yasothan, U.; Kirkpatrick, P. Pasireotide. Nat. Rev. Drug Discov. 2012, 11, 597–598. [Google Scholar] [CrossRef]
- Crider, A.M. Recent Advances in the Development of Nonpeptide Somatostatin Receptor Ligands. Mini Rev. Med. Chem. 2002, 2, 507–517. [Google Scholar] [CrossRef]
- Correia, J.D.; Paulo, A.; Raposinho, P.D.; Santos, I. Radiometallated peptides for molecular imaging and targeted therapy. Dalton Trans. 2011, 40, 6144–6167. [Google Scholar] [CrossRef]
- Jamous, M.; Haberkorn, U.; Mier, W. Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases. Molecules 2013, 18, 3379–3409. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. An Overview of Bioactive Peptides for in vivo Imaging and Therapy in Human Diseases. Mini Rev. Med. Chem. 2017, 17, 758–770. [Google Scholar] [CrossRef]
- Cutler, C.S.; Hennkens, H.M.; Sisay, N.; Huclier-Markai, S.; Jurisson, S.S. Radiometals for Combined Imaging and Therapy. Chem. Rev. 2013, 113, 858–883. [Google Scholar] [CrossRef]
- Ramogida, C.F.; Orvig, C. Tumour Targeting with Radiometals for Diagnosis and Therapy. Chem. Commun. 2013, 49, 4720–4739. [Google Scholar] [CrossRef] [PubMed]
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 902–956. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Hancock, R.D.; Martell, A.E. Ligand design for selective complexation of metal ions in aqueous solution. Chem. Rev. 1989, 89, 1875–1914. [Google Scholar] [CrossRef]
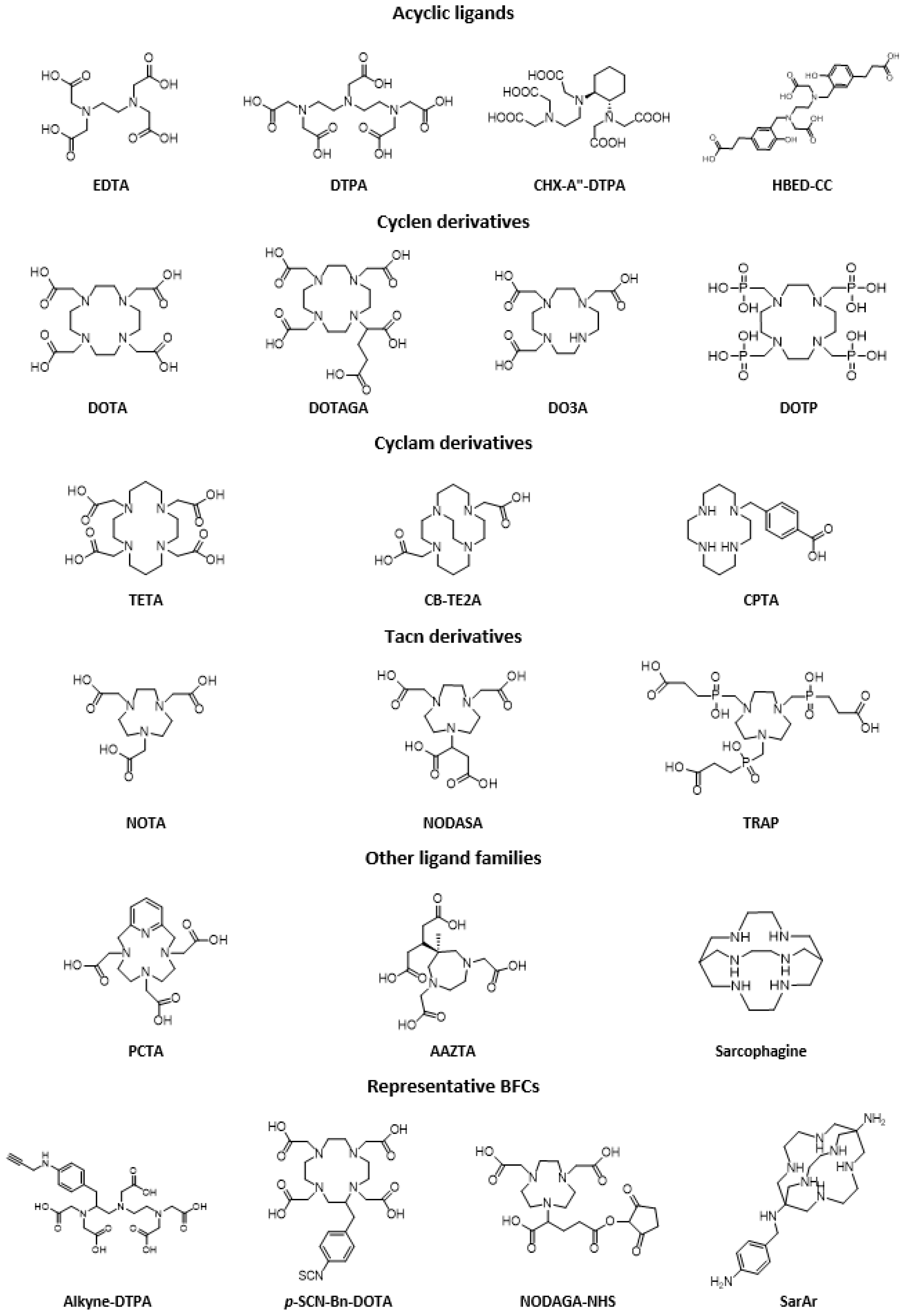
- Stasiuk, G.J.; Long, N.J. The ubiquitous DOTA and its derivatives: The impact of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid on biomedical imaging. Chem. Commun. 2013, 49, 2732–2746. [Google Scholar] [CrossRef]
- Baranyai, Z.; Tircsó, G.; Rösch, F. The Use of the Macrocyclic Chelator DOTA in Radiochemical Separations. Eur. J. Inorg. Chem. 2020, 36–56. [Google Scholar] [CrossRef]
- Sun, X.; Wuest, M.; Weisman, G.R.; Wong, E.H.; Reed, D.P.; Boswell, C.A.; Motekaitis, R.; Martell, A.E.; Welch, M.J.; Anderson, C.J. Radiolabeling and in vivo behavior of copper-64-labeled cross-bridged cyclam ligands. J. Med. Chem. 2002, 45, 469–477. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Dixit, M. Metallic radionuclides in the development of diagnostic and therapeutic radiopharmaceuticals. Dalton Trans. 2011, 40, 6112–6128. [Google Scholar] [CrossRef]
- Mushtaq, S.; Yun, S.J.; Jeon, J. Recent Advances in Bioorthogonal Click Chemistry for Efficient Synthesis of Radiotracers and Radiopharmaceuticals. Molecules 2019, 24, 3567. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef] [PubMed]
- Maecke, H.R.; Reubi, J.C. Somatostatin receptors as targets for nuclear medicine imaging and radionuclide treatment. J. Nucl. Med. 2011, 52, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R. Radiopharmaceutical development of radiolabeled peptides. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, S11–S30. [Google Scholar] [CrossRef]
- Lamberts, S.W.; Bakker, W.H.; Reubi, J.C.; Krenning, E.P. Somatostatin-receptor imaging in the localization of endocrine tumors. N. Engl. J. Med. 1990, 323, 1246–1249. [Google Scholar] [CrossRef]
- Bakker, W.H.; Krenning, E.P.; Breeman, W.A.; Kooij, P.P.; Reubi, J.C.; Koper, J.W.; de Jong, M.; Laméris, J.S.; Visser, T.J.; Lamberts, S.W. In vivo use of a radioiodinated somatostatin analogue: Dynamics, metabolism, and binding to somatostatin receptor-positive tumors in man. J. Nucl. Med. 1991, 32, 1184–1189. [Google Scholar]
- Bakker, W.H.; Krenning, E.P.; Breeman, W.A.; Koper, J.W.; Kooij, P.P.; Reubi, J.C.; Klijn, J.G.; Visser, T.J.; Docter, R.; Lamberts, S.W. Receptor scintigraphy with a radioiodinated somatostatin analogue: Radiolabeling, purification, biologic activity, and in vivo application in animals. J. Nucl. Med. 1990, 31, 1501–1509. [Google Scholar]
- Bakker, W.H.; Albert, R.; Bruns, C.; Breeman, W.A.; Hofland, L.J.; Marbach, P.; Pless, J.; Pralet, D.; Stolz, B.; Koper, J.W.; et al. [111In-DTPA-D-Phe1]-octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: Synthesis, radiolabeling and in vitro validation. Life Sci. 1991, 49, 1583–1591. [Google Scholar] [CrossRef]
- Bakker, W.H.; Krenning, E.P.; Reubi, J.C.; Breeman, W.A.; Setyono-Han, B.; de Jong, M.; Kooij, P.P.; Bruns, C.; van Hagen, P.M.; Marbach, P.; et al. In vivo application of [111In-DTPA-D-Phe1]-octreotide for detection of somatostatin receptor-positive tumors in rats. Life Sci. 1991, 49, 1593–1601. [Google Scholar] [CrossRef]
- Krenning, E.P.; Bakker, W.H.; Kooij, P.P.; Breeman, W.A.; Oei, H.Y.; de Jong, M.; Reubi, J.C.; Visser, T.J.; Bruns, C.; Kwekkeboom, D.J.; et al. Somatostatin receptor scintigraphy with indium-111-DTPA-D-Phe-1-octreotide in man: Metabolism, dosimetry and comparison with iodine-123-Tyr-3-octreotide. J. Nucl. Med. 1992, 33, 652–658. [Google Scholar] [PubMed]
- Otte, A.; Jermann, E.; Behe, M.; Goetze, M.; Bucher, H.C.; Roser, H.W.; Heppeler, A.; Mueller-Brand, J.; Maecke, H.R. DOTATOC: A powerful new tool for receptor-mediated radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 792–795. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Forrer, F.; Uusijärvi, H.; Waldherr, C.; Cremonesi, M.; Bernhardt, P.; Mueller-Brand, J.; Maecke, H.R. A comparison of 111In-DOTATOC and 111In-DOTATATE: Biodistribution and dosimetry in the same patients with metastatic neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Schmitt, J.S.; Ginj, M.; Mäcke, H.R.; Bernard, B.F.; Krenning, E.; De Jong, M.; Wenger, S.; Reubi, J.C. DOTA-NOC, a high-affinity ligand of somatostatin receptor subtypes 2, 3 and 5 for labeling with various radiometals. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1338–1347. [Google Scholar] [CrossRef]
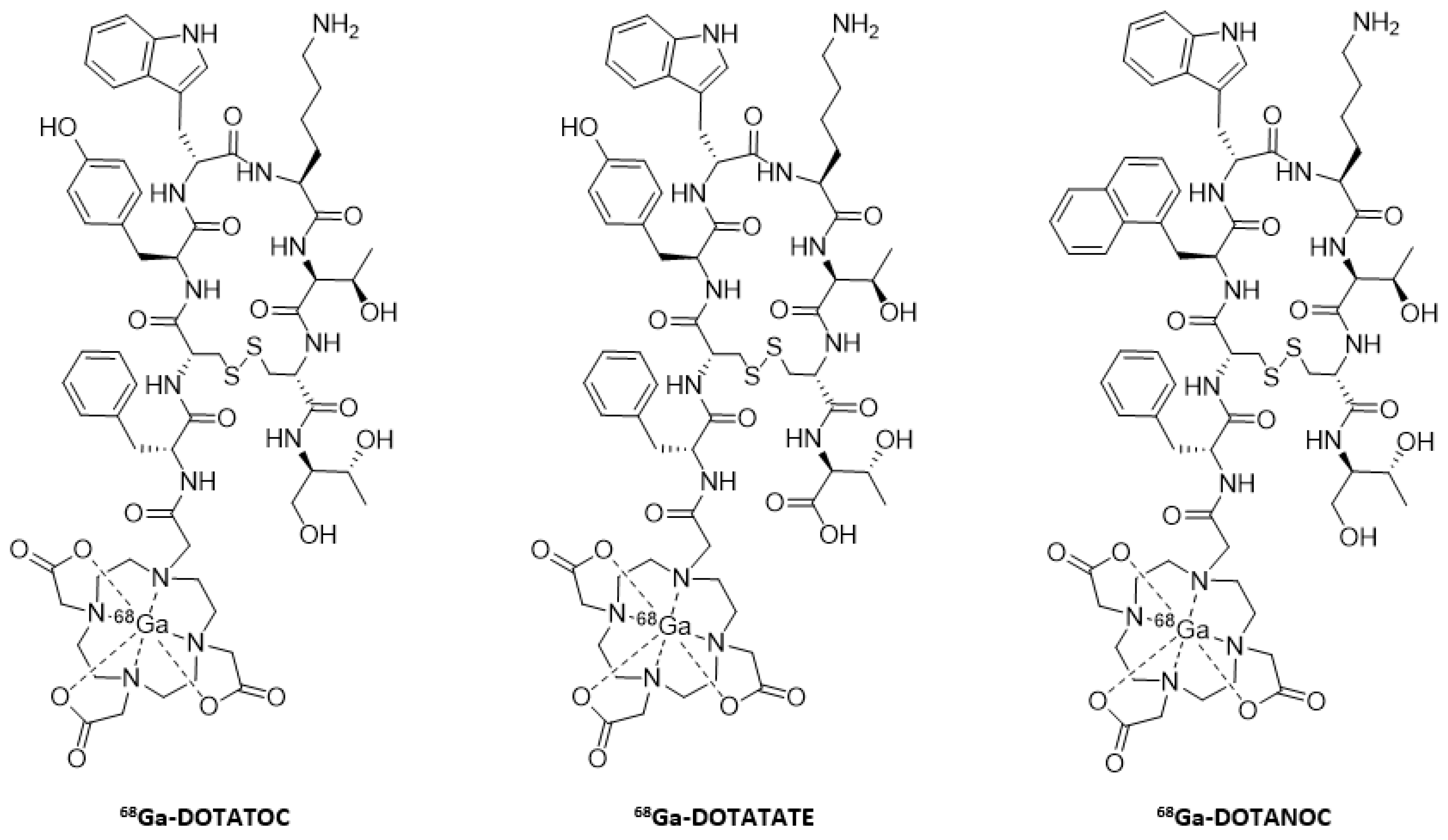
- Virgolini, I.; Ambrosini, V.; Bomanji, J.B.; Baum, R.P.; Fanti, S.; Gabriel, M.; Papathanasiou, N.D.; Pepe, G.; Oyen, W.; De Cristoforo, C.; et al. Procedure guidelines for PET/CT tumour imaging with 68Ga-DOTA-conjugated peptides: 68Ga-DOTA-TOC, 68Ga-DOTA-NOC, 68Ga-DOTA-TATE. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2004–2010. [Google Scholar] [CrossRef]
- Hofmann, M.; Maecke, H.; Börner, R.; Weckesser, E.; Schöffski, P.; Oei, L.; Schumacher, J.; Henze, M.; Heppeler, A.; Meyer, J.; et al. Biokinetics and imaging with the somatostatin receptor PET radioligand 68Ga-DOTATOC: Preliminary data. Eur. J. Nucl. Med. 2001, 28, 1751–1757. [Google Scholar] [CrossRef]
- Hofman, M.S.; Lau, W.F.; Hicks, R.J. Somatostatin receptor imaging with 68Ga DOTATATE PET/CT: Clinical utility, normal patterns, pearls, and pitfalls in interpretation. Radiographics 2015, 35, 500–516. [Google Scholar] [CrossRef]
- Wild, D.; Mäcke, H.R.; Waser, B.; Reubi, J.C.; Ginj, M.; Rasch, H.; Müller-Brand, J.; Hofmann, M. 68Ga-DOTANOC: A first compound for PET imaging with high affinity for somatostatin receptor subtypes 2 and 5. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 724. [Google Scholar] [CrossRef]
- Antunes, P.; Ginj, M.; Walter, M.A.; Chen, J.; Reubi, J.C.; Maecke, H.R. Influence of different spacers on the biological profile of a DOTA-Somatostatin analogue. Bioconjugate Chem. 2007, 18, 84–92. [Google Scholar] [CrossRef]
- Eisenwiener, K.P.; Prata, M.I.; Buschmann, I.; Zhang, H.W.; Santos, A.C.; Wenger, S.; Reubi, J.C.; Mäcke, H.R. NODAGATOC, a new chelator-coupled somatostatin analogue labeled with [67/68Ga] and [111In] for SPECT, PET, and targeted therapeutic applications of somatostatin receptor (hsst2) expressing tumors. Bioconjugate Chem. 2002, 13, 530–541. [Google Scholar] [CrossRef]
- Laznickova, A.; Laznicek, M.; Trejtnar, F.; Maecke, H.R.; Eisenwiener, K.P.; Reubi, J.C. Biodistribution of two octreotate analogs radiolabeled with indium and yttrium in rats. Anticancer Res. 2010, 30, 2177–2184. [Google Scholar] [PubMed]
- Ginj, M.; Chen, J.; Walter, M.A.; Eltschinger, V.; Reubi, J.C.; Maecke, H.R. Preclinical evaluation of new and highly potent analogues of octreotide for predictive imaging and targeted radiotherapy. Clin. Cancer Res. 2005, 11, 1136–1145. [Google Scholar] [PubMed]
- Boubaker, A.; Prior, J.O.; Willi, J.P.; Champendal, M.; Kosinski, M.; Bischof-Delaloye, A.; Maecke, H.R.; Ginj, M.; Baechler, S.; Buchegger, F. Biokinetics and dosimetry of 111In-DOTA-NOC-ATE compared with 111In-DTPA-octreotide. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1868–1875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, M.T.; Cullinane, C.; Waldeck, K.; Roselt, P.; Hicks, R.J.; Blower, P.J. Rapid kit-based 68Ga-labeling and PET imaging with THP-Tyr3-octreotate: A preliminary comparison with DOTA-Tyr3octreotate. EJNMMI Res. 2015, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Mueller, A.; Tamma, M.L.; Nicolas, G.; Rink, H.R.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Radiolabeled bicyclic somatostatin-based analogs: A novel class of potential radiotracers for SPECT/PET of neuroendocrine tumors. J. Nucl. Med. 2010, 51, 1771–1779. [Google Scholar] [CrossRef][Green Version]
- Ginj, M.; Zhang, H.; Eisenwiener, K.P.; Wild, D.; Schulz, S.; Rink, H.; Cescato, R.; Reubi, J.C.; Maecke, H.R. New pansomatostatin ligands and their chelated versions: Affinity profile, agonist activity, internalization, and tumor targeting. Clin. Cancer Res. 2008, 14, 2019–2027. [Google Scholar] [CrossRef][Green Version]
- Liu, F.; Liu, T.; Xu, X.; Guo, X.; Li, N.; Xiong, C.; Li, C.; Zhu, H.; Yang, Z. Design, Synthesis, and Biological Evaluation of 68Ga-DOTA-PA1 for Lung Cancer: A Novel PET Tracer for Multiple Somatostatin Receptor Imaging. Mol. Pharm. 2018, 15, 619–628. [Google Scholar] [CrossRef]
- Tatsi, A.; Maina, T.; Cescato, R.; Waser, B.; Krenning, E.P.; de Jong, M.; Cordopatis, P.; Reubi, J.C.; Nock, B.A. [111In-DOTA]Somatostatin-14 analogs as potential pansomatostatin-like radiotracers—First results of a preclinical study. EJNMMI Res. 2012, 2, 25. [Google Scholar] [CrossRef]
- Maina, T.; Cescato, R.; Waser, B.; Tatsi, A.; Kaloudi, A.; Krenning, E.P.; de Jong, M.; Nock, B.A.; Reubi, J.C. [111In-DOTA]LTT-SS28, a first pansomatostatin radioligand for in vivo targeting of somatostatin receptor-positive tumors. J. Med. Chem. 2014, 57, 6564–6571. [Google Scholar] [CrossRef]
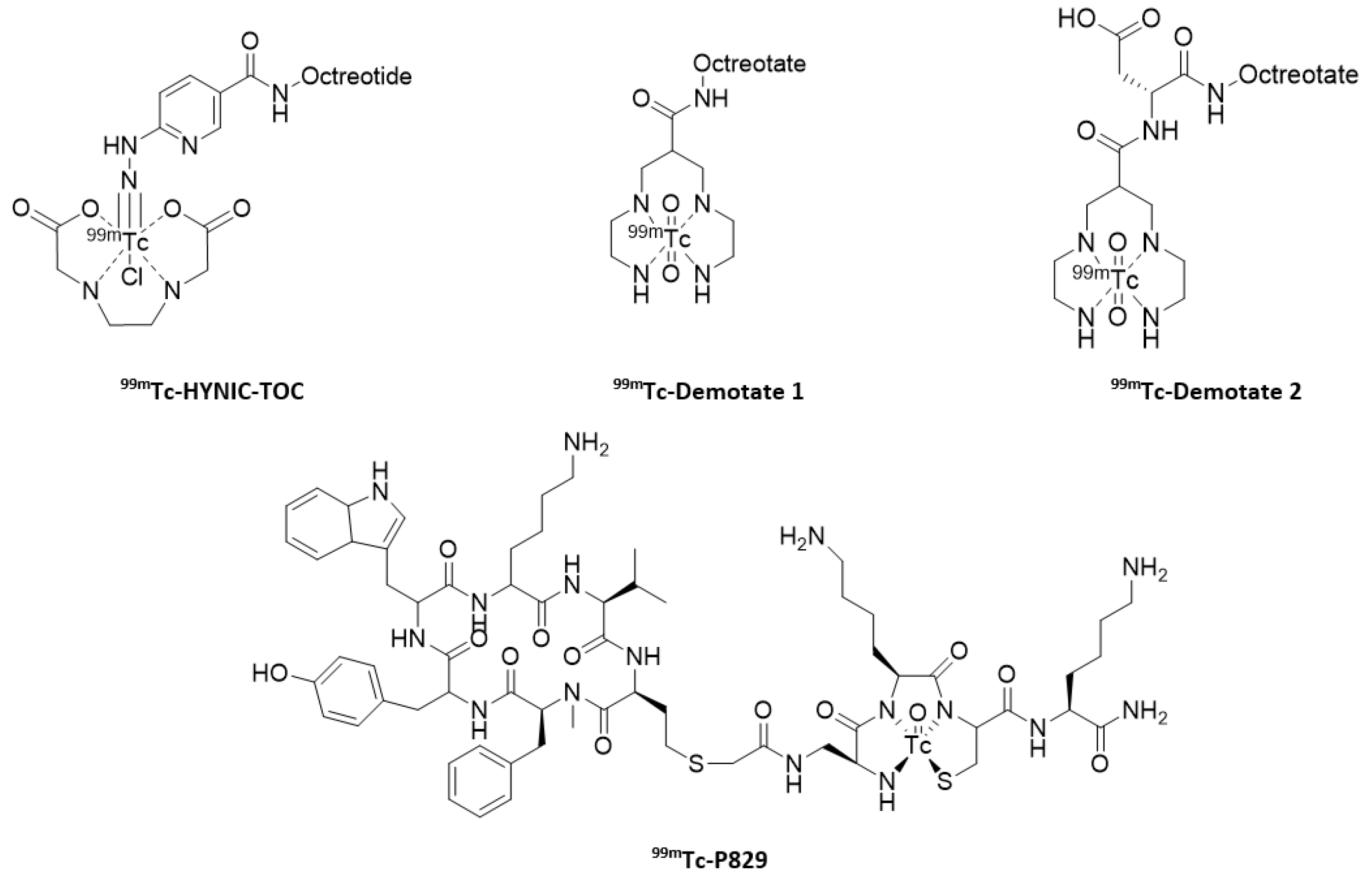
- Pearson, D.A.; Lister-James, J.; McBride, W.J.; Wilson, D.M.; Martel, L.J.; Civitello, E.R.; Taylor, J.E.; Moyer, B.R.; Dean, R.T. Somatostatin receptor-binding peptides labeled with technetium-99m: Chemistry and initial biological studies. J. Med. Chem. 1996, 39, 1361–1371. [Google Scholar] [CrossRef]
- Decristoforo, C.; Mather, S.J. Preparation, 99mTc-labeling and in vitro characterization of HYNIC and N3S modified RC-160 and [Tyr3]octreotide. Bioconjugate Chem. 1999, 10, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Stolz, B.; Albert, R.; Bruns, C.; Koch, P.; Mäcke, H. Synthesis, radiochemistry and biological evaluation of a new somatostatin analogue (SDZ 219-387) labeled with technetium-99m. Eur. J. Nucl. Med. 1994, 21, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.L.; Kolan, H.; Li, J.; Wiaderkiewicz, R.; Pallela, V.R.; Duggaraju, R.; Schally, A.V. Radiolabeled somatostatin analogues in prostate cancer. Nucl. Med. Biol. 1997, 24, 105–113. [Google Scholar] [CrossRef]
- Abiraj, K.; Ursillo, S.; Tamma, M.L.; Rylova, S.N.; Waser, B.; Constable, E.C.; Fani, M.; Nicolas, G.P.; Reubi, J.C.; Maecke, H.R. The tetraamine chelator outperforms HYNIC in a new technetium-99m-labeled somatostatin receptor 2 antagonist. EJNMMI Res. 2018, 8, 75. [Google Scholar] [CrossRef]
- Spradau, T.W.; Edwards, W.B.; Anderson, C.J.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis and biological evaluation of Tc-99m-cyclopentadienyltricarbonyltechnetium-labeled octreotide. Nucl. Med. Biol. 1999, 26, 1–7. [Google Scholar] [CrossRef]
- Makris, G.; Kuchuk, M.; Gallazzi, F.; Jurisson, S.S.; Smith, C.J.; Hennkens, H.M. Somatostatin receptor targeting with hydrophilic [99mTc/186Re]Tc/Re-tricarbonyl NODAGA and NOTA complexes. Nucl. Med. Biol. 2019, 71, 39–46. [Google Scholar] [CrossRef]
- Abrams, M.J.; Juweid, M.; tenKate, C.I.; Schwartz, D.A.; Hauser, M.M.; Gaul, F.E.; Fuccello, A.J.; Rubin, R.H.; Strauss, H.W.; Fischman, A.J. Technetium-99m-human polyclonal IgG radiolabeled via the hydrazino nicotinamide derivative for imaging focal sites of infection in rats. J. Nucl. Med. 1990, 31, 2022–2028. [Google Scholar]
- Mikolajczak, R.; Maecke, H.R. Radiopharmaceuticals for somatostatin receptor imaging. Nucl. Med. Rev. Cent. East Eur. 2016, 19, 126–132. [Google Scholar] [CrossRef]
- Decristoforo, C.; Melendez-Alafort, L.; Sosabowski, J.K.; Mather, S.J. 99mTc-HYNIC-[Tyr3]octreotide for imaging somatostatin receptor positive tumors: Preclinical evaluation and comparison with 111In-octreotide. J. Nucl. Med. 2000, 41, 1114–1119. [Google Scholar]
- Gabriel, M.; Decristoforo, C.; Donnemiller, E.; Ulmer, H.; Watfah Rychlinski, C.; Mather, S.J.; Moncayo, R. An intrapatient comparison of 99mTc-EDDA/HYNIC-TOC with 111In-DTPA-Octreotide for diagnosis of somatostatin receptor expressing tumors. J. Nucl. Med. 2003, 44, 708–716. [Google Scholar]
- Cwikla, J.B.; Mikolajczak, R.; Pawlak, D.; Buscombe, J.R.; Nasierowska-Guttmejer, A.; Bator, A.; Maecke, H.R.; Walecki, J. Initial direct comparison of 99mTc-TOC and 99mTc-TATE in identifying sites of disease in patients with proven GEP NETs. J. Nucl. Med. 2008, 49, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Decristoforo, C.; Maina, T.; Nock, B.; Gabriel, M.; Cordopatis, P.; Moncayo, R. 99mTc-Demotate 1: First data in tumour patients-results of a pilot/phase I study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Nock, B.A.; Cordopatis, P.; Bernard, B.F.; Breeman, W.A.; van Gameren, A.; van den Berg, R.; Reubi, J.C.; Krenning, E.P.; de Jong, M. [99mTc]Demotate 2 in the detection of sst2-positive tumours: A preclinical comparison with [111In]DOTA-tate. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, I.; Leimer, M.; Handmaker, H.; Lastoria, S.; Bischof, C.; Muto, P.; Pangerl, T.; Gludovacz, D.; Peck-Radosavljevic, M.; Lister-James, J.; et al. Somatostatin receptor subtype specificity and in vivo binding of a novel tumor tracer, 99mTc-P829. Cancer Res. 1998, 58, 1850–1859. [Google Scholar]
- Lebtahi, R.; Le Cloirec, J.; Houzard, C.; Daou, D.; Sobhani, I.; Sassolas, G.; Mignon, M.; Bourguet, P.; Le Guludec, D. Detection of neuroendocrine tumors: 99mTc-P829 scintigraphy compared with 111In-pentetreotide scintigraphy. J. Nucl. Med. 2002, 43, 889–895. [Google Scholar]
- Blum, J.E.; Handmaker, H.; Rinne, N.A. The utility of a somatostatin-type receptor binding peptide radiopharmaceutical (P829) in the evaluation of solitary pulmonary nodules. Chest 1999, 115, 224–232. [Google Scholar] [CrossRef]
- Bååth, M.; Kolbeck, K.G.; Danielsson, R. Somatostatin receptor scintigraphy with 99mTc-Depreotide (NeoSpect) in discriminating between malignant and benign lesions in the diagnosis of lung cancer: A pilot study. Acta Radiol. 2004, 45, 833–839. [Google Scholar] [CrossRef]
- Axelsson, R.; Herlin, G.; Bååth, M.; Aspelin, P.; Kolbeck, K.G. Role of scintigraphy with technetium-99m depreotide in the diagnosis and management of patients with suspected lung cancer. Acta Radiol. 2008, 49, 295–302. [Google Scholar] [CrossRef]
- Menda, Y.; Kahn, D. Somatostatin receptor imaging of non-small cell lung cancer with 99mTc depreotide. Semin. Nucl. Med. 2002, 32, 92–96. [Google Scholar] [CrossRef]
- Van Den Bossche, B.; D’haeninck, E.; Bacher, K.; Thierens, H.; Van Belle, S.; Dierckx, R.A.; Van de Wiele, C. Biodistribution and dosimetry of 99mTc-depreotide (P829) in patients suffering from breast carcinoma. Cancer Biother. Radiopharm. 2004, 19, 776–783. [Google Scholar] [CrossRef]
- Briganti, V.; Cuccurullo, V.; Di Stasio, G.D.; Mansi, L. Gamma Emitters in Pancreatic Endocrine Tumors Imaging in the PET Era: Is there a Clinical Space for 99mTc-peptides? Curr. Radiopharm. 2019, 12, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Uccelli, L.; Martini, P. A Picture of Modern Tc-99m Radiopharmaceuticals: Production, Chemistry, and Applications in Molecular Imaging. Appl. Sci. 2019, 9, 2526. [Google Scholar] [CrossRef]
- Boschi, A.; Martini, P.; Janevik-Ivanovska, E.; Duatti, A. The emerging role of copper-64 radiopharmaceuticals as cancer theranostics. Drug Discov. Today 2018, 23, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Dehdashti, F.; Cutler, P.D.; Schwarz, S.W.; Laforest, R.; Bass, L.A.; Lewis, J.S.; McCarthy, D.W. 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J. Nucl. Med. 2001, 42, 213–221. [Google Scholar]
- Sprague, J.E.; Peng, Y.; Sun, X.; Weisman, G.R.; Wong, E.H.; Achilefu, S.; Anderson, C.J. Preparation and biological evaluation of copper-64-labeled Tyr3-octreotate using a cross-bridged macrocyclic chelator. Clin. Cancer Res. 2004, 10, 8674–8682. [Google Scholar] [CrossRef]
- Edwards, W.B.; Fields, C.G.; Anderson, C.J.; Pajeau, T.S.; Welch, M.J.; Fields, G.B. Generally Applicable, Convenient Solid-Phase Synthesis and Receptor Affinities of Octreotide Analogs. J. Med. Chem. 1994, 37, 3749–3757. [Google Scholar] [CrossRef]
- Paterson, B.M.; Roselt, P.; Denoyer, D.; Cullinane, C.; Binns, D.; Noonan, W.; Jeffery, C.M.; Price, R.I.; White, J.M.; Hicks, R.J.; et al. PET imaging of tumours with a 64Cu labeled macrobicyclic cage amine ligand tethered to Tyr3-octreotate. Dalton Trans. 2014, 43, 1386–1396. [Google Scholar] [CrossRef]
- Marciniak, A.; Brasuń, J. Somatostatin analogues labeled with copper radioisotopes: Currrent status. J. Radioanal. Nucl. Chem. 2017, 313, 279–289. [Google Scholar] [CrossRef]
- Pfeifer, A.; Knigge, U.; Binderup, T.; Mortensen, J.; Oturai, P.; Loft, A.; Berthelsen, A.K.; Langer, S.W.; Rasmussen, P.; Elema, D.; et al. 64Cu-DOTATATE PET for Neuroendocrine Tumors: A Prospective Head-to-Head Comparison with 111In-DTPA-Octreotide in 112 Patients. J. Nucl. Med. 2015, 56, 847–854. [Google Scholar] [CrossRef]
- Johnbeck, C.B.; Knigge, U.; Loft, A.; Berthelsen, A.K.; Mortensen, J.; Oturai, P.; Langer, S.W.; Elema, D.R.; Kjaer, A. Head-to-head comparison of 64Cu-DOTATATE and 68Ga-DOTATOC PET/CT: A prospective study of 59 patients with neuroendocrine tumors. J. Nucl. Med. 2017, 58, 451–458. [Google Scholar] [CrossRef]
- Carlsen, E.A.; Johnbeck, C.B.; Binderup, T.; Loft, M.; Pfeifer, A.; Mortensen, J.; Oturai, P.; Loft, A.; Berthelsen, A.K.; Langer, S.W.; et al. 64Cu-DOTATATE PET/CT and prediction of overall and progression-free survival in patients with neuroendocrine neoplasms. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Andersen, T.L.; Baun, C.; Olsen, B.B.; Dam, J.H.; Thisgaard, H. Improving Contrast and Detectability: Imaging with [55Co]Co-DOTATATE in Comparison with [64Cu]Cu-DOTATATE and [68Ga]Ga-DOTATATE. J. Nucl. Med. 2020, 61, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Revheim, M.; Raynor, W.; Zehetner, W.; Knoll, P.; Zandieh, S.; Alavi, A. 64Cu-DOTATOC PET-CT in Patients with Neuroendocrine Tumors. Oncol. Ther. 2019, 8, 125–131. [Google Scholar] [CrossRef]
- Hicks, R.J.; Jackson, P.; Kong, G.; Ware, R.E.; Hofman, M.S.; Pattison, D.A.; Akhurst, T.A.; Drummond, E.; Roselt, P.; Callahan, J.; et al. 64Cu-SARTATE PET Imaging of Patients with Neuroendocrine Tumors Demonstrates High Tumor Uptake and Retention, Potentially Allowing Prospective Dosimetry for Peptide Receptor Radionuclide Therapy. J. Nucl. Med. 2019, 60, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Delpassand, E.S.; Ranganathan, D.; Wagh, N.; Shafie, A.; Gaber, A.; Abbasi, A.; Kjaer, A.; Tworowska, I.; Núñez, R. 64Cu-DOTATATE PET/CT for Imaging Patients with Known or Suspected Somatostatin Receptor-Positive Neuroendocrine Tumors: Results of the First US Prospective, Reader-Blinded Clinical Trial. J. Nucl. Med. 2020, 61, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Heppeler, A.; André, J.P.; Buschmann, I.; Wang, X.; Reubi, J.C.; Hennig, M.; Kaden, T.A.; Maecke, H.R. Metal-ion-dependent biological properties of a chelator-derived somatostatin analogue for tumour targeting. Chem. Eur. J. 2008, 14, 3026–3034. [Google Scholar] [CrossRef] [PubMed]
- Thisgaard, H.; Olsen, B.B.; Dam, J.H.; Bollen, P.; Mollenhauer, J.; Høilund-Carlsen, P.F. Evaluation of cobalt-labeled octreotide analogs for molecular imaging and Auger electron-based radionuclide therapy. J. Nucl. Med. 2014, 55, 1311–1316. [Google Scholar] [CrossRef]
- Thisgaard, H.; Olesen, M.; Dam, J.H. Radiosynthesis of Co-55- and Co-58m-labeled DOTATOC for positron emission tomography imaging and targeted radionuclide therapy. J. Label. Compd. Radiopharm. 2011, 54, 758–762. [Google Scholar] [CrossRef]
- Müller, C.; Domnanich, K.A.; Umbricht, C.A.; van der Meulen, N.P. Scandium and terbium radionuclides for radiotheranostics: Current state of development towards clinical application. Br. J. Radiol. 2018, 91, 20180074. [Google Scholar] [CrossRef]
- Pruszyński, M.; Majkowska-Pilip, A.; Loktionova, N.S.; Eppard, E.; Roesch, F. Radiolabeling of DOTATOC with the long-lived positron emitter 44Sc. Appl. Radiat. Isot. 2012, 70, 974–979. [Google Scholar] [CrossRef]
- Müller, C.; Vermeulen, C.; Johnston, K.; Köster, U.; Schmid, R.; Türler, A.; van der Meulen, N.P. Preclinical in vivo application of 152Tb-DOTANOC: A radiolanthanide for PET imaging. EJNMMI Res. 2016, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; van der Meulen, N.P.; Muller, C.; Klette, I.; Kulkarni, H.R.; Türler, A.; Schibli, R.; Baum, R.P. First-in-human PET/CT imaging of metastatic neuroendocrine neoplasms with cyclotron-produced 44Sc-DOTATOC: A proof-of-concept study. Cancer Biother. Radiopharm. 2017, 32, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Singh, A.; Benešová, M.; Vermeulen, C.; Gnesin, S.; Köster, U.; Johnston, K.; Müller, D.; Senftleben, S.; Kulkarni, H.R.; et al. Clinical evaluation of the radiolanthanide terbium-152: First-in-human PET/CT with 152Tb-DOTATOC. Dalton Trans. 2017, 46, 14638–14646. [Google Scholar] [CrossRef] [PubMed]
- Koumarianou, E.; Pawlak, D.; Korsak, A.; Mikolajczak, R. Comparison of receptor affinity of natSc-DOTA-TATE versus natGa-DOTA-TATE. Nucl. Med. Rev. Cent. East Eur. 2011, 14, 85–89. [Google Scholar] [CrossRef]
- Domnanich, K.A.; Müller, C.; Farkas, R.; Schmid, R.M.; Ponsard, B.; Schibli, R.; Türler, A.; van der Meulen, N.P. 44Sc for labeling of DOTA- and NODAGA-functionalized peptides: Preclinical in vitro and in vivo investigations. EJNMMI Radiopharm. Chem. 2017, 1, 8. [Google Scholar] [CrossRef]
- Sinnes, J.P.; Nagel, J.; Rösch, F. AAZTA5/AAZTA5-TOC: Synthesis and radiochemical evaluation with 68Ga, 44Sc and 177Lu. EJNMMI Radiopharm. Chem. 2019, 4, 18. [Google Scholar] [CrossRef]
- Müller, C.; Fischer, E.; Behe, M.; Köster, U.; Dorrer, H.; Reber, J.; Haller, S.; Cohrs, S.; Blanc, A.; Grünberg, J.; et al. Future prospects for SPECT imaging using the radiolanthanide terbium-155—Production and preclinical evaluation in tumor-bearing mice. Nucl. Med. Biol. 2014, 41, e58–e65. [Google Scholar] [CrossRef]
- Walrand, S.; Jamar, F.; Mathieu, I.; De Camps, J.; Lonneux, M.; Sibomana, M.; Labar, D.; Michel, C.; Pauwels, S. Quantitation in PET using isotopes emitting prompt single gammas: Application to yttrium-86. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 354–361. [Google Scholar] [CrossRef]
- Helisch, A.; Förster, G.J.; Reber, H.; Buchholz, H.G.; Arnold, R.; Göke, B.; Weber, M.M.; Wiedenmann, B.; Pauwels, S.; Haus, U.; et al. Pre-therapeutic dosimetry and biodistribution of 86Y-DOTA-Phe1-Tyr3-octreotide versus 111In-pentetreotide in patients with advanced neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 1386–1392. [Google Scholar] [CrossRef]
- Clifford, T.; Boswell, C.A.; Biddlecombe, G.B.; Lewis, J.S.; Brechbiel, M.W. Validation of a novel CHX-A “derivative suitable for peptide conjugation: Small animal PET/CT imaging using yttrium-86-CHX-A”-octreotide. J. Med. Chem. 2006, 49, 4297–4304. [Google Scholar] [CrossRef]
- Jamar, F.; Barone, R.; Mathieu, I.; Walrand, S.; Labar, D.; Carlier, P.; de Camps, J.; Schran, H.; Chen, T.; Smith, M.C.; et al. 86Y-DOTA0-D-Phe1-Tyr3-octreotide (SMT487)—A phase 1 clinical study: Pharmacokinetics, biodistribution and renal protective effect of different regimens of amino acid co-infusion. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczak, R.; van der Meulen, N.P.; Lapi, S.E. Radiometals for imaging and theranostics, current production, and future perspectives. J. Label. Compd. Radiopharm. 2019, 62, 615–634. [Google Scholar] [CrossRef] [PubMed]
- do Carmo, S.J.C.; Scott, P.J.H.; Alves, F. Production of radiometals in liquid targets. EJNMMI Radiopharm. Chem. 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Talip, Z.; Favaretto, C.; Geistlich, S.; Meulen, N.P.V. A Step-by-Step Guide for the Novel Radiometal Production for Medical Applications: Case Studies with 68Ga, 44Sc, 177Lu and 161Tb. Molecules 2020, 25, 966. [Google Scholar] [CrossRef]
- Waldmann, C.M.; Stuparu, A.D.; van Dam, R.M.; Slavik, R. The search for an alternative to [68Ga]Ga-DOTA-TATE in neuroendocrine tumor theranostics: Current state of 18F-labeled somatostatin analog development. Theranostics 2019, 9, 1336–1347. [Google Scholar] [CrossRef]
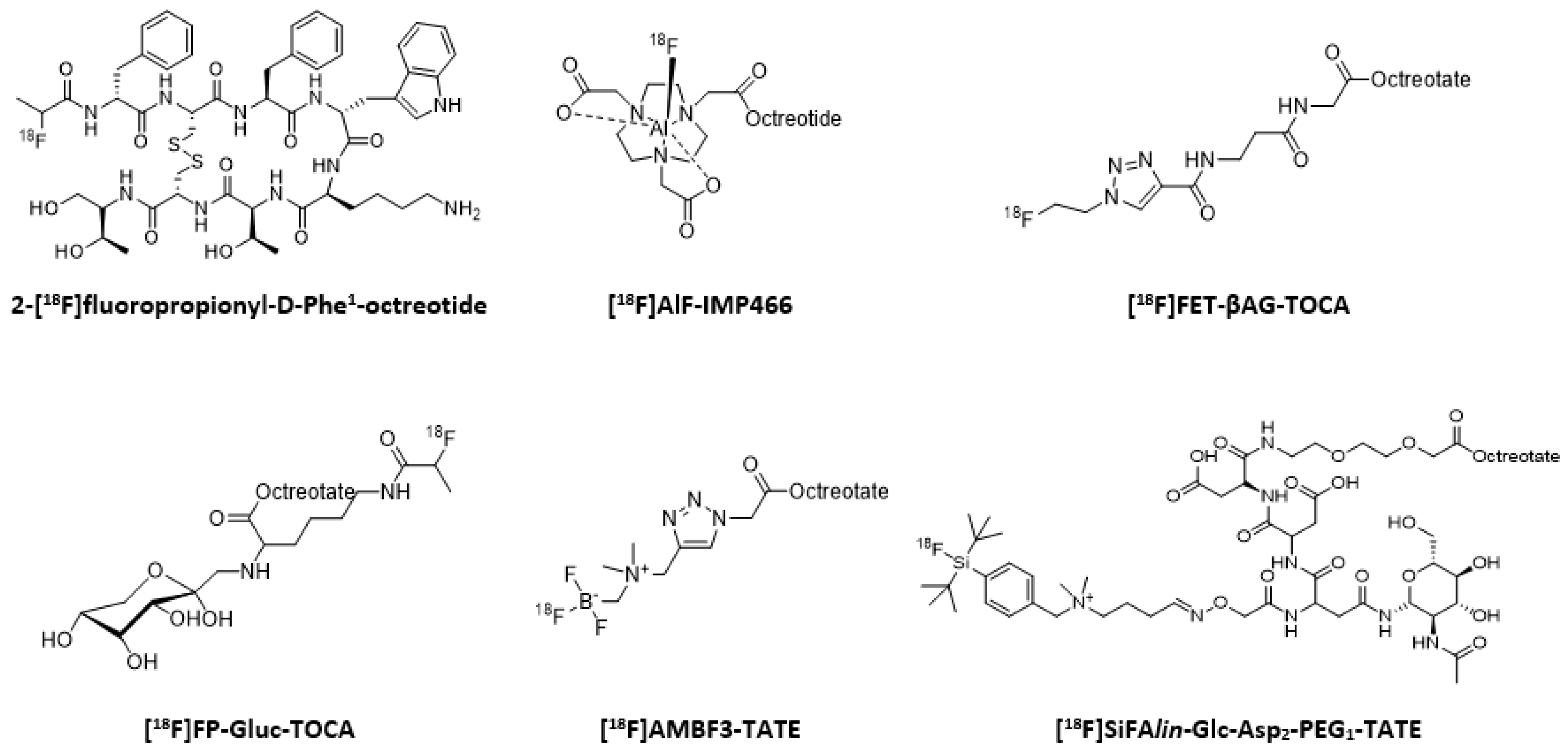
- Guhlke, S.; Wester, H.J.; Bruns, C.; Stöcklin, G. (2-[18F]Fluoropropionyl-(D)phe1)-octreotide, a potential radiopharmaceutical for quantitative somatostatin receptor imaging with PET: Synthesis, Radiolabeling, in vitro validation and biodistribution in mice. Nucl. Med. Biol. 1994, 21, 819–825. [Google Scholar] [CrossRef]
- Hostetler, E.D.; Edwards, W.B.; Anderson, C.J.; Welch, M.J. Synthesis of 4-[18F]fluorobenzoyl octreotide and biodistribution in tumour-bearing Lewis rats. J. Label. Compd Radiopharm. 1999, 42, S720–S722. [Google Scholar]
- Wester, H.J.; Schottelius, M.; Poethko, T.; Bruus-Jensen, K.; Schwaiger, M. Radiolabeled Carbohydrated Somatostatin Analogs: A Review of the Current Status. Cancer Biother. Radiopharm. 2004, 19, 231–244. [Google Scholar] [CrossRef]
- Maschauer, S.; Heilmann, M.; Wängler, C.; Schirrmacher, R.; Prante, O. Radiosynthesis and preclinical evaluation of 18F-fluoroglycosylated octreotate for somatostatin receptor imaging. Bioconjugate Chem. 2016, 27, 2707–2714. [Google Scholar] [CrossRef]
- Liu, Z.; Pourghiasian, M.; Bénard, F.; Pan, J.; Lin, K.S.; Perrin, D.M. Preclinical evaluation of a high-affinity 18F-trifluoroborate octreotate derivative for somatostatin receptor imaging. J. Nucl. Med. 2014, 55, 1499–1505. [Google Scholar] [CrossRef]
- Niedermoser, S.; Chin, J.; Wängler, C.; Kostikov, A.; Bernard-Gauthier, V.; Vogler, N.; Soucy, J.P.; McEwan, A.J.; Schirrmacher, R.; Wängler, B. In vivo evaluation of 18F-SiFAlin-Modified TATE: A potential challenge for 68Ga-DOTATATE, the clinical gold standard for somatostatin receptor imaging with PET. J. Nucl. Med. 2015, 56, 1100–1105. [Google Scholar] [CrossRef]
- Allott, L.; Dubash, S.; Aboagye, E.O. [18F]FET-βAG-TOCA: The Design, Evaluation and Clinical Translation of a Fluorinated Octreotide. Cancers 2020, 12, 865. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Eek, A.; Joosten, L.; Oyen, W.J.G.; Goldenberg, D.M.; Boerman, O.C. A novel facile method of labeling octreotide with 18F-fluorine. J. Nucl. Med. 2010, 51, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Meisetschlaeger, G.; Poethko, T.; Stahl, A.; Wolf, I.; Scheidhauer, K.; Schottelius, M.; Herz, M.; Wester, H.J.; Schwaiger, M. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J. Nucl. Med. 2006, 47, 566–573. [Google Scholar]
- Ilhan, H.; Lindner, S.; Todica, A.; Cyran, C.C.; Tiling, R.; Auernhammer, C.J.; Spitzweg, C.; Boeck, S.; Unterrainer, M.; Gildehaus, F.J.; et al. Biodistribution and first clinical results of 18F-SiFAlin-TATE PET: A novel 18F-labeled somatostatin analog for imaging of neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, E.; Cleeren, F.; Tshibangu, T.; Koole, M.; Serdons, K.; Dekervel, J.; Van Cutsem, E.; Verslype, C.; Van Laere, K.; Bormans, G.; et al. [18F]AlF-NOTA-octreotide PET imaging: Biodistribution, dosimetry and first comparison with [68Ga]Ga-DOTATATE in neuroendocrine tumour patients. Eur. J. Nucl. Med. Mol. Imaging 2020. [Google Scholar] [CrossRef]
- Kemerink, G.J.; Visser, M.G.; Franssen, R.; Beijer, E.; Zamburlini, M.; Halders, S.G.; Brans, B.; Mottaghy, F.M.; Teule, G.J. Effect of the positron range of 18F, 68Ga and 124I on PET/CT in lung-equivalent materials. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 940–948. [Google Scholar] [CrossRef]
- Uccelli, L.; Martini, P.; Cittanti, C.; Carnevale, A.; Missiroli, L.; Giganti, M.; Bartolomei, M.; Boschi, A. Therapeutic Radiometals: Worldwide Scientific Literature Trend Analysis (2008–2018). Molecules 2019, 24, 640. [Google Scholar] [CrossRef]
- Cremonesi, M.; Ferrari, M.E.; Bodei, L.; Chiesa, C.; Sarnelli, A.; Garibaldi, C.; Pacilio, M.; Strigari, L.; Summers, P.E.; Orecchia, R.; et al. Correlation of dose with toxicity and tumour response to 90Y- and 177Lu-PRRT provides the basis for optimization through individualized treatment planning. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2426–2441. [Google Scholar] [CrossRef]
- Otte, A.; Mueller-Brand, J.; Dellas, S.; Nitzsche, E.U.; Herrmann, R.; Maecke, H.R. Yttrium-90 labeled somatostatin-analogue for cancer treatment. Lancet 1998, 351, 417–418. [Google Scholar] [CrossRef]
- Otte, A.; Herrmann, R.; Heppeler, A.; Behe, M.; Jermann, E.; Powell, P.; Maecke, H.R.; Muller, J. Yttrium-90 DOTATOC: First clinical results. Eur. J. Nucl. Med. 1999, 26, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Vinjamuri, S.; Gilbert, T.M.; Banks, M.; McKane, G.; Maltby, P.; Poston, G.; Weissman, H.; Palmer, D.H.; Vora, J.; Pritchard, D.M.; et al. Peptide Receptor Radionuclide Therapy With 90Y-DOTATATE/90Y-DOTATOC in Patients with Progressive Metastatic Neuroendocrine Tumours: Assessment of Response, Survival and Toxicity. Br. J. Cancer 2013, 108, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, I.; Britton, K.; Buscombe, J.; Moncayo, R.; Paganelli, G.; Riva, P. In- and Y-DOTA-lanreotide: Results and implications of the MAURITIUS trial. Semin. Nucl. Med. 2002, 32, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Cremonesi, M.; Grana, C.M.; Chinol, M.; Baio, S.M.; Severi, S.; Paganelli, G. Yttrium-labeled peptides for therapy of NET. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, S93–S102. [Google Scholar] [CrossRef]
- Gabriel, M.; Nilica, B.; Kaiser, B.; Virgolini, I.J. Twelve-Year Follow-up After Peptide Receptor Radionuclide Therapy. J. Nucl. Med. 2019, 60, 524–529. [Google Scholar] [CrossRef]
- Baum, R.P.; Kluge, A.W.; Kulkarni, H.; Schorr-Neufing, U.; Niepsch, K.; Bitterlich, N.; van Echteld, C.J. [177Lu-DOTA]0-D-Phe1-Tyr3-Octreotide (177Lu-DOTATOC) For Peptide Receptor Radiotherapy in Patients with Advanced Neuroendocrine Tumours: A Phase-II Study. Theranostics 2016, 6, 501–510. [Google Scholar] [CrossRef]
- Esser, J.P.; Krenning, E.P.; Teunissen, J.J.; Kooij, P.P.; van Gameren, A.L.; Bakker, W.H.; Kwekkeboom, D.J. Comparison of [177Lu-DOTA0,Tyr3]octreotate and [177Lu-DOTA0,Tyr3]octreotide: Which peptide is preferable for PRRT? Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1346–1351. [Google Scholar] [CrossRef]
- de Jong, M.; Breeman, W.A.; Valkema, R.; Bernard, B.F.; Krenning, E.P. Combination radionuclide therapy using 177Lu- and 90Y-labeled somatostatin analogs. J. Nucl. Med. 2005, 46, 13S–17S. [Google Scholar]
- Kunikowska, J.; Królicki, L.; Hubalewska-Dydejczyk, A.; Mikołajczak, R.; Sowa-Staszczak, A.; Pawlak, D. Clinical results of radionuclide therapy of neuroendocrine tumours with 90Y-DOTATATE and tandem 90Y/177Lu-DOTATATE: Which is a better therapy option? Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1788–1797. [Google Scholar] [CrossRef]
- Kunikowska, J.; Zemczak, A.; Kołodziej, M.; Gut, P.; Łoń, I.; Pawlak, D.; Mikołajczak, R.; Kamiński, G.; Ruchała, M.; Kos-Kudła, B.; et al. Tandem peptide receptor radionuclide therapy using 90Y/177Lu-DOTATATE for neuroendocrine tumors efficacy and side-effects—Polish multicenter experience. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 922–933. [Google Scholar] [CrossRef]
- Brabander, T.; Nonnekens, J.; Hofland, J. The next generation of peptide receptor radionuclide therapy. Endocr. Relat. Cancer 2019, 26, C7–C11. [Google Scholar] [CrossRef] [PubMed]
- Adant, S.; Shah, G.M.; Beauregard, J.M. Combination treatments to enhance peptide receptor radionuclide therapy of neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef]
- Werner, R.A.; Bluemel, C.; Allen-Auerbach, M.S.; Higuchi, T.; Herrmann, K. 68Gallium- and 90Yttrium-/177Lutetium: “theranostic twins” for diagnosis and treatment of NETs. Ann. Nucl. Med. 2015, 29, 1–7. [Google Scholar] [CrossRef]
- Waseem, N.; Aparici, C.M.; Kunz, P.L. Evaluating the Role of Theranostics in Grade 3 Neuroendocrine Neoplasms. J. Nucl. Med. 2019, 60, 882–891. [Google Scholar] [CrossRef]
- Sorbye, H.; Kong, G.; Grozinsky-Glasberg, S. PRRT in high-grade gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Endocr. Relat. Cancer 2020, 27, R67–R77. [Google Scholar] [CrossRef]
- Mak, I.Y.F.; Hayes, A.R.; Khoo, B.; Grossman, A. Peptide Receptor Radionuclide Therapy as a Novel Treatment for Metastatic and Invasive Phaeochromocytoma and Paraganglioma. Neuroendocrinology 2019, 109, 287–298. [Google Scholar] [CrossRef]
- Vyakaranam, A.R.; Crona, J.; Norlén, O.; Granberg, D.; Garske-Román, U.; Sandström, M.; Fröss-Baron, K.; Thiis-Evensen, E.; Hellman, P.; Sundin, A. Favorable Outcome in Patients with Pheochromocytoma and Paraganglioma Treated with 177Lu-DOTATATE. Cancers 2019, 11, 909. [Google Scholar] [CrossRef]
- Lepareur, N.; Lacœuille, F.; Bouvry, C.; Hindré, F.; Garcion, E.; Chérel, M.; Noiret, N.; Garin, E.; Knapp, F.F.R., Jr. Rhenium-188 Labeled Radiopharmaceuticals: Current Clinical Applications in Oncology and Promising Perspectives. Front. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Zamora, P.O.; Gulhke, S.; Bender, H.; Diekmann, D.; Rhodes, B.A.; Biersack, H.J.; Knapp, F.F., Jr. Experimental radiotherapy of receptor-positive human prostate adenocarcinoma with 188Re-RC-160, a directly-radiolabeled somatostatin analogue. Int. J. Cancer 1996, 65, 214–220. [Google Scholar] [CrossRef]
- Zamora, P.O.; Bender, H.; Gulhke, S.; Marek, M.J.; Knapp, F.F., Jr.; Rhodes, B.A.; Biersack, H.J. Pre-clinical experience with Re-188-RC-160, a radiolabeled somatostatin analog for use in peptide-targeted radiotherapy. Anticancer Res. 1997, 17, 1803–1808. [Google Scholar] [PubMed]
- Arteaga de Murphy, C.; Pedraza-López, M.; Ferro-Flores, G.; Murphy-Stack, E.; Chávez-Mercado, L.; Ascencio, J.A.; García-Salinas, L.; Hernández-Gutiérrez, S. Uptake of 188Re-beta-naphthyl-peptide in cervical carcinoma tumours in athymic mice. Nucl. Med. Biol. 2001, 28, 319–326. [Google Scholar] [CrossRef]
- Molina-Trinidad, E.M.; de Murphy, C.A.; Ferro-Flores, G.; Murphy-Stack, E.; Jung-Cook, H. Radiopharmacokinetic and dosimetric parameters of 188Re-lanreotide in athymic mice with induced human cancer tumors. Int. J. Pharm. 2006, 310, 125–130. [Google Scholar] [CrossRef]
- Molina-Trinidad, E.M.; de Murphy, C.A.; Jung-Cook, H.; Stack, E.M.; Pedraza-Lopez, M.; Morales-Marquez, J.L.; Serrano, G.V. Therapeutic 188Re-lanreotide: Determination of radiopharmacokinetic parameters in rats. J. Pharm. Pharmacol. 2010, 62, 456–461. [Google Scholar] [CrossRef]
- Cyr, J.E.; Pearson, D.A.; Wilson, D.M.; Nelson, C.A.; Guaraldi, M.; Azure, M.T.; Lister-James, J.; Dinkelborg, L.M.; Dean, R.T. Somatostatin receptor-binding peptides suitable for tumour radiotherapy with Re-188 or Re-186. Chemistry and initial biological studies. J. Med. Chem. 2007, 50, 1354–1364. [Google Scholar] [CrossRef]
- Edelman, M.J.; Clamon, G.; Kahn, D.; Magram, M.; Lister-James, J.; Line, B.R. Targeted radiopharmaceutical therapy for advanced lung cancer: Phase I trial of rhenium Re188 P2045, a somatostatin analog. J. Thorac. Oncol. 2009, 4, 1550–1554. [Google Scholar] [CrossRef]
- Nelson, C.A.; Azure, M.T.; Adams, C.T.; Zinn, K.R. The somatostatin analog 188Re-P2045 inhibits the growth of AR42J pancreatic tumor xenografts. J. Nucl. Med. 2014, 55, 2020–2025. [Google Scholar] [CrossRef][Green Version]
- Champion, C.; Quinto, M.A.; Morgat, C.; Zanotti-Fregonara, P.; Hindié, E. Comparison between Three Promising ß-emitting Radionuclides, 67Cu, 47Sc and 161Tb, with Emphasis on Doses Delivered to Minimal Residual Disease. Theranostics 2016, 6, 1611–1618. [Google Scholar] [CrossRef]
- De Jong, M.; Breeman, W.A.; Bernard, B.F.; Rolleman, E.J.; Hofland, L.J.; Visser, T.J.; Setyono-Han, B.; Bakker, W.H.; van der Pluijm, M.E.; Krenning, E.P. Evaluation in vitro and in rats of 161Tb-DTPA-octreotide, a somatostatin analogue with potential for intraoperative scanning and radiotherapy. Eur. J. Nucl. Med. 1995, 22, 608–616. [Google Scholar] [CrossRef]
- Loveless, C.S.; Radford, L.L.; Ferran, S.J.; Queern, S.L.; Shepherd, M.R.; Lapi, S.E. Photonuclear production, chemistry, and in vitro evaluation of the theranostic radionuclide 47Sc. EJNMMI Res. 2019, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Doligalski, M.L.; Tichacek, C.J.; Pandya, D.N.; Budzevich, M.M.; El-Haddad, G.; Khushalani, N.I.; Moros, E.G.; McLaughlin, M.L.; Wadas, T.J.; et al. Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules 2019, 24, 4314. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.; Norenberg, J.; Anderson, T.; Atcher, R. A comparison of high- versus low-linear energy transfer somatostatin receptor targeted radionuclide therapy in vitro. Cancer Biother. Radiopharm. 2005, 20, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Norenberg, J.P.; Anderson, T.L.; Prossnitz, E.R.; Stabin, M.G.; Atcher, R.W. Somatostatin-receptor-targeted α-emitting 213Bi is therapeutically more effective than β--emitting 177Lu in human pancreatic adenocarcinoma cells. Nucl. Med. Biol. 2007, 34, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; de Blois, E.; Morgenstern, A.; Bruchertseifer, F.; de Jong, M.; Breeman, W.; Konijnenberg, M. In Vitro comparison of 213Bi- and 177Lu-radiation for peptide receptor radionuclide therapy. PLoS ONE 2017, 12, e0181473. [Google Scholar] [CrossRef]
- Norenberg, J.P.; Krenning, B.J.; Konings, I.R.H.M.; Kusewitt, D.F.; Nayak, T.K.; Anderson, T.L.; de Jong, M.; Garmestani, K.; Brechbiel, M.W.; Kvols, L.K. 213Bi-[DOTA0, Tyr3]Octreotide Peptide Receptor Radionuclide Therapy of Pancreatic Tumors in a Preclinical Animal Model. Clin. Cancer Res. 2006, 12, 897–903. [Google Scholar] [CrossRef]
- Miederer, M.; Henriksen, G.; Alke, A.; Mossbrugger, I.; Quintanilla-Martinez, L.; Senekowitsch-Schmidtke, R.; Essler, M. Preclinical evaluation of the alpha-particle generator nuclide 225Ac for somatostatin receptor radiotherapy of neuroendocrine tumors. Clin. Cancer Res. 2008, 14, 3555–3561. [Google Scholar] [CrossRef]
- Chan, H.S.; Konijnenberg, M.W.; de Blois, E.; Koelewijn, S.; Baum, R.P.; Morgenstern, A.; Bruchertseifer, F.; Breeman, W.A.; de Jong, M. Influence of tumour size on the efficacy of targeted alpha therapy with 213Bi-[DOTA0,Tyr3]-octreotate. EJNMMI Res. 2016, 6, 6. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Zhang, J.; Singh, A.; Kulkarni, H.R.; Schuchardt, C.; Müller, D.; Wester, H.J.; Maina, T.; Rösch, F.; van der Meulen, N.P.; Müller, C.; et al. From Bench to Bedside-The Bad Berka Experience with First-in-Human Studies. Semin. Nucl. Med. 2019, 49, 422–437. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 934–946. [Google Scholar] [CrossRef] [PubMed]
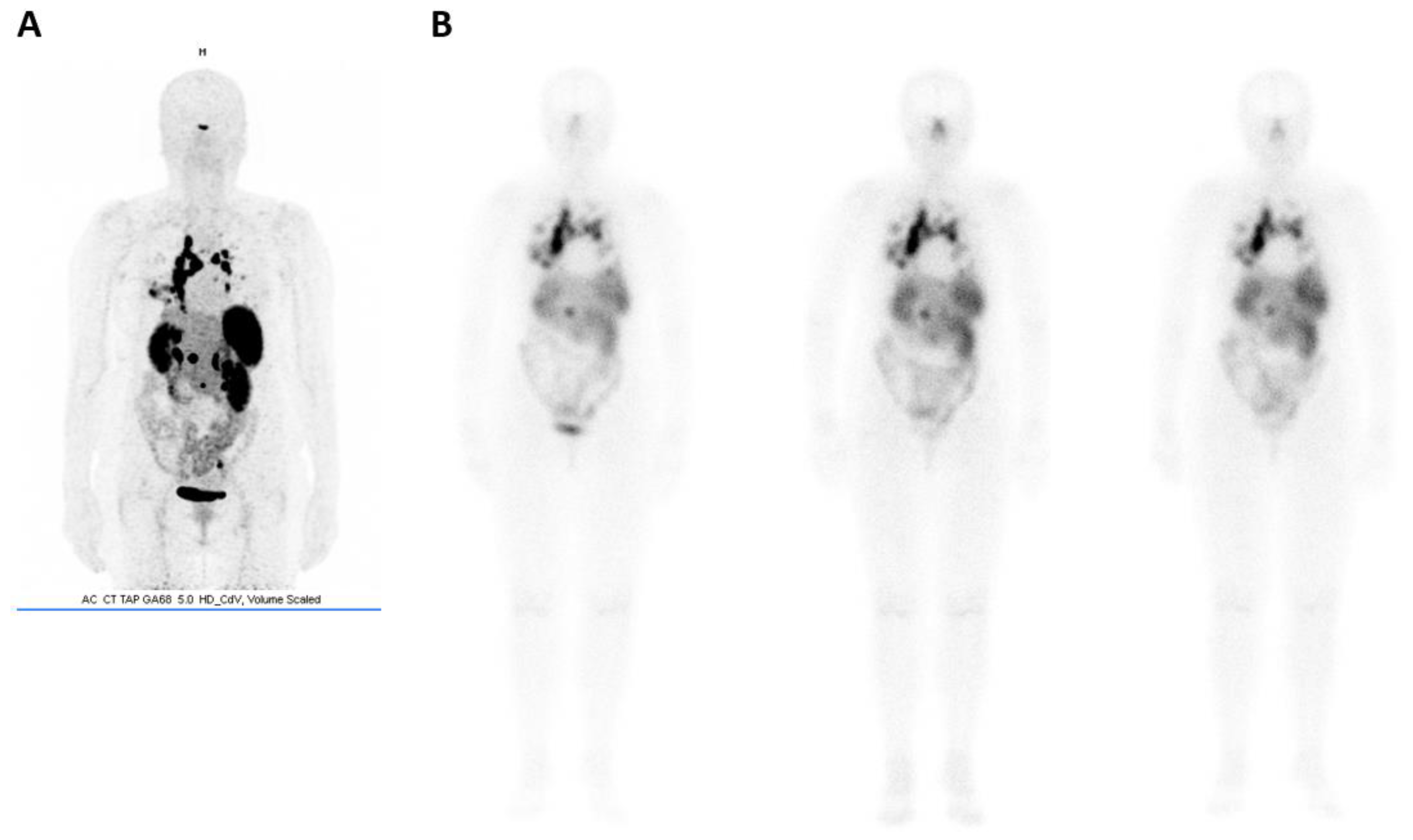
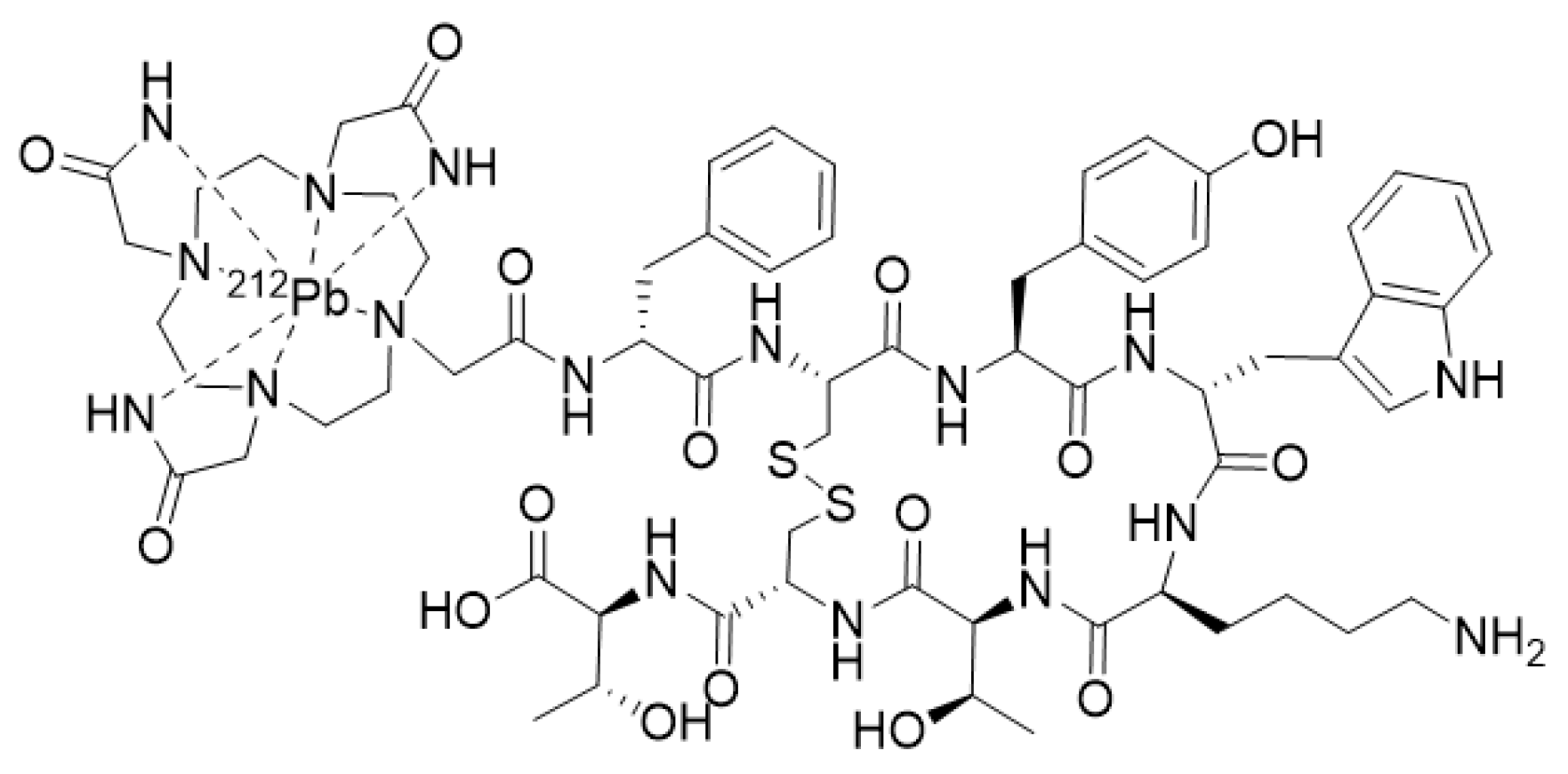
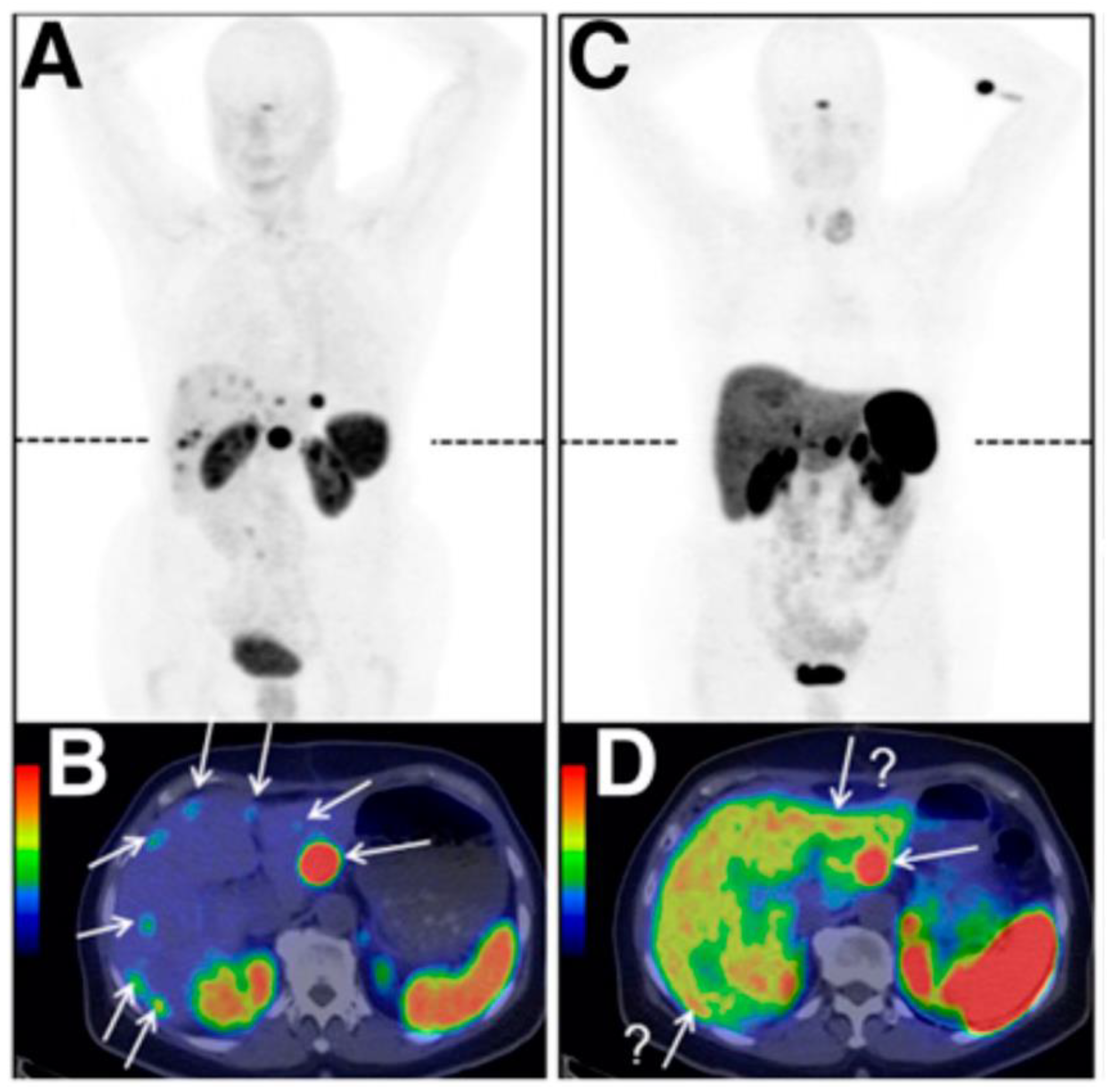
- Stallons, T.A.R.; Saidi, A.; Tworowska, I.; Delpassand, E.S.; Torgue, J.J. Preclinical Investigation of 212Pb-DOTAMTATE for Peptide Receptor Radionuclide Therapy in a Neuroendocrine Tumor Model. Mol. Cancer Ther. 2019, 18, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Tworowska, I.; Delpassand, E.S.; Bolek, L.; Shanoon, F.; Sgouros, G.; Frey, E.; He, B.; Muzammil, A.; Ghaly, M.; Stallons, T.; et al. Targeted Alpha-emitter Therapy of Neuroendocrine Tumors using 212Pb-octreotate (AlphaMedix™). J. Med. Imaging Radiat. Sci. 2019, 50, S34. [Google Scholar] [CrossRef]
- Guérard, F.; Gestin, J.F.; Brechbiel, M.W. Production of [211At]-astatinated radiopharmaceuticals and applications in targeted α-particle therapy. Cancer Biother. Radiopharm. 2013, 28, 1–20. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Boskovitz, A.; Shankar, S.; Zalutsky, M.R. Radioiodine and 211At-labeled guanidinomethyl halobenzoyl octreotate conjugates: Potential peptide radiotherapeutics for somatostatin receptor-positive cancers. Peptides 2004, 25, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; Affleck, D.J.; Schottelius, M.; Wester, H.; Friedman, H.S.; Zalutsky, M.R. Synthesis and evaluation of glycosylated octreotate analogues labeled with radioiodine and 211At via a tin precursor. Bioconjugate Chem. 2006, 17, 195–203. [Google Scholar] [CrossRef]
- Zhao, B.; Qin, S.; Chai, L.; Lu, G.; Yang, Y.; Cai, H.; Yuan, X.; Fan, S.; Huang, Q.; Yu, F. Evaluation of astatine-211-labeled octreotide as a potential radiotherapeutic agent for NSCLC treatment. Bioorg. Med. Chem. 2018, 26, 1086–1091. [Google Scholar] [CrossRef]
- Krenning, E.P.; Valkema, R.; Kooij, P.P.; Breeman, W.A.; Bakker, W.H.; de Herder, W.W.; van Eijck, C.H.; Kwekkeboom, D.J.; de Jong, M.; Pauwels, S. Scintigraphy and radionuclide therapy with [indium-111-labeled-diethyltriamine penta-acetic acid-D-Phe1]-octreotide. Ital. J. Gastroenterol. Hepatol. 1999, 31, S219–S223. [Google Scholar]
- Valkema, R.; De Jong, M.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Lugtenburg, P.J.; De Jong, F.H.; Christiansen, A.; Kam, B.L.; De Herder, W.W.; et al. Phase I study of peptide receptor radionuclide therapy with [In-DTPA]octreotide: The Rotterdam experience. Semin. Nucl. Med. 2002, 32, 110–122. [Google Scholar] [CrossRef]
- Anthony, L.B.; Woltering, E.A.; Espenan, G.D.; Cronin, M.D.; Maloney, T.J.; McCarthy, K.E. Indium-111-pentetreotide prolongs survival in gastroenteropancreatic malignancies. Semin. Nucl. Med. 2002, 32, 123–132. [Google Scholar] [CrossRef]
- Buscombe, J.R.; Caplin, M.E.; Hilson, A.J. Long-term efficacy of high-activity 111In-pentetreotide therapy in patients with disseminated neuroendocrine tumors. J. Nucl. Med. 2003, 44, 1–6. [Google Scholar] [PubMed]
- Lewington, V.J. Targeted radionuclide therapy for neuroendocrine tumours. Endocr. Relat. Cancer 2003, 10, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Capello, A.; Krenning, E.; Bernard, B.; Reubi, J.C.; Breeman, W.; de Jong, M. 111In-labeled somatostatin analogues in a rat tumour model: Somatostatin receptor status and effects of peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 1288–1295. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Tamma, M.L.; Cescato, R.; Maecke, H.R.; Reubi, J.C. Highly efficient in vivo agonist induced internalization of sst2 receptors in somatostatin target tissues. J. Nucl. Med. 2009, 50, 936941. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Schulz, S.; Waser, B.; Eltschinger, V.; Rivier, J.E.; Wester, H.J.; Culler, M.; Ginj, M.; Liu, Q.; Schonbrunn, A.; et al. Internalization of sst2, sst3, and sst5 receptors: Effects of somatostatin agonists and antagonists. J. Nucl. Med. 2006, 47, 502–511. [Google Scholar] [PubMed]
- Fani, M.; Peitl, P.K.; Velikyan, I. Current status of radiopharmaceuticals for the theranostics of neuroendocrine neoplasms. Pharmaceuticals 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Mäcke, H.; Rivier, J. Highly Increased 125I-JR11 Antagonist Binding In Vitro Reveals Novel Indications for sst2 Targeting in Human Cancers. J. Nucl. Med. 2017, 58, 300–306. [Google Scholar] [CrossRef]
- Dude, I.; Zhang, Z.; Rousseau, J.; Hundal-Jabal, N.; Colpo, N.; Merkens, H.; Lin, K.S.; Bénard, F. Evaluation of agonist and antagonist radioligands for somatostatin receptor imaging of breast cancer using positron emission tomography. EJNMMI Radiopharm. Chem. 2017, 2, 4. [Google Scholar] [CrossRef]
- Rylova, S.N.; Stoykow, C.; Del Pozzo, L.; Abiraj, K.; Tamma, M.L.; Kiefer, Y.; Fani, M.; Maecke, H.R. The somatostatin receptor 2 antagonist 64Cu-NODAGA-JR11 outperforms 64Cu-DOTA-TATE in a mouse xenograft model. PLoS ONE 2018, 13, e0195802. [Google Scholar] [CrossRef]
- Krebs, S.; Pandit-Taskar, N.; Reidy, D.; Beattie, B.J.; Lyashchenko, S.K.; Lewis, J.S.; Bodei, L.; Weber, W.A.; O’Donoghue, J.A. Biodistribution and radiation dose estimates for 68Ga-DOTA-JR11 in patients with metastatic neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Behe, M.; Brink, I.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J. Nucl. Med. 2011, 52, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef]
- Reubi, J.C.; Erchegyi, J.; Cescato, R.; Waser, B.; Rivier, J.E. Switch from antagonist to agonist after addition of a DOTA chelator to a somatostatin analog. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 15511558. [Google Scholar] [CrossRef]
- Radford, L.; Gallazzi, F.; Watkinson, L.; Carmack, T.; Berendzen, A.; Lewis, M.R.; Jurisson, S.S.; Papagiannopoulou, D.; Hennkens, H.M. Synthesis and evaluation of a 99mTc tricarbonyl-labeled somatostatin receptor-targeting antagonist peptide for imaging of neuroendocrine tumors. Nucl. Med. Biol. 2017, 47, 4–9. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Fischer, R.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: A pilot study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin Receptor Antagonists for Imaging and Therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef]
- Mansi, R.; Fani, M. Design and development of the theranostic pair 177Lu-OPS201/68Ga-OPS202 for targeting somatostatin receptor expressing tumors. J. Label. Compd. Radiopharm. 2019, 62, 635–645. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Beykan, S.; Bouterfa, H.; Kaufmann, J.; Bauman, A.; Lassmann, M.; Reubi, J.C.; Rivier, J.E.F.; Maecke, H.R.; Fani, M.; et al. Safety, Biodistribution, and Radiation Dosimetry of 68Ga-OPS202 in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase I Imaging Study. J. Nucl. Med. 2018, 59, 909–914. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Schreiter, N.; Kaul, F.; Uiters, J.; Bouterfa, H.; Kaufmann, J.; Erlanger, T.E.; Cathomas, R.; Christ, E.; Fani, M.; et al. Sensitivity Comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase II Imaging Study. J. Nucl. Med. 2018, 59, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gönen, M.; Lohrmann, C.; et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef] [PubMed]
- Rangger, C.; Haubner, R. Radiolabeled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology. Pharmaceuticals 2020, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Villard, L.; Romer, A.; Marincek, N.; Brunner, P.; Koller, M.T.; Schindler, C.; Ng, Q.K.T.; Mäcke, H.R.; Müller-Brand, J.; Rochlitz, C.; et al. Cohort study of somatostatin-based radiopeptide therapy with [90Y-DOTA]-TOC versus [90Y-DOTA]-TOC plus [177Lu-DOTA]-TOC in neuroendocrine cancers. J. Clin. Oncol. 2012, 30, 1100–1106. [Google Scholar] [CrossRef]
- Gill, M.R.; Falzone, N.; Du, Y.; Vallis, K.A. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 2017, 18, e414–e423. [Google Scholar] [CrossRef]
- Hartrampf, P.E.; Hänscheid, H.; Kertels, O.; Schirbel, A.; Kreissl, M.C.; Flentje, M.; Sweeney, R.A.; Buck, A.K.; Polat, B.; Lapa, C. Long-term results of multimodal peptide receptor radionuclide therapy and fractionated external beam radiotherapy for treatment of advanced symptomatic meningioma. Clin. Transl. Radiat. Oncol. 2020, 22, 29–32. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Approaches to multireceptor targeting: Hybrid radioligands, radioligand cocktails, and sequential radioligand applications. J. Nucl. Med. 2017, 58, 10S–16S. [Google Scholar] [CrossRef]
- Ghosh, S.C.; Rodriguez, M.; Carmon, K.S.; Voss, J.; Wilganowski, N.L.; Schonbrunn, A.; Azhdarinia, A. A Modular Dual-Labeling Scaffold That Retains Agonistic Properties for Somatostatin Receptor Targeting. J. Nucl. Med. 2017, 58, 1858–1864. [Google Scholar] [CrossRef]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of Theranostics: An Outlook on Precision Oncology in Nuclear Medicine. J. Nucl. Med. 2019, 60, 13S–19S. [Google Scholar] [CrossRef]
- Jones, W.; Griffiths, K.; Barata, P.C.; Paller, C.J. PSMA Theranostics: Review of the Current Status of PSMA-Targeted Imaging and Radioligand Therapy. Cancers 2020, 12, 1367. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | SSTR Expression | Ref |
|---|---|---|
| Astrocytoma | + | [17] |
| Breast carcinoma | + (SSTR2) | [11] |
| Cholangiocarcinoma | + (SSTR2) | [18] |
| Colorectal carcinoma | - | [17] |
| Endometrial carcinoma | - | [17] |
| Ependymoma | + (SSTR1, SSTR5) | [11] |
| Esophageal carcinoma | - | [17] |
| Ewing sarcoma | - | [17] |
| Exocrine pancreatic tumor | - | [17] |
| Gastric carcinoma | + (SSTR1 > SSTR2, SSTR5) | [11] |
| Gastrinoma | + (SSTR2) | [17] |
| Glioblastoma | - | [17] |
| Growth hormone-producing pituitary adenoma | + (SSTR2, SSTR5) | [17] |
| Gut carcinoid | + (SSTR2 > SSTR1, SSTR5) | [17] |
| Hepatocellular carcinoma | + (SSTR2, SSTR5) | [19] |
| Insulinoma | + (SSTR1, SSTR2, SSTR3) | [20] |
| Leiomyoma | + | [17] |
| Lymphoma | + (SSTR2) | [11] |
| Medullary thyroid carcinoma | + (SSTR2) | [11] |
| Medulloblastoma | + (SSTR2) | [17] |
| Meningioma | + (SSTR2) | [17] |
| Neuroblastoma | + (SSTR2) | [17] |
| Non-functioning pituitary adenoma | + (SSTR3 > SSTR2) | [17] |
| Non-small cell lung cancer | - | [17] |
| Ovarian carcinoma | + | [17] |
| Paraganglioma | + (SSTR2) | [17] |
| Pheochromocytoma | + (SSTR1, SSTR2) | [17] |
| Prostate carcinoma | + (SSTR1) | [17] |
| Renal cell carcinoma | + (SSTR2) | [11] |
| Small cell lung cancer | + (SSTR2) | [17] |
| Urinary bladder carcinoma | - | [17] |
| Radionuclide | Half-Life (h) | Type of Emission | Energy of Emitted Radiation (keV) | Source | Application |
|---|---|---|---|---|---|
| 99mTc | 6.01 | γ | 140 | Generator | SPECT imaging |
| 111In | 67.4 | γ | 172, 245 | Cyclotron | SPECT imaging |
| 18F | 1.83 | β+ | 634 | Cyclotron | PET imaging |
| 64Cu | 12.7 | β+/γ/β- | 653 | Cyclotron | PET imaging |
| 68Ga | 1.1 | β+ | 1190 | Generator/Cyclotron | PET imaging |
| 90Y | 64.1 | β− | 2284 | Generator | Therapy |
| 177Lu | 160.8 | β−/γ | 497 | Cyclotron | Therapy |
| 188Re | 17 | β−/γ | 2118 | Generator | Therapy |
| 211At | 7.2 | α | 5870 | Cyclotron | Therapy |
| 225Ac | 238 | α | 5830 | Generator | Therapy |
| Peptide | Peptidic Sequence |
|---|---|
| OC Octreotide | d-Phe1-cyclo(Cys2-Phe3-d-Trp4-Lys5-Thr6-Cys7)Thr(ol)8 |
| LAN Lanreotide | β-d-Nal1-cyclo(Cys2-Tyr3-d-Trp4-Lys5-Val6-Cys7)Thr8-NH2 |
| VAP Vapreotide | d-Phe1-cyclo(Cys2-Phe3-d-Trp4-Lys5-Val6-Cys7)Trp8-NH2 |
| TOC [Tyr3]-Octreotide | d-Phe1-cyclo(Cys2-Tyr3-d-Trp4-Lys5-Thr6-Cys7)Thr(ol)8 |
| TATE [Tyr3]-Octreotate | d-Phe1-cyclo(Cys2-Tyr3-d-Trp4-Lys5-Thr6-Cys7)Thr8 |
| NOC [1-Nal3]-Octreotide | d-Phe1-cyclo(Cys2-1-Nal3-d-Trp4-Lys5-Thr6-Cys7)Thr(ol)8 |
| NOC-ATE [1-Nal3, Thr8]-Octreotide | d-Phe1-cyclo(Cys2-1-Nal3-d-Trp4-Lys5-Thr6-Cys7)Thr8 |
| BOC [BzThi3]-Octreotide | d-Phe1-cyclo(Cys2-BzThi3-d-Trp4-Lys5-Thr6-Cys7)Thr(ol)8 |
| BOC-ATE [BzThi3, Thr8]-Octreotide | d-Phe1-cyclo(Cys2-BzThi3-d-Trp4-Lys5-Thr6-Cys7)Thr8 |
| Antagonist Peptide | Peptidic Sequence |
|---|---|
| Sst2-ANT (BASS) | p-NO2-Phe1-cyclo(d-Cys2-Tyr3-d-Trp4-Lys5-Thr6-Cys7)d-Tyr8-NH2 |
| LM3 | p-Cl-Phe1-cyclo(d-Cys2-Tyr3-d-Aph4(Cbm)-Lys5-Thr6-Cys7)d-Tyr8-NH2 |
| JR10 | p-NO2-Phe1-cyclo(d-Cys2-Tyr3-d-Aph4(Cbm)-Lys5-Thr6-Cys7)d-Tyr8-NH2 |
| JR11 (Satoreotide) | p-Cl-Phe1-cyclo(d-Cys2-Aph3(Hor)-d-Aph4(Cbm)-Lys5-Thr6-Cys7)d-Tyr8-NH2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules 2020, 25, 4012. https://doi.org/10.3390/molecules25174012
Eychenne R, Bouvry C, Bourgeois M, Loyer P, Benoist E, Lepareur N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules. 2020; 25(17):4012. https://doi.org/10.3390/molecules25174012
Chicago/Turabian StyleEychenne, Romain, Christelle Bouvry, Mickael Bourgeois, Pascal Loyer, Eric Benoist, and Nicolas Lepareur. 2020. "Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy" Molecules 25, no. 17: 4012. https://doi.org/10.3390/molecules25174012
APA StyleEychenne, R., Bouvry, C., Bourgeois, M., Loyer, P., Benoist, E., & Lepareur, N. (2020). Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules, 25(17), 4012. https://doi.org/10.3390/molecules25174012