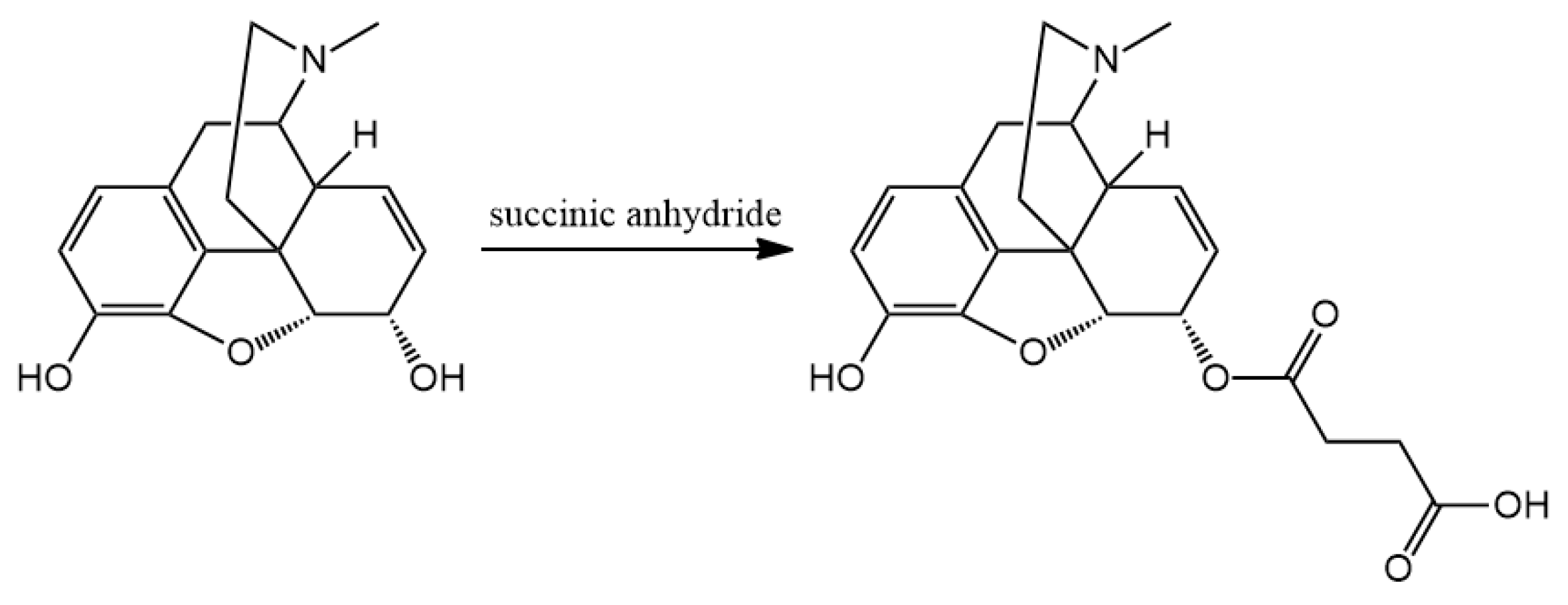
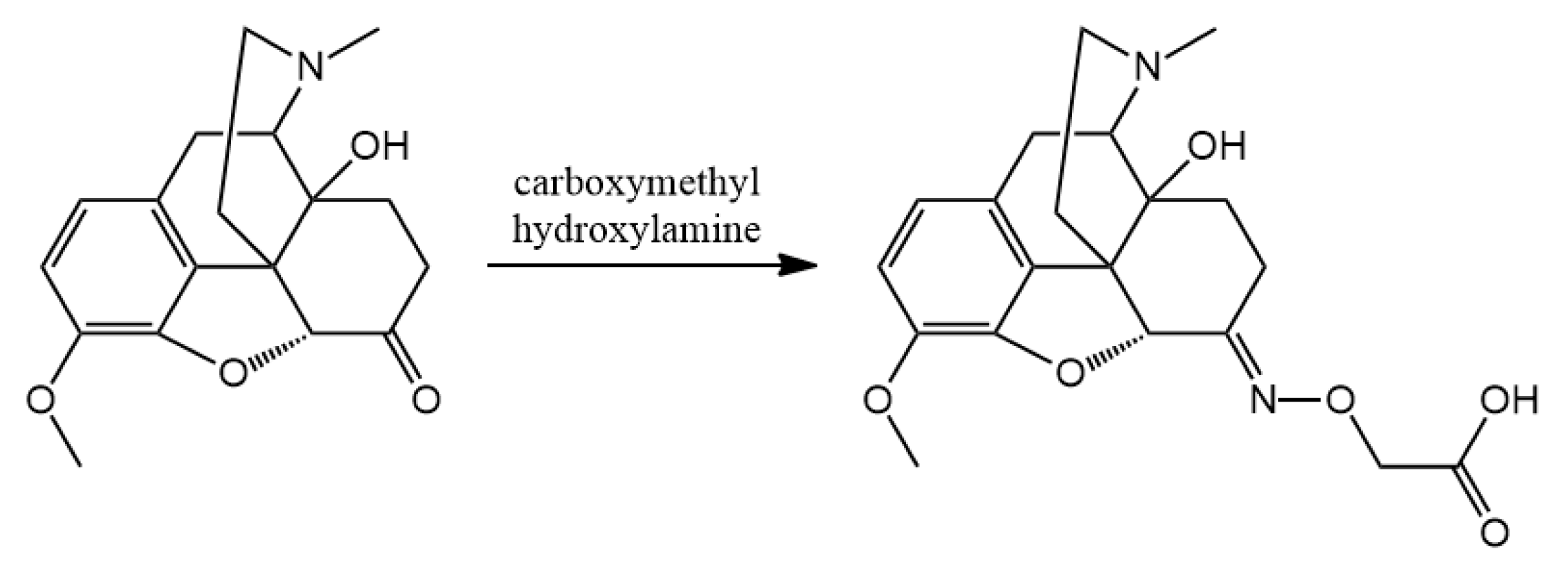
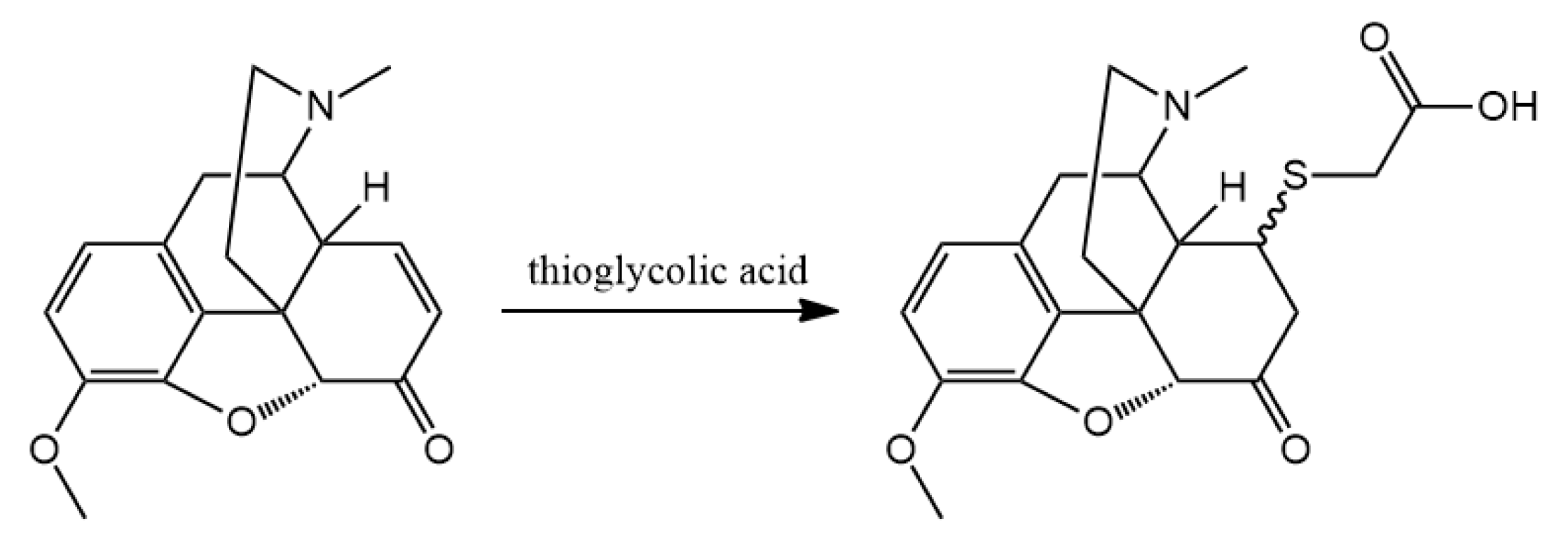
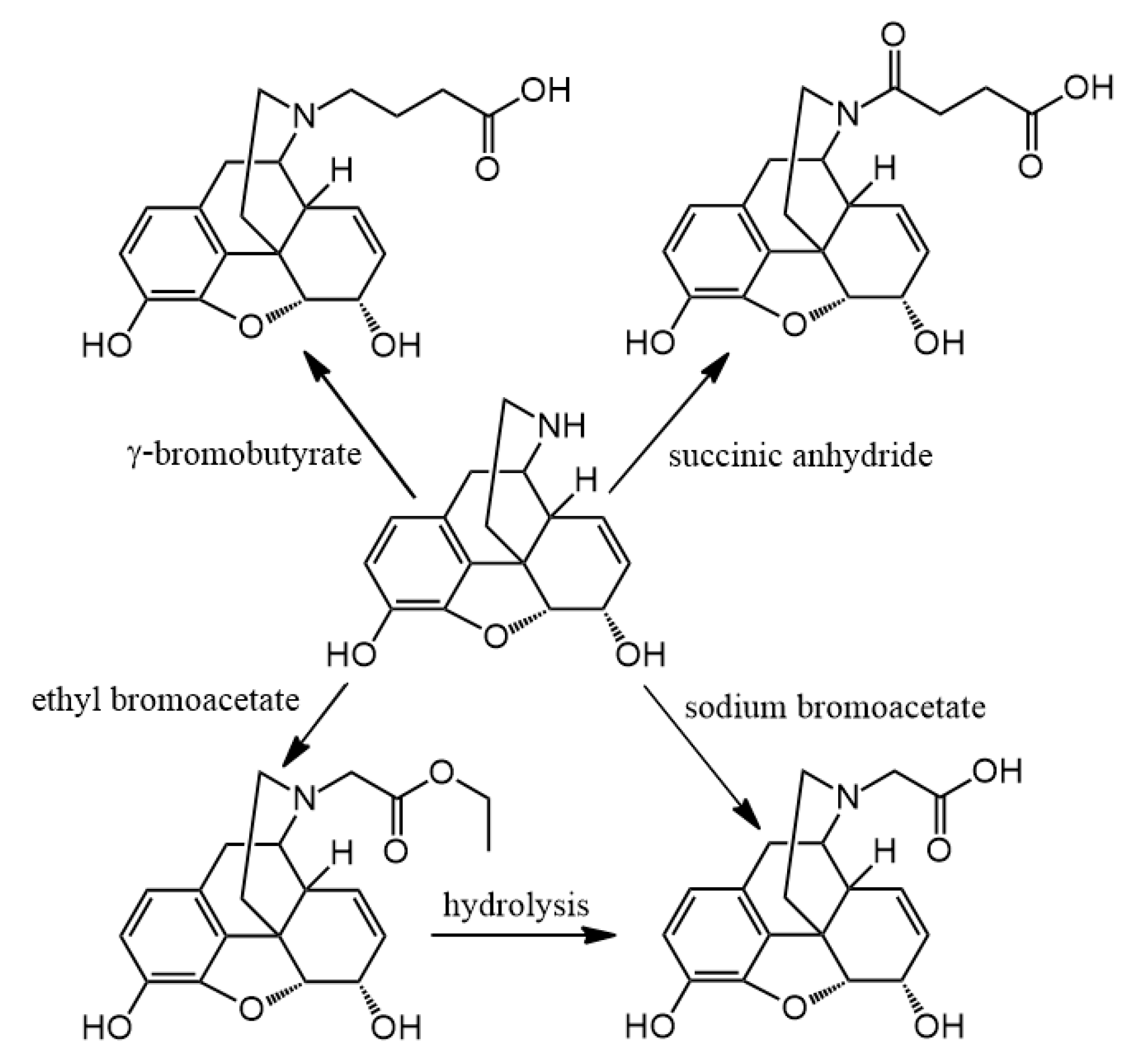
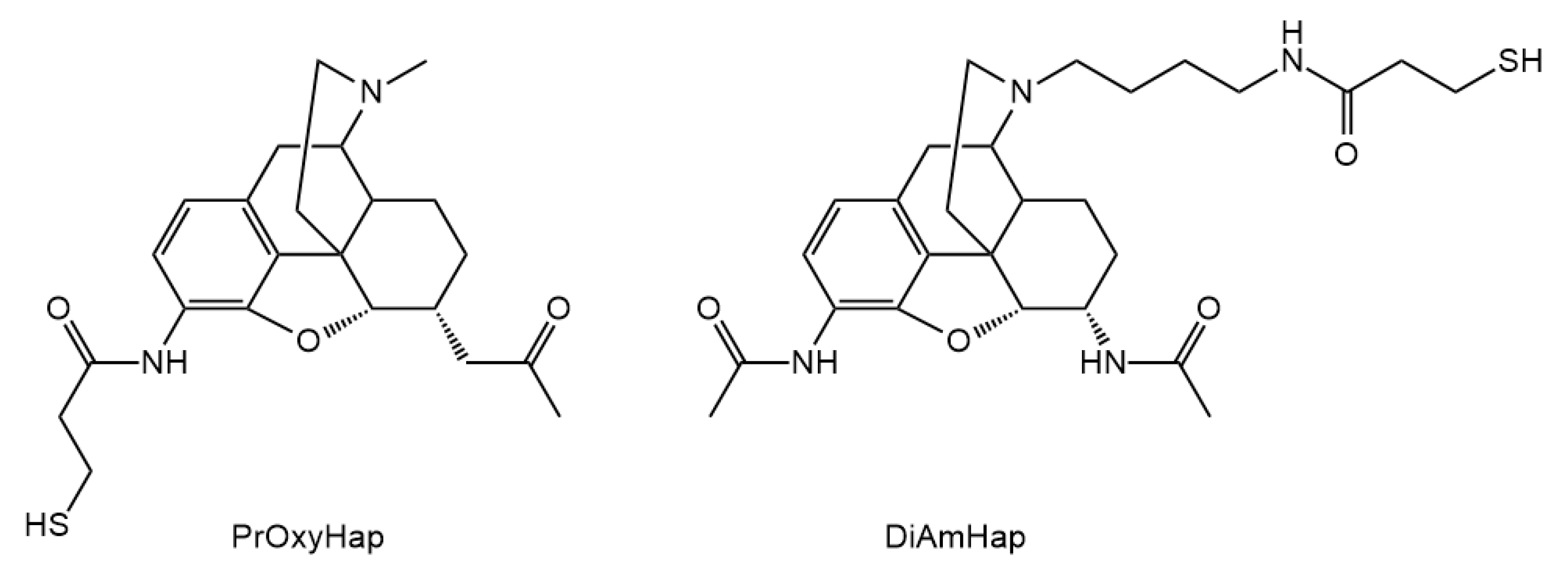
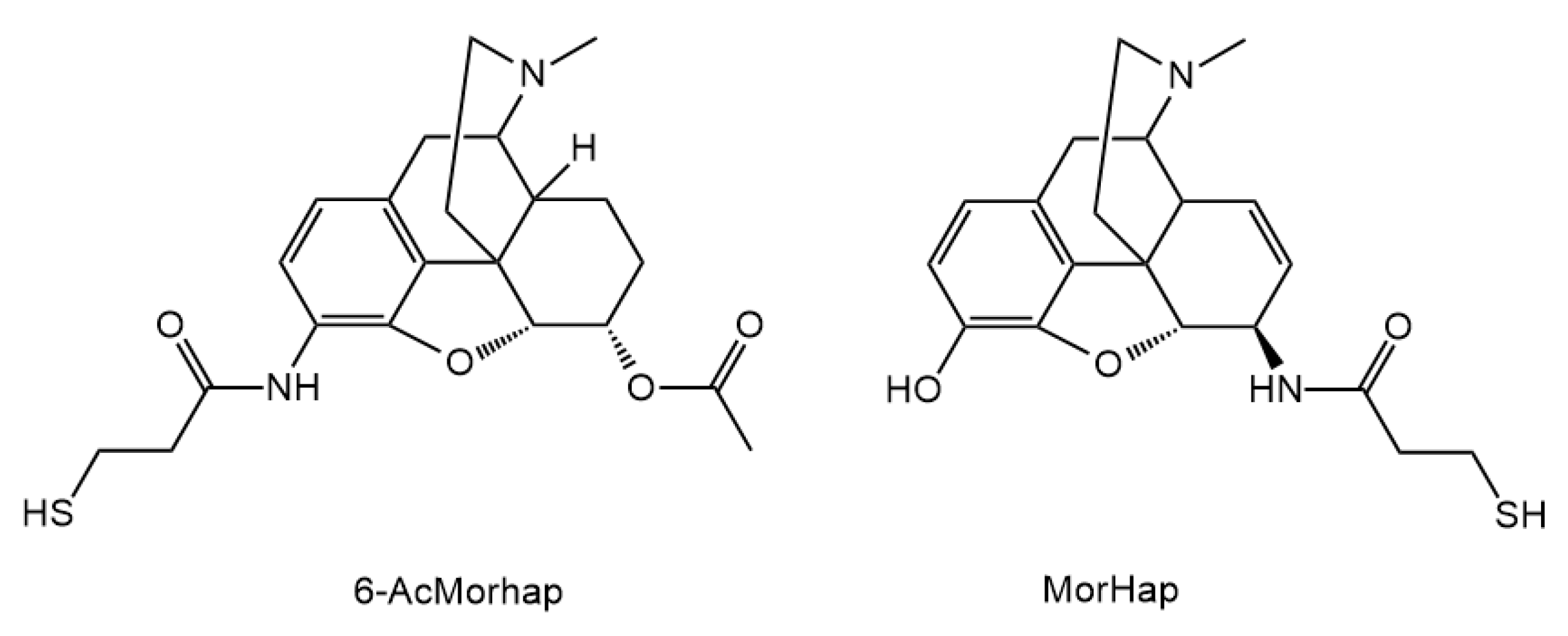
3.2. Hapten Synthesis
Altogether 36 compounds have been synthetized and most of them are new molecules, only
N-carboxymethyl-normorphine ethyl ester, and
N-carboxyethyl-norcodeine ethyl ester were previously synthesized [
42,
43]. The synthesis of nor-compounds (normorphine, dihydronormorphine, norcodeine, dihydronorcodeine, noroxymorphone and noroxycodone) by
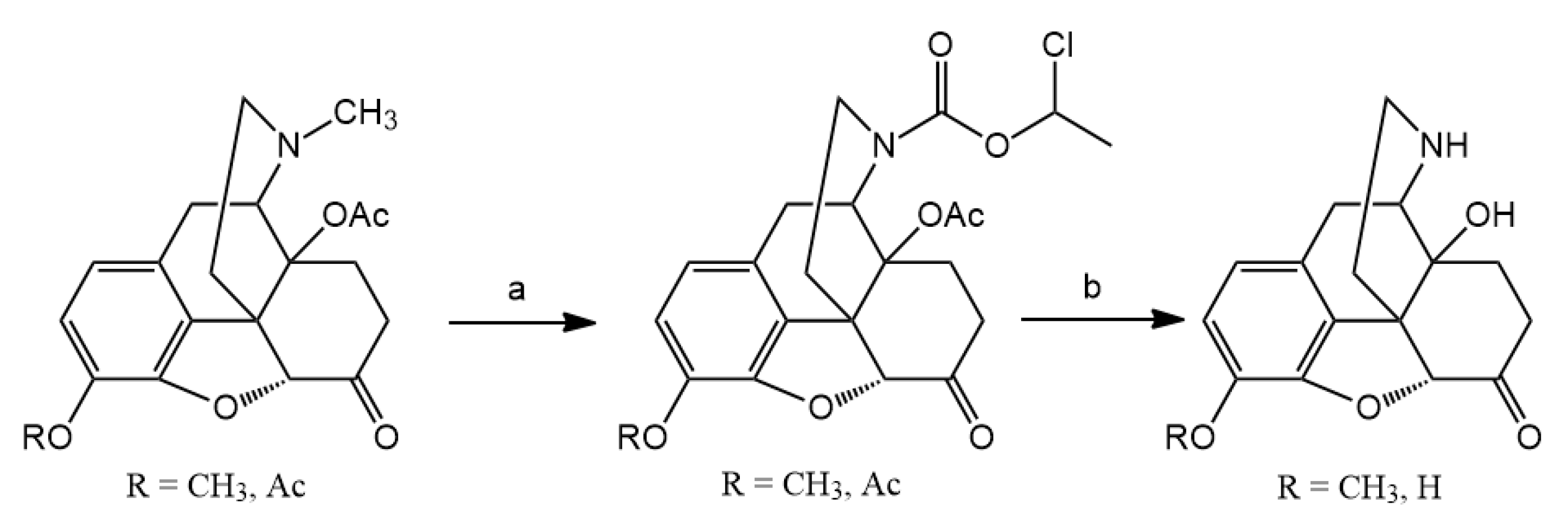
N-demethylation with α-chloroethyl chloroformate was accomplished. Nor-compounds were converted to
N-carboxyethyl methyl derivatives with ethyl bromoacetate and nor-compounds were also converted to
N-carboxyethyl ethyl derivatives with the reaction of ethyl acrylate. The
N-substituted esters were hydrolyzed in order to obtain compounds with free carboxylic acid on nitrogen. The coupling of these free carboxylic groups with amino acid esters was attempted but these reactions were unsuccessful and the expected products were could not be isolated. Therefore, a new reaction was elaborated in order to obtain hapten coupled amino acid esters. The conversion of all nor-compounds was achieved by the reaction with
N-chloroacetyl amino acid esters. Finally, the alkaline hydrolysis of these
N-acetylglycine-normorphinan ethyl esters was achieved too.
3.2.1. N-Demethylation Reactions
Codeine or dihydrocodeine (3 mmol) was dissolved in dried 1,2-dichloroethane (30 mL), then sodium hydrogen carbonate (0.84 g) was added to the solution. To this ice-cold mixture, α-chloroethyl chloroformate (10 mmol) was added dropwise and the reaction mixture was stirred at room temperature for 30 min., then heated at 90 °C overnight. The resulting suspension was cooled to room temperature and inorganic salts were filtered off. The filtrate was evaporated under reduced pressure and the residue was dissolved in methanol and the solution was heated at 60 °C for 6 h. Methanol was removed under reduced pressure to yield a crystalline solid, the hydrochloride salt of norcodeine or dihydronorcodeine. Free base was liberated with 10% sodium hydroxide (pH = 9) and was extracted with chloroform. The chloroform extract was dried (sodium sulfate) and evaporated to result in the nor-compound.
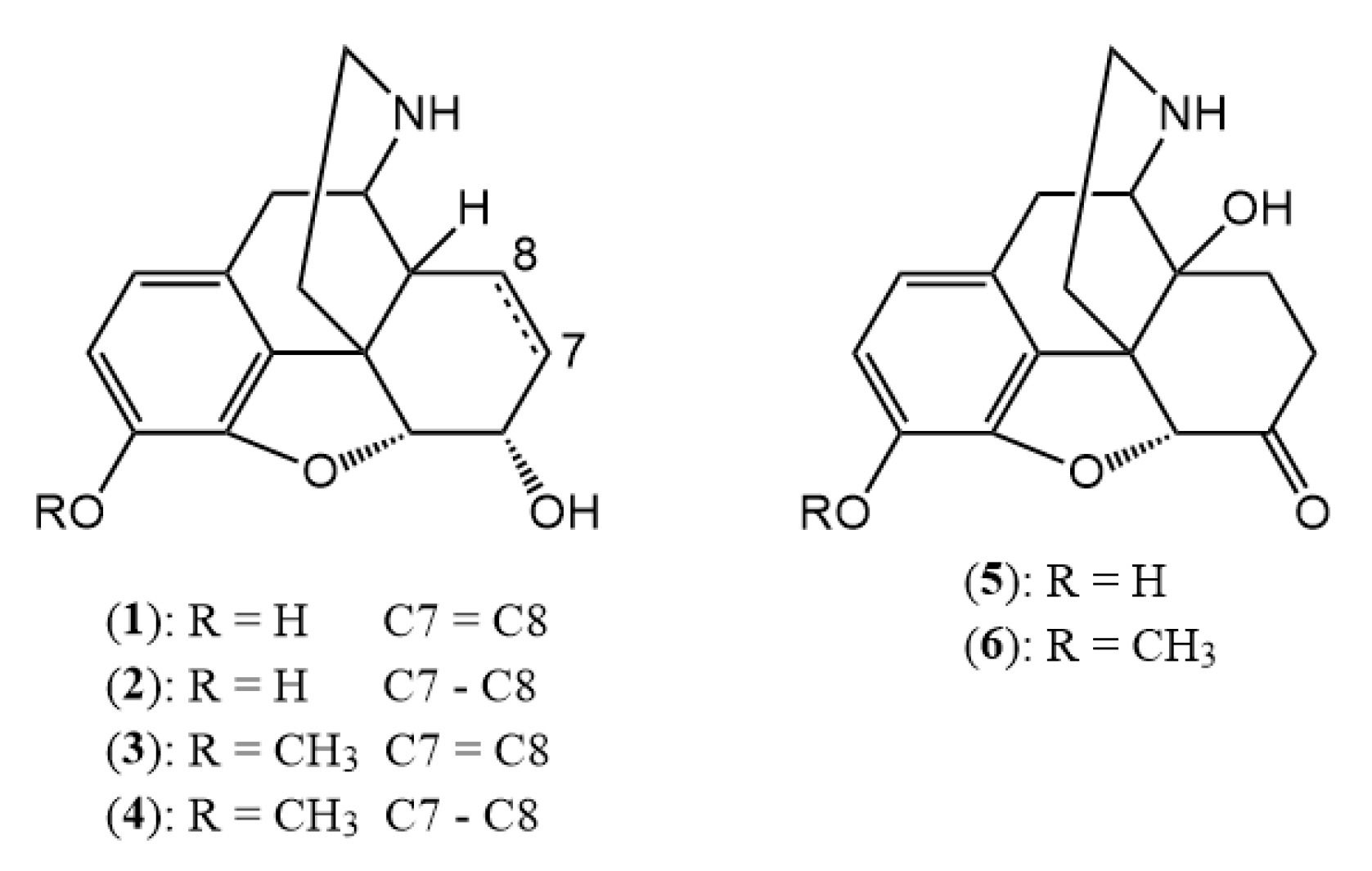
Norcodeine (3) yield: 87%, m.p.: 185 °C (acetone); 1H-NMR (600 MHz, DMSO-d6) δ 6.60 (d, J = 8.2 Hz, H-2), 6.44 (d, J = 8.2 Hz, H-1), 5.52 (dp, J = 9.7, 1.5 Hz, H-7), 5.21 (dt, J = 9.8, 2.8 Hz, H-8), 4.64 (dd, J = 5.9, 1.4 Hz, H-5), 4.09 (dh, J = 5.4, 2.6 Hz, H-6), 3.72 (s, OCH3), 3.46 (dd, J = 5.9, 3.3 Hz, H-9), 2.71 (m, H-10), 2.67 (m, H-16), 2.65 (m, H-10), 2.46 (q, J = 2.8 Hz, H-14), 1.82 (td, J = 12.2, 5.5 Hz, H-15), 1.59 (dt, J = 12.3, 2.4 Hz, H-15); 13C-NMR (150 MHz, DMSO-d6) δ 147.7 (C4), 141.6 (C3), 133.7 (C7), 131.8 (C12), 129.1 (C8), 128.3 (C11), 118.7 (C1), 113.6 (C2), 93.1 (C5), 66.8 (C6), 56.5 (OCH3), 51.5 (C9), 44.3 (C13), 41.1 (C14), 38.4 (C16), 36.9 (C15), 31.3 (C10)
Dihydronorcodeine (4) yield: 83%, m.p.: 194 °C (ethanol); 1H-NMR (600 MHz, DMSO-d6) δ 6.69 (d, J = 8.1 Hz, H-2), 6.53 (d, J = 8.1 Hz, H-1), 4.43 (d, J = 4.9 Hz, H-5), 3.81 (m, H-6), 3.76 (s, OCH3), 3.17 (s, H-9), 2.81 (dd, J = 18.1, 6.2 Hz, H-10), 2.61 (m, H-10), 2.59 (m, H-16), 2.54 (m, H-16), 2.06 (ddd, J = 11.8, 6.7, 2.8 Hz, H-14), 1.66 (td, J = 12.3, 5.2 Hz, H-15), 1.45 (m, H-15), 1.36 (m, H-8), 1.32 (m, H-7), 1.18 (m, H-7), 0.83 (tt, J = 11.9, 6.1 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 147.5 (C4), 141.2 (C3), 130.9 (C12), 127.8 (C11), 118.5 (C1), 114.4 (C2), 91.1 (C5), 66.3 (C6), 56.7 (OCH3), 52.3 (C9), 43.0 (C13), 39.1 (C14), 38.4 (C16), 38.1 (C15), 30.5 (C10), 26.4 (C7), 20.2 (C8)
(Dihydro)normorphine was prepared by N-demethylation of diacetyl (dihydro)morphine by means of α-chloro-ethyl chloroformate using the above-mentioned procedure and the carbamate was treated with methanol at 60 °C to afford the diacetyl (dihydro)normorphine hydrochloride salt. (Dihydro)normorphine was obtained by hydrolysis of diacetyl (dihydro)normorphine × HCl in 5% HCl at 100 °C for 4 h. (Dihydro)normorphine base was precipitated with 25% ammonia solution (pH = 9–10) and filtered off.
Normorphine (1) yield: 84%, m.p.: 273–275 °C; 1H-NMR (600 MHz, DMSO-d6) δ 6.46 (d, J = 8.0 Hz, H-2), 6.35 (d, J = 8.1 Hz, H-1), 5.55 (dp, J = 9.9, 1.5 Hz, H-7), 5.20 (dt, J = 9.8, 2.8 Hz, H-8), 4.67 (dd, J = 6.1, 1.3 Hz, H-5), 4.07 (dd, J = 6.1, 3.1 Hz, H-6), 3.72 (m, H-9), 2.88 (dd, J = 13.2, 4.7 Hz, H-16), 2.73 (m, H-10), 2.72 (m, H-16), 2.55 (p, J = 2.8 Hz, H-14), 1.92 (td, J = 13.0, 5.0 Hz, H-15), 1.68 (m, H-15), 1.20 (s, H-10); 13C-NMR (150 MHz, DMSO-d6) δ 146.7 (C4), 139.2 (C3), 134.5 (C7), 130.7 (C12), 127.6 (C8), 124.7 (C11), 119.1 (C1), 117.1 (C2), 92.1 (C5), 66.4 (C6), 51.4 (C9), 43.6 (C13), 39.6 (C14), 37.9 (C16), 35.0 (C15), 29.2 (C10)
Dihydronormorphine (2) yield: 80%, m.p.: 265 °C (ethanol); 1H-NMR (600 MHz, DMSO-d6) δ 6.53 (d, J = 7.9 Hz, H-2), 6.42 (d, J = 8.0 Hz, H-1), 4.42 (d, J = 4.9 Hz, H-5), 3.81 (ddd, J = 8.6, 4.9, 3.5 Hz, H-6), 3.23 (dd, J = 6.2, 2.8 Hz, H-9), 2.79 (dd, J = 18.2, 6.3 Hz, H-10), 2.64 (m, H-16), 2.62 (m, H-10), 2.57 (m, J-16), 2.07 (ddd, J = 11.6, 6.7, 2.9 Hz, H-14), 1.69 (td, J = 12.5, 5.0 Hz, H-15), 1.48 (ddd, J = 12.5, 3.7, 1.5 Hz, H-15), 1.37 (m, H-8), 1.31 (m, H-7), 1.19 (m, H-7), 0.83 (tt, J = 12.2, 6.2 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 146.4 (C4), 138.4 (C3), 130.3 (C12), 125.4 (C11), 118.4 (C1), 117.2 (C2), 90.5 (C5), 66.4 (C6), 52.3 (C9), 43.1 (C13), 38.6 (C14), 38.3 (C16), 37.7 (C15), 29.9 (C10), 26.3 (C7), 20.1 (C8)
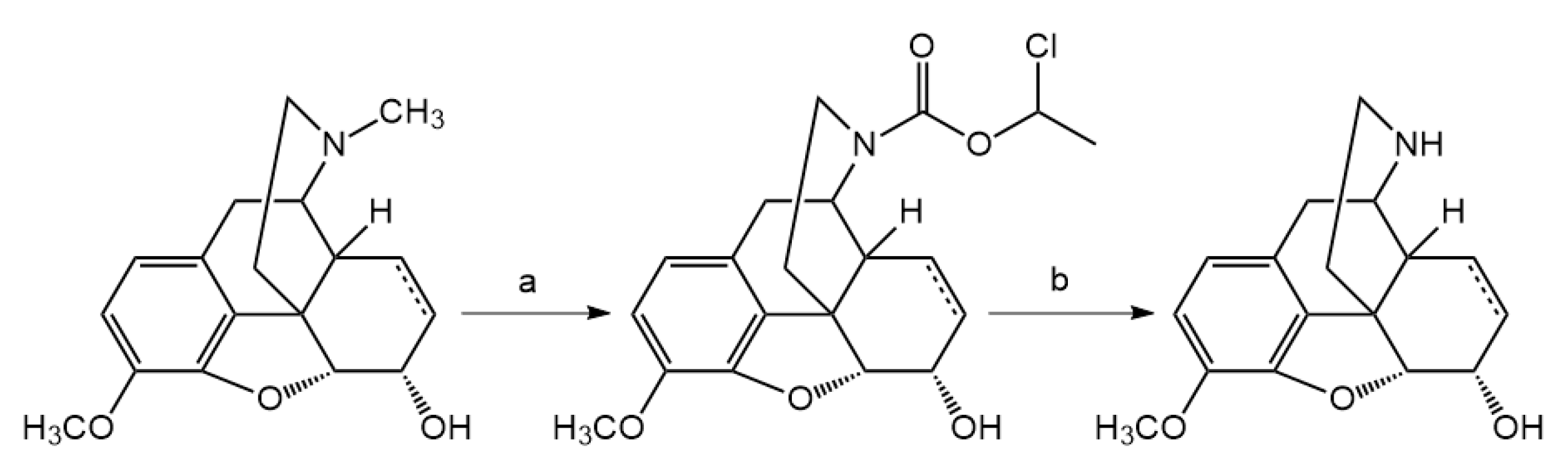
14-O-Acetyloxycodone (2 mmol) was N-demethylated with α-chloro-ethyl chloroformate (10 mmol) for 16 h. The reaction was monitored by thin-layer chromatography to check the conversion, if it was necessary, another 5 mmol portion of α-chloro-ethyl chloroformate was added. The carbamate intermediate was decomposed in methanol to yield the hydrochloride salt of 14-O-acetylnoroxycodone. Acid hydrolysis in refluxing 10% HCl for 6 h resulted in the hydrochloride salt of noroxycodone. The free base of noroxycodone was liberated from the acid solution with 10% sodium hydroxide (pH = 10) and it was extracted with chloroform. The chloroform extract was dried under sodium sulfate and after evaporation of the solution, afforded the oily noroxycodone, which was rubbed with diethyl ether to produce crystalline material.
Noroxycodone (6) yield: 86%, m.p.: 163–166 °C; 1H-NMR (600 MHz, DMSO-d6) δ 6.70 (d, J = 8.2 Hz, H-2), 6.62 (d, J = 8.2 Hz, H-1), 4.70 (s, H-5), 3.78 (s, OCH3), 2.96 (m, H-10), 2.94 (m, H-9), 2.91 (m, H-7), 2.88 (m, H-10), 2.58 (dd, J = 13.3, 4.7 Hz, H-16), 2.34 (td, J = 12.6, 3.2 Hz, H-16), 2.30 (td, J = 12.0, 4.5 Hz, H-15), 2.08 (dt, J = 14.1, 3.2 Hz, H-7), 1.74 (ddd, J = 13.6, 5.1, 2.9 Hz, H-8), 1.40 (td, J = 14.1, 3.4 Hz, H-8), 1.14 (dd, J = 12.1, 3.0 Hz, H-15); 13C-NMR (150 MHz, DMSO-d6) δ 208.9 (C6), 144.9 (C4), 142.3 (C3), 130.3 (C12), 126.8 (C11), 119.5 (C1), 115.1 (C2), 90.5 (C5), 70.1 (C14), 57.3 (C9), 56.8 (OCH3), 51.0 (C13), 37.9 (C16), 36.3 (C7), 32.2 (C10), 32.0 (C8), 30.2 (C15)
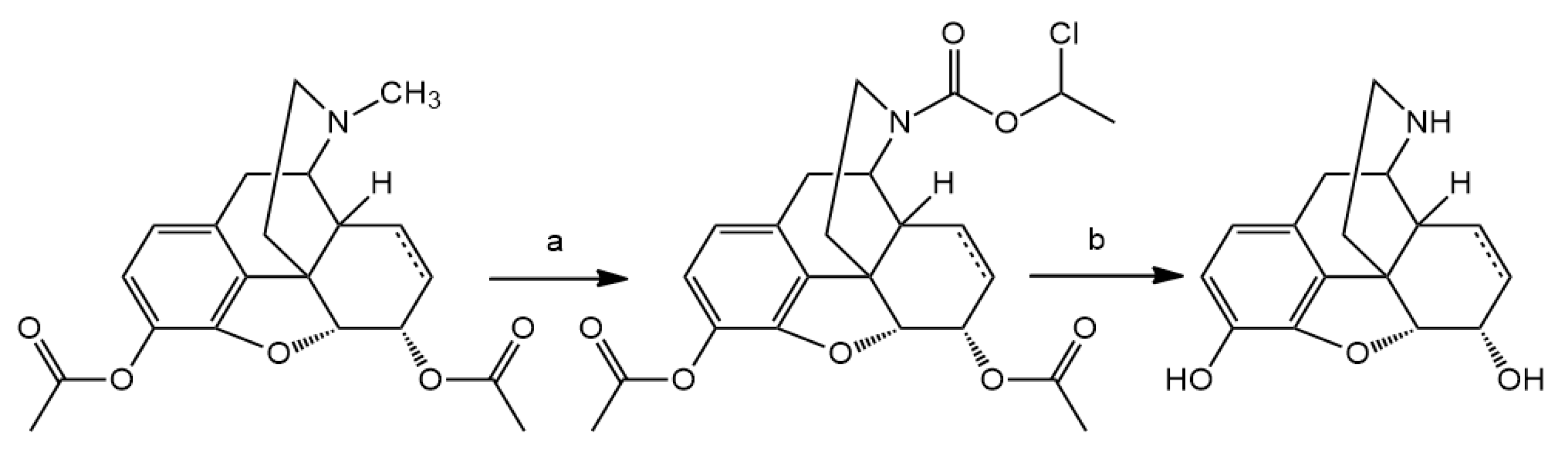
N-Demethylation of 3,14-di-O-acetyloxymorphone yielded the hydrochloride salt of 3,14-di-O-acetylnoroxymorphone, which was hydrolyzed in 10% HCl solution. The noroxymorphone base was precipitated from the acid solution with 25% ammonia solution to yield noroxymorphone which is pure, for further reactions.
Noroxymorphone (5) yield: 81%, m.p.: >280 °C; 1H-NMR (600 MHz, DMSO-d6) δ 6.52 (d, J = 8.1 Hz, H-2), 6.49 (d, J = 8.1 Hz, H-1), 4.64 (s, H-5), 2.97 (m, H-9), 2.91 (m, H-10), 2.90 (m, H-7), 2.83 (m, H-10), 2.62 (dd, J = 13.1, 4.6 Hz, H-16), 2.37 (td, J = 12.8, 3.6 Hz, H-16), 2.29 (dd, J = 12.3, 4.8 Hz, H-15), 2.07 (dt, J = 14.2, 3.2 Hz, H-7), 1.74 (ddd, J = 13.6, 5.2, 2.9 Hz, H-8), 1.42 (td, J = 14.0, 3.5 Hz, H-8), 1.15 (dd, J = 12.1, 3.2 Hz, H-15); 13C-NMR (150 MHz, DMSO-d6) δ 209.1 (C6), 143.9 (C4), 139.8 (C3), 130.0 (C12), 124.4 (C11), 119.4 (C1), 117.6 (C2), 89.9 (C5), 70.1 (C14), 57.3 (C9), 50.9 (C13), 37.8 (C16), 36.3 (C7), 31.83 (C10), 31.77 (C8), 30.0 (C15)
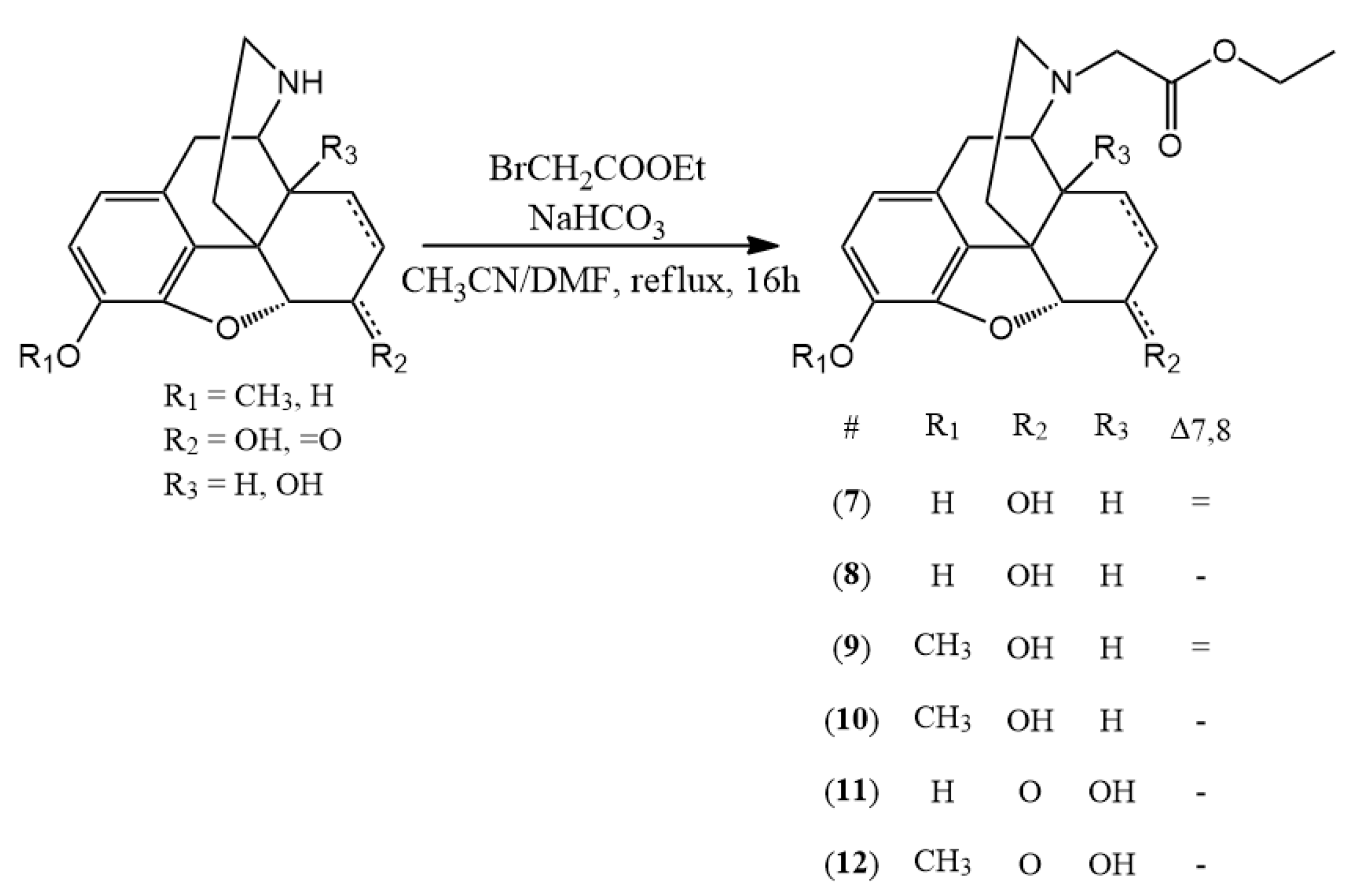
3.2.2. General Synthesis of N-Carboxymethyl-Nor-Compound Ethyl Esters
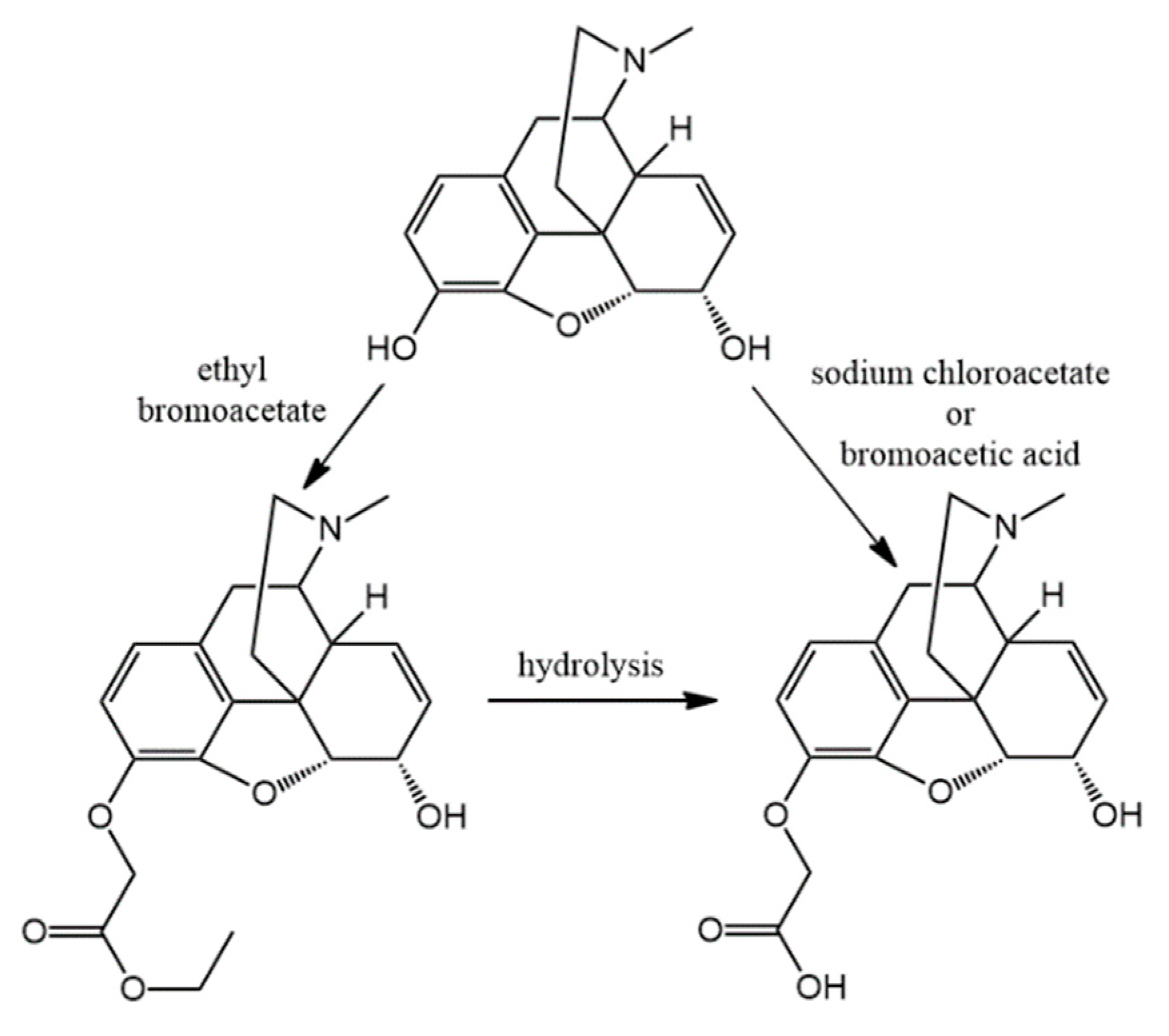
The appropriate normorphine derivative (2 mmol) was dissolved in 30 mL of acetonitrile. In the presence of sodium bicarbonate (10 mmol) ethyl bromoacetate (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 16 h. TLC monitoring showed that conversion was complete. The inorganic salts were filtered and the solvent was evaporated. Water (30 mL) was added to the residue and the pH was set around nine with cc. ammonia. Then it was extracted with chloroform (3 × 25 mL) and after unifying the organic phases, it was dried on sodium sulfate. After filtration the chloroform was evaporated. If it was necessary, column chromatography was used. (chloroform: methanol 9:1)
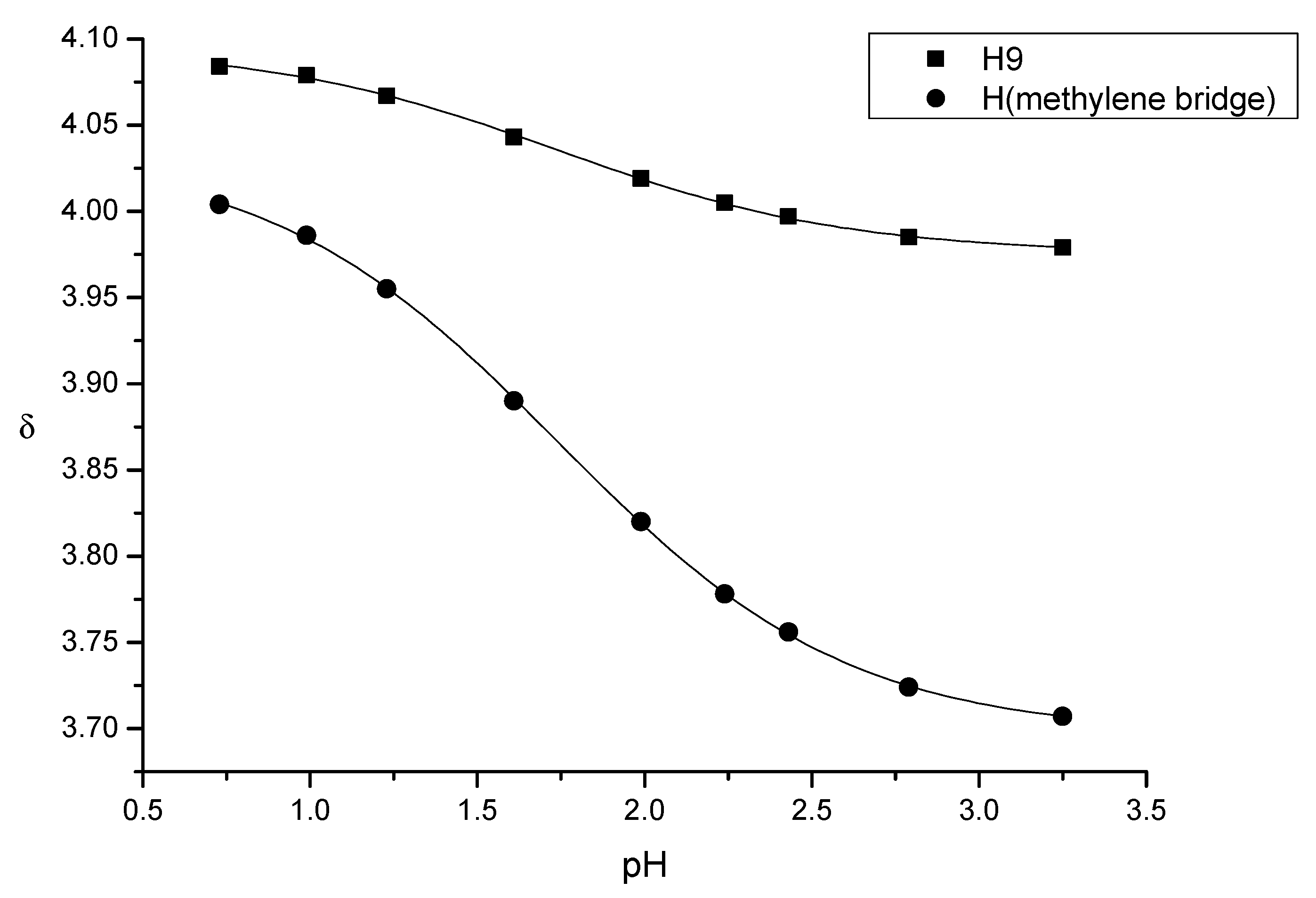
(7) N-carboxymethyl-normorphine ethyl ester (ethyl 2-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 86%, m.p.: 116 °C, HR-MS [M + H+]: calculated: 358.1649, measured: 358.1636; 1H-NMR (600 MHz, DMSO-d6) δ 6.40 (d, J = 8.0 Hz, H-2), 6.31 (d, J = 8.0 Hz, H-1), 5.49 (m, H-7), 5.19 (dt, J = 9.7, 2.8 Hz, H-8), 4.64 (dd, J = 6.1, 1.3 Hz, H-5), 4.09 (dd, J = 6.1, 3.1 Hz, H-6), 4.06 (q, J = 7.1 Hz, ester CH2), 3.39 (s, CH2 bridge), 3.35 (m, H-9), 3.23 (d, J = 16.5 Hz, CH2 bridge), 2.77 (d, J = 18.5 Hz, H-10) 2.58 (m, H-16), 2.55 (m, H-14), 2.31 (td, J = 12.2, 3.4 Hz, H-16), 2.26 (dd, J = 18.5, 6.4 Hz, H-10), 1.98 (m, H-15), 1.58 (dt, J = 11.0, 2.2 Hz, H-15), 1.16 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 170.9 (ester C=O), 146.6 (C4), 138.9 (C3), 133.8 (C7), 131.3 (C12), 128.6 (C8), 125.7 (C11), 118.9 (C1), 116.7 (C2), 91.9 (C5), 66.7 (C6), 60.4 (ester CH2), 57.2 (C9), 56.5 (CH2 bridge), 44.9 (C16), 43.5 (C13), 40.66 (C14), 35.6 (C15), 22.4 (C10), 14.5 (ester CH3)
(8) N-carboxymethyl-dihydronormorphine ethyl ester (ethyl 2-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 84%, m.p.: 86 °C, HR-MS [M + H+]: calculated: 360.1805, measured: 360.1789; 1H-NMR (600 MHz, DMSO-d6) δ 6.49 (d, J = 8.0 Hz, H-2), 6.38 (d, J = 8.0 Hz, H-1), 4.41 (d, J = 4.8 Hz, H-5), 4.04 (q, J = 7.1 Hz, ester CH2), 3.79 (dd, J = 9.1, 4.4 Hz, H-6), 3.31 (s, (CH2 bridge), 3.15 (d, J = 16.4 Hz, (CH2 bridge), 3.02 (dd, J = 6.0, 2.8 Hz, H-9), 2.71 (d, J = 18.3 Hz, H-10), 2.49 (m, H-16) 2.31 (dd, J = 18.4, 6.0 Hz, H-10), 2.18–2.09 (m, H-14, H-15), 1.78 (d, J = 5.1 Hz, H-15), 1.44–1.38 (m, H-15), 1.35 (dd, J = 13.4, 6.8 Hz, H-8), 1.28 (ddt, J = 12.8, 6.5, 3.9 Hz, H-7), 1.15 (t, J = 7.1 Hz, ester CH3), 1.11 (m, H-7), 0.83 (tt, J = 12.5, 6.4 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 171.0 (ester C=O), 146.4 (C4), 138.4 (C3), 130.4 (C12), 125.4 (C11), 118.4 (C1), 117.1 (C2), 90.3 (C5), 66.5 (C6), 60.4 (ester CH2), 58.3 (C9), 56.8 (CH2 bridge), 44.9 (C16), 42.2 (C13), 38.2 (C14), 37.4 (C15), 26.0 (C7), 21.9 (C10), 19.9 (C8), 14.6 (ester CH3)
(9) N-carboxymethyl-norcodeine ethyl ester (ethyl 2-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 90%, m.p.: oil, HR-MS [M + H+]: calculated: 372.1805, measured: 372.1800; 1H-NMR (600 MHz, DMSO-d6) δ 6.58 (d, J = 8.2 Hz, H-2), 6.42 (d, J = 8.2 Hz, H-1), 5.49 (ddt, J = 9.8, 3.2, 1.5 Hz, H-7), 5.19 (dt, J = 9.8, 2.8 Hz, H-8), 4.66 (dd, J = 6.0, 1.3 Hz, H-5), 4.10 (tq, J = 5.7, 2.7 Hz, H-6), 4.05 (q, J = 7.1 Hz, ester CH2), 3.70 (d, J = 2.2 Hz, CH2 bridge), 3.69 (s, OCH3), 3.23 (d, J = 16.6 Hz, CH2 bridge), 2.81 (d, J = 18.6 Hz, H-10), 2.57 (dp, J = 8.8, 3.3, 2.6 Hz, H-16), 2.56 (m, H-14), 2.34–2.25 (m, H-10, H-16), 1.98 (td, J = 12.6, 5.0 Hz, H-15), 1.62–1.53 (m, H-15), 1.16 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 170.6 (ester C=O), 147.2 (C4), 141,7 (C3), 133.9 (C7), 131.6 (C12), 128.7 (C8), 127.5 (C11), 118.9 (C1), 113.7 (C2), 92.5 (C5), 67.0 (C6), 60.5 (ester CH2), 57.3 (C9), 56.6 (CH2 bridge), 56.5 (OCH3), 44.9 (C16), 43.5 (C13), 40.6 (C14), 35.6 (C15), 22.6 (C10), 14.6 (ester CH3)
(10) N-carboxymethyl-dihydronorcodeine ethyl ester (ethyl 2-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 88%, m.p.: oil, HR-MS [M + H+]: calculated: 374.1961, measured: 374.1959; 1H-NMR (600 MHz, DMSO-d6) δ 6.66 (d, J = 8.2 Hz, H-2), 6.50 (d, J = 8.2 Hz, H-1), 4.43 (dd, J = 7.8, 4.9 Hz, H-5), 4.04 (q, J = 7.1 Hz, ester CH2), 3.79 (tt, J = 8.7, 4.1 Hz, H-6), 3.72 (s, OCH3), 3.32 (s, CH2 bridge), 3.16 (d, J = 16.4 Hz, CH2 bridge), 3.04 (dd, J = 5.9, 2.7 Hz, H-9), 2.75 (d, J = 18.4 Hz, H-10), 2.51–2.48 (m, H-16), 2.34 (dd, J = 18.4, 6.0 Hz, H-10), 2.17–2.09 (m, H-16, H-14), 1.79 (td, J = 12.4, 5.0 Hz, H-15), 1.40 (ddd, J = 12.5, 3.6, 1.7 Hz, H-15), 1.34 (dd, J = 13.3, 6.7 Hz, H-8), 1.29 (ddd, J = 13.4, 7.2, 4.2 Hz, H-7), 1.15 (t, J = 7.1 Hz, ester CH3), 1.13–1.08 (m, H-7), 0.84 (tt, J = 11.9, 6.1 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 170.9 (ester C=O), 147.4 (C4), 141.3 (C3), 130.7 (C12), 127.3 (C11), 118.4 (C1), 114.4 (C2), 90.7 (C5), 66.3 (C6), 60.3 (ester CH2), 58.2 (C9), 56.8 (CH2 bridge), 56.7 (OCH3), 44.9 (C16), 42.4 (C13), 38.4 (C14), 37.3 (C15), 26.1 (C7), 22.0 (C10), 19.8 (C8), 14.5 (ester CH3)
(11) N-carboxymethyl-noroxymorphone ethyl ester (ethyl 2-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 82%, m.p.: 160 °C, HR-MS [M + H+]: calculated: 374.1598, measured: 374.1588; 1H-NMR (600 MHz, DMSO-d6) δ 6.53 (d, J = 8.1 Hz, H-2), 6.50 (d, J = 8.1 Hz, H-1), 4.74 (s, H-5), 4.12–4.06 (m, ester CH2), 3.43 (d, J = 17.1 Hz, CH2 bridge), 3.31 (d, J = 17.2 Hz, CH2 bridge), 2.97 (d, J = 18.5 Hz, H-10), 2.90 (d, J = 5.9 Hz, H-9), 2.89–2.82 (m, H-7), 2.57 (d, J = 5.9 Hz, H-10), 2.49 (m, H-16), 2.38–2.29 (m, H-15), 2.18 (td, J = 12.0, 3.6 Hz, H-16), 2.06 (dt, J = 14.2, 3.2 Hz, H-7), 1.71 (ddd, J = 13.4, 5.0, 3.0 Hz, H-8), 1.40 (td, J = 14.1, 3.4 Hz, H-8), 1.28–1.22 (m, H-15), 1.18 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 209.0 (C6), 171.3 (ester C=O), 143.8 (C4), 139.8 (C3), 129.6 (C12), 123.6 (C11), 119.5 (C1), 117.6 (C2), 89.7 (C5), 70.4 (C14), 62.8 (C9), 60.7 (ester CH2), 55.6 (CH2 bridge), 50.3 (C13), 43.9 (C16), 36.2 (C7), 31.5 (C8), 30.6 (C15), 24.3 (C10), 14.5 (ester CH3)
(12) N-carboxymethyl-noroxycodone ethyl ester (ethyl 2-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetate) yield: 87%, m.p.: 144 °C, HR-MS [M + H+]: calculated: 388.1755, measured: 388.1752; 1H-NMR (600 MHz, DMSO-d6) δ 6.72 (d, J = 8.2 Hz, H-2), 6.63 (d, J = 8.2 Hz, H-1), 4.81 (s, H-5), 4.15–4.03 (m, ester CH2), 3.75 (s, OCH3), 3.44 (d, J = 17.1 Hz, CH2 bridge), 3.32 (d, CH2 bridge), 3.01 (d, J = 18.6 Hz, H-10), 2.92 (d, J = 5.7 Hz, H-9), 2.87 (td, J = 14.4, 5.0 Hz, H-7), 2.59 (dd, J = 18.7, 5.8 Hz, H-10), 2.52–2.48 (m, H-16), 2.34 (td, J = 12.5, 5.3 Hz, H-15), 2.17 (td, J = 12.0, 3.6 Hz, H-16), 2.06 (dt, J = 14.1, 3.2 Hz, H-7), 1.72 (ddd, J = 13.5, 5.0, 3.0 Hz, H-8), 1.43–1.34 (m, H-8), 1.30–1.23 (m, H-15), 1.18 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 208.8 (C6), 171.3 (ester C=O), 144.8 (C4), 142.4 (C3), 129,9 (C12), 125.8 (C11), 119.7 (C1), 115.2 (C2), 90.2 (C5), 70.3 (C14), 62.8 (C9), 60.7 (ester CH2), 56.8 (OCH3), 55.7 (CH2 bridge), 50.2 (C13), 43.8 (C16), 36.3 (C7), 31.6 (C8), 30.5 (C15), 24.3 (C16), 14.5 (ester CH3)
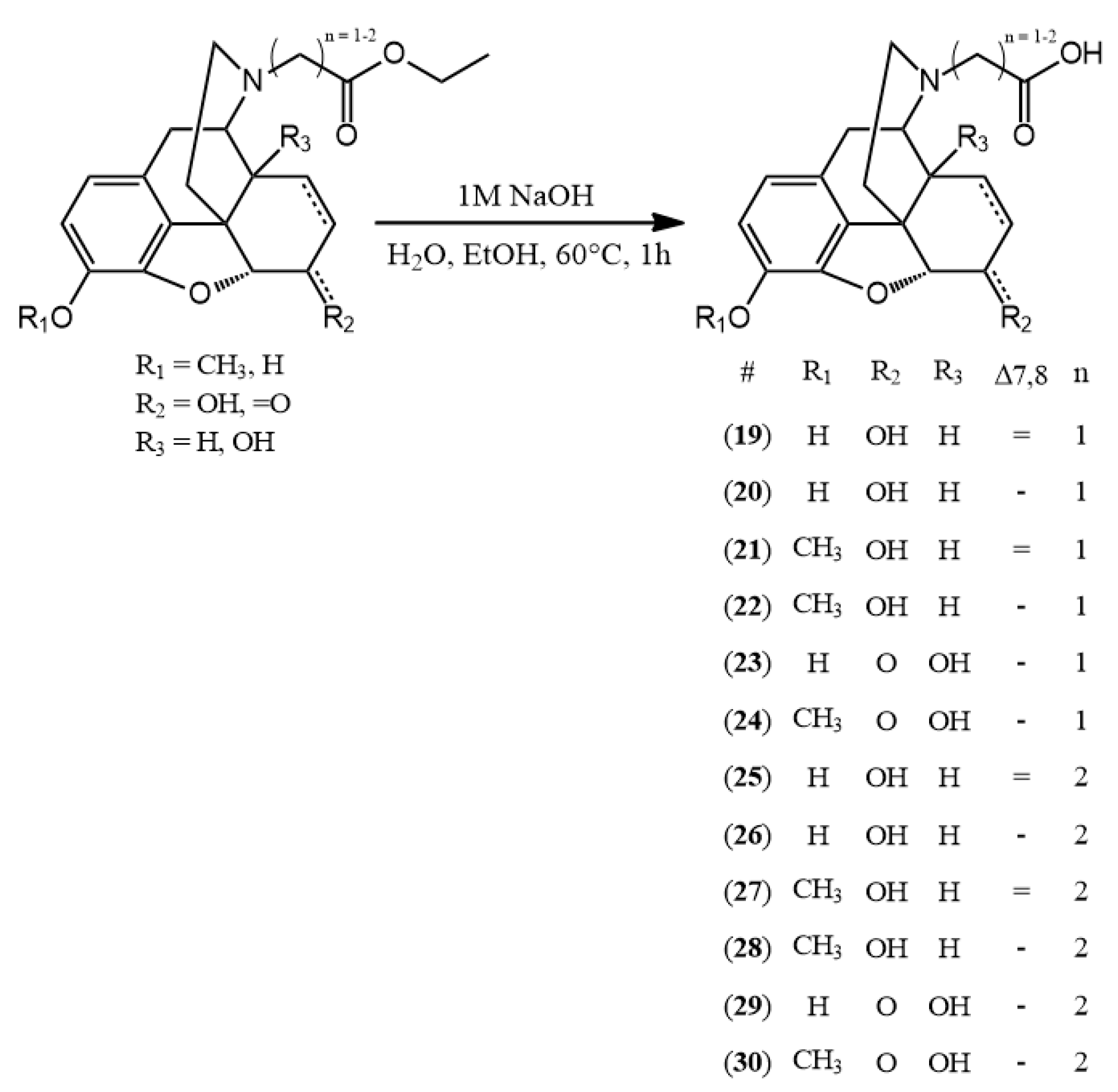
3.2.3. General Synthesis of N-Carboxymethyl-Nor-Compounds
The N-carboxymethyl-nor-compound ethyl ester (0.5 mmol) was added to the mixture of ethanol (1 g) and water (2.5 mL). To this solution, sodium hydroxide (1 M, 0.5 mmol) was added and the reaction mixture was heated for 1 h at 60 °C. The hydrolysis reaction was monitored by TLC and upon complete disappearance of the starting material, the pH was adjusted to 3–4 with 10% HCl solution. Then the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
(19) N-carboxymethyl-normorphine HCl (2-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 92%, m.p.: > 270 °C decomp., HR-MS [M + H+]: calculated: 330.1336, measured: 330.1332; 1H-NMR (600 MHz, D2O) δ 6.61 (d, J = 8.2 Hz, H-2), 6.53 (d, J = 8.2 Hz, H-1), 5.60 (m, H-7), 5.24 (d, J = 10.0 Hz, H-8), 4.92 (d, J = 6.5 Hz, H-5), 4.27 (m, H-9), 4.25 (m, H-6), 4.00 (m, CH2 bridge) 3.48 (q, J = 7.1 Hz, H-16), 3.11 (d, J = 20.1 Hz, H-10), 2.99 (d, J = 18.1 Hz, H-14), 2.80 (dd, J = 20.0, 6.8 Hz, H-10), 2.39–2.19 (m, H-15), 2.02 (d, J = 14.6 Hz, H-13); 13C-NMR (150 MHz, D2O) δ 169.2 (ester C=O), 145.5 (C4), 137.8 (C3), 132.9 (C7), 129.0 (C12), 125.4 (C8), 123.0 (C11), 120.0 (C1), 117.6 (C2), 89.9 (C5), 65.5 (C6), 60.2 (C9), 54.9 (CH2 bridge), 47.1 (C16), 41.8 (C13), 38.1 (C14), 32.2 (C15), 21.9 (C16)
(20) N-carboxymethyl-dihydronormorphine HCl (2-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 93%, m.p.: > 315 °C decomp., HR-MS [M + H+]: calculated: 332.1492, measured: 332.1482; 1H-NMR (600 MHz, D2O) δ 6.69 (d, J = 7.9 Hz, H-2), 6.62 (d, J = 8.2 Hz, H-1), 4.64 (m, H-5), 4.07–3.83 (m, H-6, H-9, CH2 bridge), 3.34 (dd, J = 13.4, 4.4 Hz, H-16), 3.06 (d, J = 20.2 Hz, H-10), 2.91 (dd, J = 19.9, 6.0 Hz, H-10), 2.82 (ddd, J = 17.3, 13.9, 7.6 Hz, H-16), 2.47 (t, J = 19.4 Hz, H-14), 2.08 (td, J = 13.9, 5.0 Hz, H-15), 1.78 (dd, J = 14.7, 3.7 Hz, H-15), 1.44 (q, J = 6.2 Hz, H-7, H-8), 0.99–0.83 (m, H-8); 13C-NMR (150 MHz, D2O) δ 168.3 (carboxylic C=O), 145.2 (C4), 137.1 (C3), 128.4 (C12), 122.6 (C11), 119.8 (C1), 117.8 (C2), 89.0 (C5), 66.1 (C6), 61.5 (C9), 55.0 (CH2 bridge), 47.5 (C16), 40.2 (C13), 38.3 (C14), 34.1 (C15), 26.0 (C7), 21.4 (C10), 17.7 (C8)
(21) N-carboxymethyl-norcodeine HCl (2-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 95%, m.p.: > 260 °C decomp., HR-MS [M + H+]: calculated: 344.1492, measured: 344.1481; 1H-NMR (600 MHz, D2O) δ 6.79 (d, J = 8.3 Hz, H-2), 6.65 (d, J = 8.3 Hz, H1), 5.68–5.54 (m, H-7), 5.33–5.18 (m, H-8), 5.03–4.87 (m, H-5), 4.39–3.94 (m, H-9, H-6, CH2 bridge), 3.72 (s, OCH3), 3.55–3.40 (m, H-16), 3.16 (d, J = 20.1 Hz, H-10), 3.10–2.73 (m, H-14, H-10), 2.31 (s, H-15), 2.05 (d, J = 14.9 Hz, H-15); 13C-NMR (150 MHz, D2O) δ 168.8 (carboxylic C=O), 146.4 (C4), 141.9 (C3), 133.1 (C7), 128.9 (C12), 125.5 (C8), 124.0 (C11), 120.4 (C1), 114.7 (C2), 90.6 (C5), 65.9 (C6), 60.5 (C9), 56.4 (OCH3) 54.8 (CH2 bridge), 46.8 (C16), 41.7 (C13), 38.4 (C14), 32.4 (C15), 21.9
(22) N-carboxymethyl-dihydronorcodeine HCl (2-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 90%, m.p.: > 275 °C decomp., HR-MS [M + H+]: calculated: 346.1649, measured: 346.1644; 1H-NMR (600 MHz, D2O) δ 6.85 (d, J = 8.2 Hz, H-2), 6.72 (d, J = 8.2 Hz, H-1), 4.69 (d, J = 5.4 Hz, H-5), 4.16–3.79 (m, H-6, H-9, CH2 bridge), 3.80–3.70 (s, OCH3), 3.38–3.29 (m, H-16), 3.09 (d, J = 20.2 Hz, H-10), 2.99–2.87 (m, H-10), 2.80 (t, J = 12.6 Hz, H-16), 2.46 (d, J = 11.6 Hz, H-14), 2.08 (dd, J = 14.7, 10.0 Hz, H-15), 1.78 (d, J = 14.0 Hz, H-15), 1.45 (d, J = 8.7 Hz, H-7, H-8), 0.98–0.87 (m, H-8); 13C-NMR (150 MHz, D2O) δ 168.5 (carboxylic C=O), 145.8 (C4), 141.3 (C3), 127.8 (C12), 123.4 (C11), 119.9 (C1), 115.0 (C2), 89.2 (C5), 66.0 (C6), 61.5 (C9), 56.7 (OCH3), 55.1 (CH2 bridge), 47.4 (C16), 40.1 (C13), 38.2 (C14), 34.1 (C15), 26.0 (C7), 21.4 (C10), 17.6 (C8)
(23) N-carboxymethyl-noroxymorphone HCl (2-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 96%, m.p.: > 290 °C decomp., HR-MS [M + H+]: calculated: 346.1285, measured: 346.1283; 1H-NMR (600 MHz, D2O) δ 6.70 (d, J = 8.2 Hz, H-2), 6.68 (d, J = 8.3 Hz, H-1), 4.92 (s, H-5), 3.93 (d, J = 16.7 Hz, CH2 bridge), 3.88 (d, J = 6.2 Hz, H-9), 3.69 (d, J = 16.7 Hz, CH2 bridge), 3.29 (d, J = 20.0 Hz, H-10), 3.18 (dd, J = 13.1, 4.9 Hz, H-16), 3.06 (dd, J = 20.1, 6.4 Hz, H-10), 2.89 (td, J = 14.8, 5.1 Hz, H-7), 2.81 (td, J = 13.0, 4.1 Hz, H-16), 2.65 (td, J = 13.5, 5.0 Hz, H-15), 2.20 (dt, J = 14.9, 3.2 Hz, H-7), 1.96 (ddd, J = 14.6, 5.2, 2.9 Hz, H-8), 1.65 (dd, J = 12.9, 3.7 Hz, H-15), 1.60 (dd, J = 14.6, 3.6 Hz, H-8); 13C-NMR (150 MHz, D2O) δ 209.3 (C6), 168.8 (carboxylic C=O), 143.0 (C4), 138.5 (C3), 127.2 (C12), 121.7 (C11), 121.1 (C1), 118.7 (C2), 89.2 (C5), 70.7 (C14), 64.9 (C9), 54.7 (CH2 bridge), 48.6 (C13), 46.7 (C16), 34.6 (C7), 30.5 (C8), 27.5 (C15), 23.9 (C10)
(24) N-carboxymethyl-noroxycodone HCl (2-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetic acid hydrochloride) yield: 94%, m.p.: > 210 °C decomp., HR-MS [M + H+]: calculated: 360.1442, measured: 360.1435; 1H-NMR (600 MHz, D2O) δ 6.86 (d, J = 8.4 Hz, H-2), 6.78 (d, J = 8.4 Hz, H-1), 4.95 (s, H-5), 3.94–3.79 (m, H-9, CH2 bridge), 3.74 (s, OCH3), 3.63 (d, J = 16.4 Hz, CH2 bridge), 3.31 (d, J = 20.0 Hz, H-10), 3.17 (ddt, J = 13.1, 5.0, 1.4 Hz, H-16), 3.12–3.05 (m, H-10), 2.90 (td, J = 14.8, 5.1 Hz, H-7), 2.80 (td, J = 13.0, 4.1 Hz, H-16), 2.65 (td, J = 13.5, 5.0 Hz, H-15), 2.21 (dt, J = 14.9, 3.2 Hz, H-7), 1.96 (ddd, J = 14.5, 5.1, 2.9 Hz, H-8), 1.65 (dd, J = 14.2, 3.8 Hz, H-15), 1.60 (dd, J = 14.6, 3.5 Hz, H-8); 13C-NMR (150 MHz, D2O) δ 210.3 (C6), 169.2 (carboxylic C=O), 143.9 (C4), 142.4 (C3), 127.1 (C12), 122.5 (C11), 121.1 (C1), 115.6 (C2), 89.4 (C5), 70.4 (C14), 64.9 (C9), 56.7 (OCH3), 55.2 (CH2 bridge), 48.7 (C13), 46.6 (C16), 34.5 (C7), 30.5 (C8), 27.6 (C15), 23.9 (C10)
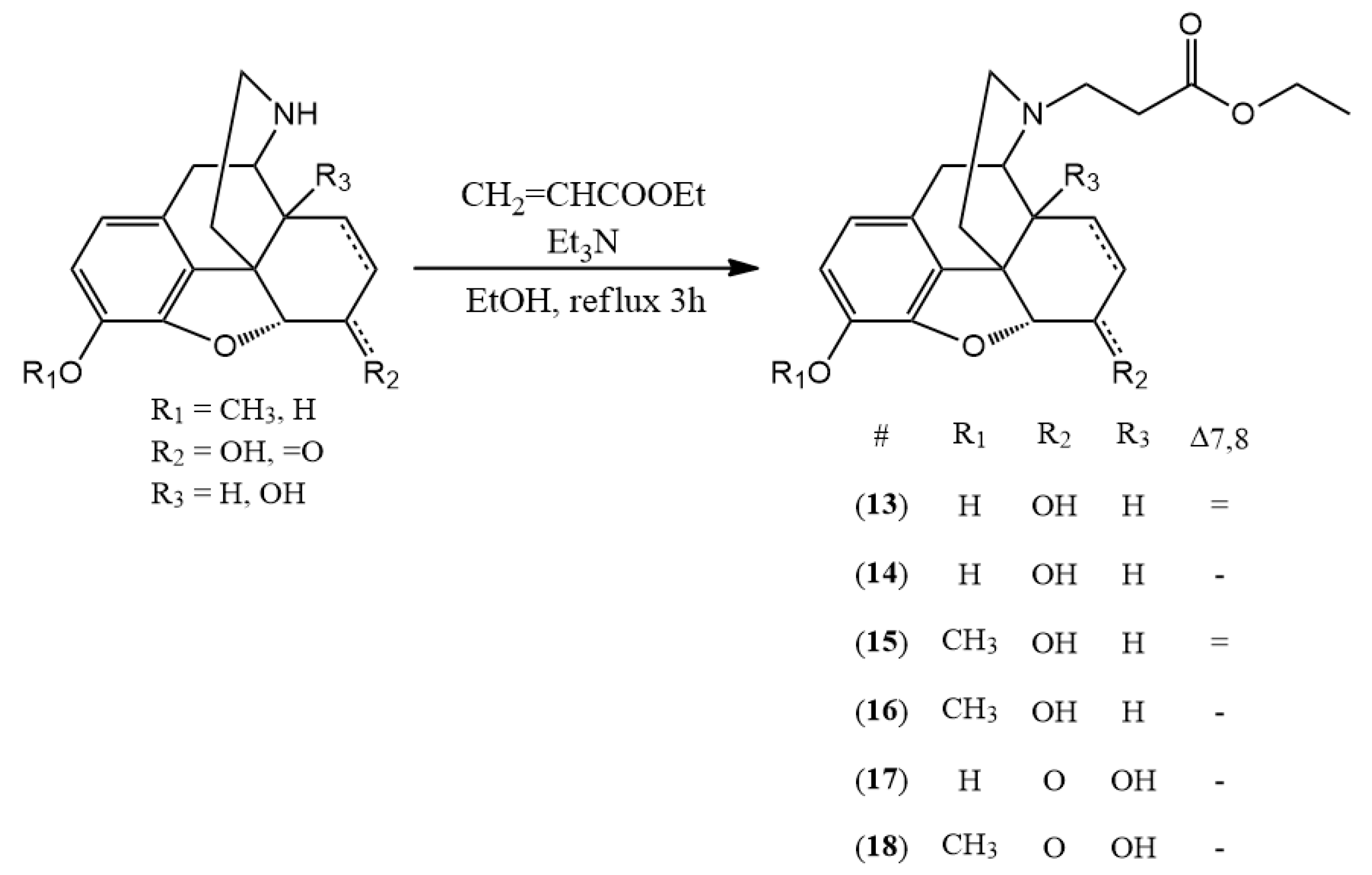
3.2.4. General Synthesis of N-Carboxyethyl-Nor-Compound Ethyl Esters
The normorphine derivative (2 mmol) was dissolved in 30 mL of ethanol. In the presence of triethylamine (1 mL) ethyl acrylate (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 3 h. After the TLC showed no more starting compound, the solvent was evaporated. If it was necessary, column chromatography was used. (chloroform:methanol 9:1)
(13) N-carboxyethyl-normorphine ethyl ester (ethyl 3-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 98%, m.p.: 158 °C, HR-MS [M + H+]: calculated: 372.1805, measured: 372.1798; 1H-NMR (600 MHz, DMSO-d6) δ 6.44 (d, J = 8.0 Hz, H-2), 6.34 (d, J = 8.1 Hz, H-1), 5.52 (dp, J = 9.7, 1.5 Hz, H-7), 5.22 (dt, J = 9.8, 2.8 Hz, H-8), 4.66 (dd, J = 6.1, 1.3 Hz, H-5), 4.15–4.00 (m, H-6, ester CH2), 3.35 (m, H-9), 2.78 (dt, J = 14.3, 5.9 Hz, H-10, ethylene bridge CH2), 2.64 (dt, J = 12.9, 6.6 Hz, ethylene bridge CH2), 2.60–2.53 (m, H-16), 2.48–2.39 (m, H-14, ethylene bridge CH2), 2.33–2.21 (m, H-16, H-10), 1.92 (td, J = 12.5, 5.0 Hz, H-15), 1.61 (ddd, J = 12.5, 3.4, 1.7 Hz, H-15), 1.19 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 172.3 (ester C=O), 146.8 (C4), 138.7 (C3), 133.9 (C7), 131.2 (C12), 128.8 (C8), 125.7 (C11), 118.9 (C1), 116.8 (C2), 92.0 (C5), 66.8 (C6), 60.3 (ester CH2), 57.1 (C9), 50.8 (ethylene bridge CH2), 44.6 (C16), 43.8 (C13), 41.6 (C14), 36.0 (C15), 33.7 (ethylene bridge CH2), 22.2 (C10), 14.7 (ester CH3)
(14) N-carboxyethyl-dihydronormorphine ethyl ester (ethyl 3-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 96%, m.p.: 120 °C, HR-MS [M + H+]: calculated: 374.1961, measured: 374.1960; 1H-NMR (600 MHz, DMSO-d6) δ 6.53 (d, J = 8.0 Hz, H-2), 6.41 (d, J = 8.0 Hz, H-1), 4.44 (d, J = 4.8 Hz, H-5), 4.06 (q, J = 7.1 Hz, ester CH2), 3.80 (dq, J = 8.7, 4.2 Hz, H-6), 3.16–2.99 (m, H-9), 2.77 (d, J = 18.5 Hz, H-10), 2.46 (d, J = 6.9 Hz, ethylene bridge CH2), 2.12 (d, J = 39.2 Hz, H-16), 1.82–1.72 (m, H-15), 1.46 (d, J = 12.4 Hz, H-15), 1.38 (dq, J = 13.5, 6.9 Hz, H-8), 1.30 (dtd, J = 13.2, 6.5, 3.4 Hz, H-7), 1.18 (td, J = 7.2, 4.2 Hz, H-7, ester CH3), 0.87 (tt, J = 12.6, 6.4 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 172.2 (ester C=O), 146.5 (C4), 138.3 (C3), 130.1 (C12), 125.2 (C11), 117.8 (C1), 116.6 (C2), 89.6 (C5), 65.8 (C6), 59.6 (ester CH2), 57.4 (C9), 49.9 (ethylene bridge CH2), 44.0 (C16), 42.7 (C13), 37.6 (C14), 36.7 (C15), 32.6 (ethylene bridge CH2), 25.4 (C7), 20.9 (C10), 19.3 (C8), 14.2 (ester CH3)
(15) N-carboxyethyl-norcodeine ethyl ester (ethyl 3-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 97%, m.p.: oil, HR-MS [M + H+]: calculated: 386.1962, measured: 386.1951; 1H-NMR (600 MHz, DMSO-d6) δ 6.61 (d, J = 8.2 Hz, H-2), 6.45 (d, J = 8.2 Hz, H-1), 5.53 (dp, J = 9.8, 1.5 Hz, H-7), 5.23 (dt, J = 9.8, 2.8 Hz, H-8), 4.67 (dd, J = 5.9, 1.3 Hz, H-5), 4.11 (dt, J = 5.7, 2.9 Hz, H-6), 4.07 (q, J = 7.1 Hz, ester CH2), 3.71 (s, OCH3), 3.36 (s, H-9), 2.86–2.74 (m, H-10, ethylene bridge), 2.64 (dt, J = 12.9, 6.7 Hz, ethylene bridge), 2.61–2.53 (m, H-16), 2.49–2.42 (m, H-14, ethylene bridge), 2.31 (dd, J = 18.6, 6.3 Hz, H-10), 2.24 (td, J = 12.2, 3.4 Hz, H-16), 1.92 (td, J = 12.5, 5.0 Hz, H-15), 1.61 (ddd, J = 12.6, 3.4, 1.7 Hz, H-15), 1.18 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 171.7 (ester C=O), 145.0 (C4), 141.2 (C3), 133.1 (C7), 130.7 (C12), 128.0 (C8), 127.0 (C11), 118.1 (C1), 113.0 (C2), 91.7 (C5), 66.2 (C6), 59.5 (ester CH2), 56.2 (C9), 55.8 (OCH3), 49.9 (ethylene bridge CH2), 43.7 (C16), 43.4 (C13), 40.2 (C14), 35.2 (C15), 32.9 (ethylene bridge CH2), 21.5 (C10), 14.0 (ester CH3)
(16) N-carboxyethyl-dihydronorcodeine ethyl ester (ethyl 3-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 93%, m.p.: oil, HR-MS [M + H+]: calculated: 388.2118, measured: 388.2111; 1H-NMR (600 MHz, DMSO-d6) δ 6.69 (d, J = 8.2 Hz, H-2), 6.53 (d, J = 8.1 Hz, H-1), 4.45 (dd, J = 6.7, 4.9 Hz, H-5), 4.06 (qd, J = 7.1, 1.2 Hz, ester CH2), 3.84–3.77 (m, H-6), 3.76 (s, OCH3), 3.04 (dd, J = 5.9, 2.8 Hz, H-9), 2.81–2.70 (m, H-10, ethylene bridge CH2), 2.59 (dq, J = 12.8, 6.6, 5.9 Hz, ethylene bridge CH2), 2.46–2.38 (m, ethylene bridge CH2), 2.36 (dd, J = 18.4, 6.0 Hz, H-10), 2.12–2.02 (m, H-14, H-16), 1.74 (ddd, J = 14.3, 11.2, 4.9 Hz, H-15), 1.44 (ddd, J = 12.4, 3.6, 1.7 Hz, H-15), 1.37 (dt, J = 13.4, 6.8 Hz, H-8), 1.31 (dq, J = 10.0, 3.5 Hz, H-7), 1.18 (t, J = 7.1 Hz, ester CH3), 1.16–1.11 (m, H-8), 0.94–0.82 (m, H-7); 13C-NMR (150 MHz, DMSO-d6) δ 172.4 (ester C=O), 147.4 (C4), 140.9 (C3), 130.7 (C12), 127.2 (C11), 118.4 (C1), 114.4 (C2), 90.7 (C5), 66.3 (C6), 60.1 (ester CH2), 57.9 (C9), 56.7 (OCH3), 50.6 (ethylene bridge CH2), 44.5 (C16), 42.6 (C13), 38.6 (C14), 37.5 (C15), 33.6 (ethylene bridge CH2), 26.1 (C7), 21.6 (C10), 19.8 (C8), 14.6 (ester CH3)
(17) N-carboxyethyl-noroxymorphone ethyl ester (ethyl 3-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 99%, m.p.: 137 °C, HR-MS [M + H+]: calculated: 388.1754, measured: 388.1740; 1H-NMR (600 MHz, DMSO-d6) δ 6.56 (d, J = 8.1 Hz, H-2), 6.52 (d, J = 8.1 Hz, H-1), 4.75 (s, H-5), 4.08 (qd, J = 7.1, 2.8 Hz, ester CH2), 2.97 (d, J = 18.5 Hz, H-10), 2.91 (d, J = 5.9 Hz, H-9), 2.90–2.84 (m, H-7), 2.75 (dt, J = 12.7, 7.2 Hz, ethylene bridge CH2), 2.67 (dt, J = 12.6, 6.2 Hz, ethylene bridge CH2), 2.60–2.53 (m, H-10), 2.53–2.51 (m, H-16), 2.27 (td, J = 12.6, 5.1 Hz, H-15), 2.08 (dt, J = 14.1, 3.2 Hz, H-7), 2.02 (td, J = 12.1, 3.7 Hz, H-16), 1.74 (ddd, J = 13.3, 5.0, 2.9 Hz, H-8), 1.43 (td, J = 14.0, 3.4 Hz, H-8), 1.32–1.25 (m, H-15), 1.21 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 208.7 (C6), 172.0 (ester C=O), 143.4 (C4), 139.4 (C3), 129.4 (C12), 123.3 (C11), 119.0 (C1), 117.2 (C2), 89.3 (C5), 69.8 (C14), 62.6 (C9), 59.9 (ester CH2), 50.1 (C13), 49.7 (ethylene bridge CH2), 42.7 (C16), 35.8 (C7), 32.9 (ethylene bridge CH2), 31.1 (C8), 30.2 (C15), 23.1 (C10), 14.2 (ester CH3)
(18) N-carboxyethyl-noroxycodone ethyl ester (ethyl 3-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoate) yield: 98%, m.p.: 119 °C, HR-MS [M + H+]: calculated: 402.1911, measured: 402.1905; 1H-NMR (600 MHz, DMSO-d6) δ 6.74 (d, J = 8.3 Hz, H-2), 6.66 (d, J = 8.2 Hz, H-1), 4.83 (d, J = 7.8 Hz, H-5), 4.09 (qd, J = 7.1, 2.8 Hz, ester CH2), 3.78 (s, OCH3), 3.02 (d, J = 18.6 Hz, H-10), 2.94 (d, J = 5.7 Hz, H-9), 2.89 (d, J = 5.0 Hz, H-7), 2.81–2.72 (m, ethylene bridge CH2), 2.68 (dt, J = 12.6, 6.2 Hz, ethylene bridge CH2), 2.59 (dd, J = 18.7, 5.8 Hz, H-10), 2.57–2.52 (m, ethylene bridge CH2), 2.28 (td, J = 12.6, 5.2 Hz, H-15), 2.08 (dt, J = 14.1, 3.2 Hz, H-7), 2.00 (td, J = 12.2, 3.7 Hz, H-16), 1.75 (ddd, J = 13.4, 5.0, 3.0 Hz, H-8), 1.46–1.37 (m, H-8), 1.33–1.26 (m, H-15), 1.21 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 208.3 (C6), 172.1 (ester C=O), 144.2 (C4), 141.9 (C3), 129.5 (C12), 125.5 (C11), 118.9 (C1), 114.4 (C2), 89.4 (C5), 69.8 (C14), 62.5 (C9), 59.4 (ester CH2), 55.9 (OCH3), 50.2 (C13), 49.2 (ethylene bridge CH2), 42.7 (C16), 35.5 (C7), 32.4 (ethylene bridge CH2), 30.8 (C8), 29.7 (C15), 22.8 (C10), 13.9 (ester CH3)
3.2.5. General Synthesis of N-Carboxyethyl-Nor-Compounds
The N-carboxyethyl-nor-compound ethyl ester (0.5 mmol) was added to the mixture of ethanol (1 g) and water (2.5 mL). To this solution, sodium hydroxide (1 M, 0.5 mmol) was added and the reaction mixture was heated for 1 h at 60 °C. The hydrolysis reaction was monitored by TLC and upon complete disappearance of the starting material the pH was adjusted to 3–4 with 10% HCl solution. Then, the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
(25) N-carboxyethyl-normorphine HCl (3-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 96%, m.p.: > 290 °C decomp., HR-MS [M + H+]: calculated: 344.1492, measured: 344.1483; 1H-NMR (600 MHz, D2O) δ 6.64 (d, J = 8.2 Hz, H-2), 6.56 (d, J = 8.2 Hz, H-1), 5.62 (d, J = 9.7 Hz, H-7), 5.27 (d, J = 10.1 Hz, H-8), 4.94 (dd, J = 6.4, 1.3 Hz, H-5), 4.29–4.18 (m, H-6, H-9), 3.48–3.33 (m, H-16, ethylene bridge CH2), 3.13 (d, J = 20.1 Hz, H-10), 2.96 (t, J = 13.2 Hz, H-16), 2.89–2.78 (m, H-14, H-10), 2.71 (td, J = 7.0, 2.7 Hz, ethylene bridge CH2), 2.22 (t, J = 13.8 Hz, H-15), 2.04 (d, J = 14.3 Hz, H-15); 13C-NMR (150 MHz, D2O) δ 175.3 (carboxylic C=O), 145.1 (C4), 137.8 (C3), 129.3 (C12), 133.1 (C7), 125.5 (C8), 123.2 (C11), 120.3 (C1), 117.6 (C2), 90.3 (C5), 65.6 (C6), 58.8 (C9), 50.7 (ethylene bridge CH2), 46.0 (C16), 41.8 (C13), 38.1 (C14), 32.5 (C15), 29.6 (ethylene bridge CH2), 21.2 (C10)
(26) N-carboxyethyl-dihydronormorphine HCl (3-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 96%, m.p.: > 295 °C decomp., HR-MS [M + H+]: calculated: 346.1649, measured: 346.1647; 1H-NMR (600 MHz, D2O) δ 6.70 (dd, J = 8.2, 0.9 Hz, H-2), 6.63 (d, J = 8.1 Hz, H-1), 4.65 (d, J = 5.5 Hz, H-5), 4.02 (td, J = 5.9, 2.8 Hz, H-6), 3.91 (dd, J = 6.4, 2.6 Hz, H-9), 3.48–3.32 (m, ethylene bridge CH2), 3.26 (dd, J = 13.1, 5.3 Hz, H-16), 3.08 (d, J = 19.7 Hz, H-10), 2.90 (dd, J = 19.9, 6.1 Hz, H-10), 2.83–2.72 (m, H-16, ethylene bridge CH2), 2.33 (ddd, J = 11.7, 5.3, 2.8 Hz, H-14), 2.01 (td, J = 13.6, 4.8 Hz, H-15), 1.83–1.76 (m, H-15), 1.51–1.38 (m, H-7, H-8), 1.00–0.86 (m, H-8); 13C-NMR (150 MHz, D2O) δ 174.2 (carboxylic C=O), 145.1 (C4), 137.3 (C3), 128.1 (C12), 122.4 (C11), 119.8 (C1), 117.9 (C2), 88.9 (C5), 66.0 (C6), 60.0 (C9), 50.0 (ethylene bridge CH2), 46.9 (C16), 40.6 (C13), 38.3 (C14), 34.0 (C15), 29.0 (ethylene bridge CH2), 25.9 (C7), 20.5 (C10), 17.6 (C8)
(27) N-carboxyethyl-norcodeine HCl (3-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 93%, m.p.: > 200 °C decomp., HR-MS [M + H+]: calculated: 358.1649, measured: 358.1634; 1H-NMR (600 MHz, D2O) δ 6.78 (d, J = 8.3 Hz, H-2), 6.65 (d, J = 8.3 Hz, H-1), 5.61 (d, J = 9.8 Hz, H-7), 5.26 (d, J = 9.9 Hz, H-8), 4.94 (dd, J = 6.4, 1.3 Hz, H-5), 4.32–4.15 (m, H-6, H-9), 3.71 (s, OCH3), 3.51 (q, J = 7.1 Hz, ethylene bridge CH2), 3.48–3.34 (m, H-16), 3.17 (d, J = 20.3 Hz, H-10), 3.00–2.78 (m, H-16, H-14, H-10), 2.75 (td, J = 7.0, 2.5 Hz, ethylene bridge CH2), 2.22 (d, J = 13.9 Hz, H-15), 2.03 (d, J = 14.4 Hz, H-15); 13C-NMR (150 MHz, D2O) δ 175.1 (carboxylic C=O), 146.3 (C4), 142.0 (C3), 133.1 (C7), 129.00 (C12), 125.6 (C8), 124.0 (C11), 120.3 (C1), 114.4 (C2), 90.5 (C5), 65.7 (C6), 58.9 (C9), 56.4 (OCH3), 50.6 (ethylene bridge CH2), 46.1 (C16), 41.9 (C13), 38.3 (C14), 32.5 (C15), 29.4 (ethylene bridge CH2), 21.2 (C10)
(28) N-carboxyethyl-dihydronorcodeine HCl (3-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 95%, m.p.: > 220 °C decomp., HR-MS [M + H+]: calculated: 360.1805, measured: 360.1791; 1H-NMR (600 MHz, D2O) δ 6.82 (d, J = 8.2 Hz, H-2), 6.70 (d, J = 8.2 Hz, H-1), 4.64 (m, H-5), 3.99 (q, J = 4.9 Hz, H-6), 3.92 (dd, J = 6.3, 2.6 Hz, H-9), 3.73 (s, OCH3), 3.43 (dt, J = 14.1, 7.2 Hz, ethylene bridge CH2), 3.37 (dt, J = 13.7, 7.0 Hz, ethylene bridge CH2), 3.25 (dd, J = 13.2, 4.7 Hz, H-16), 3.10 (d, J = 19.8 Hz, H-10), 2.90 (dd, J = 19.9, 6.2 Hz, H-10), 2.79 (td, J = 7.1, 4.4 Hz, ethylene bridge CH2), 2.71 (td, J = 13.3, 4.1 Hz, H-16), 2.35 (ddd, J = 12.0, 5.4, 2.7 Hz, H-14), 2.03 (td, J = 13.7, 4.8 Hz, H-15), 1.77–1.68 (m, H-15), 1.48–1.34 (m, H-8, H-7), 0.90 (tt, J = 11.3, 6.0 Hz, H-8); 13C-NMR (151 MHz, D2O) δ 173.8 (carboxylic C=O), 146.0 (C4), 141.5 (C3), 128.1 (C12), 123.5 (C11), 119.9 (C1), 114.9 (C2), 89.2 (C5), 66.0 (C6), 60.1 (C9), 56.6 (OCH3), 49.9 (ethylene bridge CH2), 46.9 (C16), 40.5 (C13), 38.1 (C14), 34.1 (C15), 28.9 (ethylene bridge CH2), 25.8 (C7), 20.6 (C10), 17.6 (C8)
(29) N-carboxyethyl-noroxymorphone HCl (3-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 99%, m.p.: > 280 °C decomp., HR-MS [M + H+]: calculated: 360.1442, measured: 360.1332; 1H-NMR (600 MHz, D2O) δ 6.72 (d, J = 8.2 Hz, H-2), 6.69 (d, J = 8.3 Hz, H-1), 4.93 (s, H-5), 3.74 (d, J = 6.1 Hz, H-9), 3.41 (hept, J = 6.8, 5.9 Hz, ethylene bridge CH2), 3.34–3.25 (m, H-16, H-10), 3.05 (dd, J = 19.9, 6.3 Hz, H-10), 2.89 (td, J = 14.8, 5.1 Hz, H-7), 2.85–2.66 (m, H-16, ethylene bridge CH2), 2.60 (td, J = 13.4, 4.8 Hz, H-15), 2.21 (dt, J = 14.8, 3.2 Hz, H-7), 1.95 (ddd, J = 14.5, 5.2, 3.0 Hz, H-8), 1.64 (ddd, J = 17.9, 13.7, 3.5 Hz, H-8, H-15); 13C-NMR (151 MHz, D2O) δ 211.5 (C6), 175.0 (carboxylic C=O) 143.2 (C4), 138.7 (C3), 127.2 (C12), 121.7 (C11), 120.9 (C1), 118.7 (C2), 89.5 (C5), 70.6 (C14), 64.0 (C9), 49.7 (ethylene bridge CH2), 49.1 (C13), 45.8 (C16), 34.4 (C7), 30.5 (C8), 28.2 (ethylene bridge CH2), 27.1 (C15), 23.1 (C10)
(30) N-carboxyethyl-noroxycodone HCl (3-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)propanoic acid hydrochloride) yield: 96%, m.p.: > 260 °C decomp., HR-MS [M + H+]: calculated: 374.1598, measured: 374.1597; 1H-NMR (600 MHz, D2O) δ 6.87 (d, J = 8.3 Hz, H-2), 6.78 (d, J = 8.4 Hz, H-1), 4.95 (s, H-5), 3.75 (d, J = 4.5 Hz, H-9, OCH3), 3.42 (t, J = 6.6 Hz, ethylene bridge CH2), 3.32 (d, J = 20.1 Hz, H-10, H-16), 3.08 (dd, J = 20.0, 6.3 Hz, H-10), 2.89 (td, J = 14.8, 5.1 Hz, H-7), 2.82 (dt, J = 18.2, 6.6 Hz, ethylene bridge CH2), 2.70 (td, J = 13.5, 12.9, 4.5 Hz, H-16), 2.60 (td, J = 13.4, 4.8 Hz, H-15), 2.20 (dt, J = 14.9, 3.2 Hz, H-7), 1.95 (ddd, J = 14.6, 5.2, 3.0 Hz, H-8), 1.68–1.59 (m, H-8, H-15); 13C-NMR (151 MHz, D2O) δ 211.0 (C6), 174.9 (carboxylic C=O), 144.3 (C4), 142.6 (C3), 127.1 (C12), 122.4 (C11), 121.1 (C1), 115.6 (C2), 89.4 (C5), 70.2 (C14), 64.0 (C9), 56.7 (OCH3), 49.7 (ethylene bridge CH2), 48.4 (C13), 45.9 (C16), 34.4 (C7), 30.5 (C8), 28.2 (ethylene bridge CH2), 27.2 (C15), 23.1 (C10)
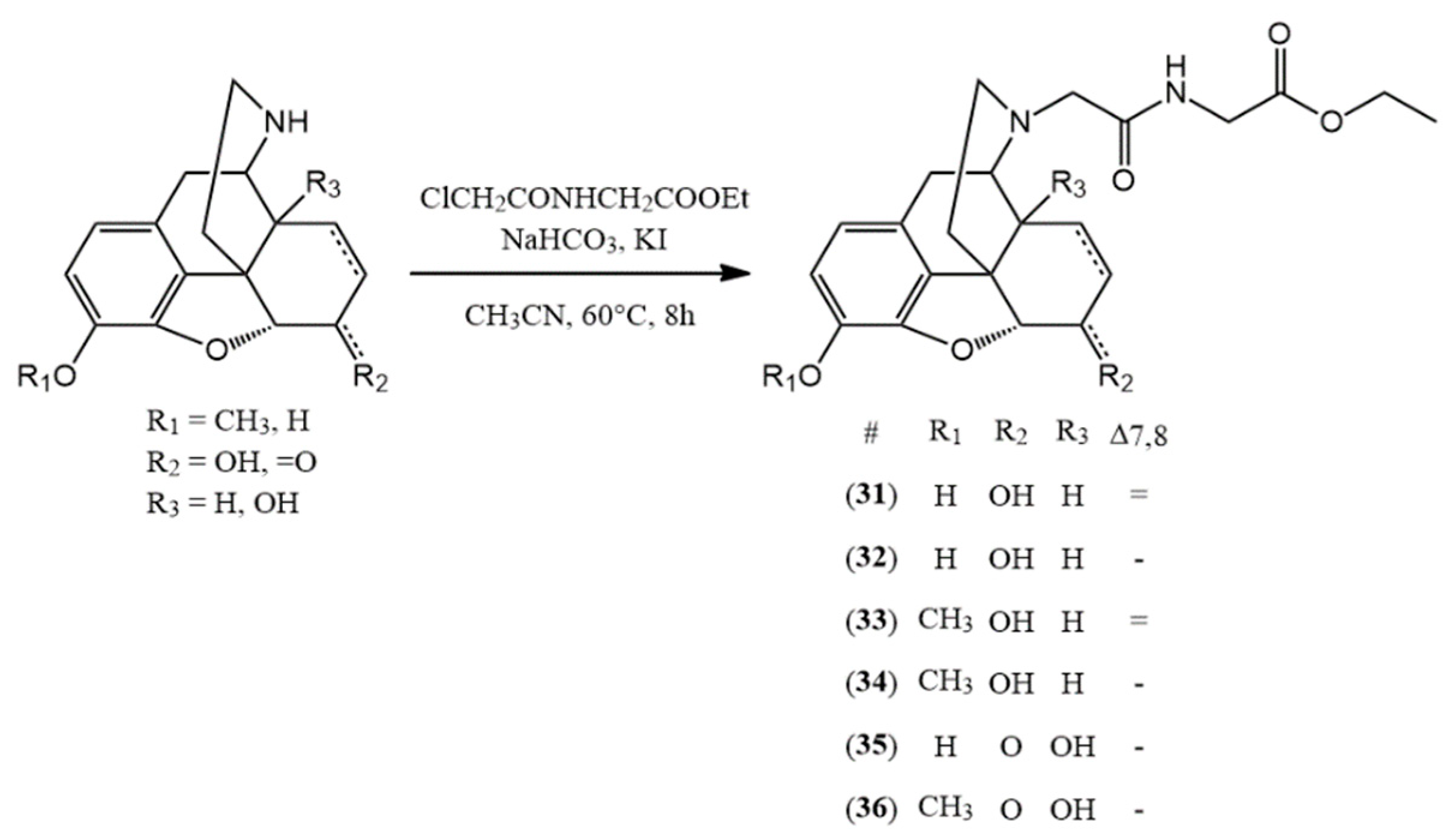
3.2.6. General Synthesis of N-Acetylglycine-Nor-Compound Ethyl Esters
The normorphine derivative (2 mmol) was dissolved in 30 mL of acetonitrile. In the presence of sodium bicarbonate (10 mmol) and potassium iodide (2.4 mmol), N-(chloroacetyl)glycine ethyl ester (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 8 h. After the TLC showed no more starting compound, the inorganic salts were filtered and the solvent was evaporated. If it was necessary, column chromatography was used. (chloroform: methanol 9:1)
(31) N-acetylglycine-normorphine ethyl ester (ethyl 2-(2-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 94%, m.p.: 105 °C, HR-MS [M + H+]: calculated: 415.1863, measured: 415.1848; 1H-NMR (600 MHz, DMSO-d6) δ 6.42 (d, J = 8.0 Hz, H-2), 6.32 (d, J = 8.1 Hz, H-1), 5.53 (dp, J = 9.9, 1.6 Hz, H-7), 5.20 (dt, J = 9.8, 2.8 Hz, H-8), 4.67 (dd, J = 6.1, 1.3 Hz, H-5), 4.06 (q, J = 7.1 Hz, ester CH2), 3.85 (qd, J = 17.4, 6.2 Hz, glycine CH2), 3.35 (dd, J = 6.1, 3.0 Hz, H-9), 3.20 (d, J = 16.1 Hz, CH2 bridge), 2.98 (d, J = 16.1 Hz, CH2 bridge), 2.76 (d, J = 18.4 Hz, H-10), 2.71 (dd, J = 4.7, 2.5 Hz, H-14), 2.55 (dd, J = 12.1, 4.6 Hz, H-16), 2.41–2.29 (m, H-10, H-16), 2.05 (td, J = 12.5, 4.9 Hz, H-15), 1.61 (d, J = 12.7 Hz, H-15), 1.16 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 171.0 (amide C=O), 170.3 (ester C=O), 146.7 (C4), 139.1 (C3), 133.8 (C7), 131.2 (C12), 128.7 (C8), 125.4 (C11), 119.0 (C1), 116.7 (C2), 91.8 (C6), 66.9 (C6), 60.8 (ester CH2), 58.9 (CH2 bridge), 57.6 (C9), 45.0 (C16), 43.5 (C13), 40.9 (glycine CH2), 40.7 (C14), 35.7 (C15), 23.3 (C10), 14.5 (ester CH3)
(32) N-acetylglycine-dihydronormorphine ethyl ester (ethyl 2-(2-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 91%, m.p.: 76 °C, HR-MS [M + H+]: calculated: 417.2020, measured: 417.2020; 1H-NMR (600 MHz, DMSO-d6) δ 6.53 (d, J = 8.0 Hz, H-2), 6.42 (d, J = 8.0 Hz, H-1), 4.47 (d, J = 4.8 Hz, H-5), 4.09 (q, J = 7.1 Hz, ester CH2), 3.88 (d, J = 6.4 Hz, glycine CH2), 3.85 (d, J = 6.0 Hz, glycine CH2), 3.83–3.78 (m, H-6), 3.19 (d, J = 16.1 Hz, CH2 bridge), 3.07 (d, J = 3.2 Hz, H-9), 2.93 (d, J = 16.0 Hz, CH2 bridge), 2.73 (d, J = 18.3 Hz, H-10), 2.48 (m, H-16), 2.38 (t, J = 1.8 Hz, H-10), 2.29 (s, H-14), 2.26–2.18 (m, H-16), 1.90 (td, J = 12.3, 4.9 Hz, H-15), 1.46 (dt, J = 10.8, 2.3 Hz, H-15), 1.40 (dq, J = 13.8, 7.1 Hz, H-8), 1.32 (td, J = 6.7, 3.6 Hz, H-7), 1.18 (m, ester CH3, H-7), 0.86 (d, J = 18.8 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 171.0 (amide C=O), 170.6 (ester C=O), 146.5 (C4), 138.5 (C3), 130.4 (C12), 125.3 (C11), 118.5 (C1), 117.2 (C2), 90.4 (C5), 66.7 (C6), 61.0 (ester CH2), 59.2 (CH2 bridge), 58.8 (C9), 45.1 (C16), 42.5 (C13), 41.0 (glycine CH2), 38.2 (C14), 37.5 (C15), 26.0 (C7), 23.0 (C10), 20.0 (C8), 14.6 (ester CH3)
(33) N-acetylglycine-norcodeine ethyl ester (ethyl 2-(2-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 90%, m.p.: oil, HR-MS [M + H+]: calculated: 429.2020, measured: 429.2011; 1H-NMR (600 MHz, DMSO-d6) δ 6.59 (d, J = 8.2, H-2), 6.44 (d, J = 8.3, H-1), 5.53 (d, J = 9.8 Hz, H-7), 5.21 (dt, J = 7.4, 3.4 Hz, H-8), 4.69 (d, J = 5.4 Hz, H-5), 4.10 (m, H-6), 4.06 (tdd, J = 7.2, 4.7, 2.6 Hz, ester CH2), 3.86 (m, glycine CH2), 3.69 (d, J = 3.2 Hz, OCH3), 3.37 (dt, J = 7.6, 3.8 Hz, H-9), 3.20 (dd, J = 16.3, 4.2 Hz, CH2 bridge), 2.98 (dd, J = 15.7, 3.8 Hz, CH2 bridge), 2.80 (dd, J = 18.5, 4.1 Hz, H-10), 2.75–2.69 (m, H-14), 2.58 (m, H-16), 2.41–2.31 (m, H-10, H-16), 2.11–1.99 (m, H-15), 1.61 (d, J = 12.9 Hz, H-15), 1.16 (ddd, J = 7.1, 4.7, 2.3 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 170.9 (amide C=O), 170.5 (ester C=O), 147.7 (C4), 141.8 (C3), 133.7 (C7), 131.5 (C12), 128.7 (C8), 127.5 (C11), 118.8 (C1), 113.8 (C2), 92.4 (C5), 67.0 (C6), 60.9 (ester CH2), 58.9 (CH2 bridge), 55.5 (OCH3), 57.7 (C9), 44.9 (C16), 43.6 (C13), 40.9 (glycine CH2), 40.6 (C14), 35.7 (C15), 23.4 (C10), 14.5 (ester CH3)
(34) N-acetylglycine-dihydronorcodeine ethyl ester (ethyl 2-(2-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 89% m.p.: 76 °C, HR-MS [M + H+]: calculated: 431.2177, measured: 431.2165; 1H-NMR (600 MHz, DMSO-d6) δ 6.70 (d, J = 8.2 Hz, H-2), 6.54 (d, J = 8.2 Hz, H-1), 4.49 (dd, J = 4.9, 3.6 Hz, H-5), 4.08 (q, J = 7.1 Hz, ester CH2), 3.88 (d, J = 6.3 Hz, glycine CH2), 3.84 (d, J = 5.9 Hz, glycine CH2), 3.82 (q, J = 3.9 Hz, H-6), 3.76 (s, OCH3), 3.20 (d, J = 16.1 Hz, CH2 bridge), 3.09 (dd, J = 6.1, 2.7 Hz, H-9), 2.93 (d, J = 16.1 Hz, CH2 bridge), 2.77 (d, J = 18.4 Hz, H-10), 2.48–2.40 (m, H-10), 2.30 (ddd, J = 11.5, 6.7, 2.7 Hz, H-14), 2.20 (td, J = 12.2, 3.4 Hz, H-16), 1.90 (td, J = 12.4, 4.9 Hz, H-15), 1.45 (dd, J = 11.6, 3.1 Hz, H-15), 1.39 (dt, J = 13.4, 6.8 Hz, H-8), 1.32 (qd, J = 6.8, 4.7 Hz, H-7), 1.18 (t, J = 7.1 Hz, ester CH3), 0.89 (tt, J = 12.7, 6.4 Hz, H-8); 13C-NMR (150 MHz, DMSO-d6) δ 171.0 (amide C=O), 170.4 (ester C=O), 147.4 (C4), 141.3 (C3), 130.6 (C12), 127.2 (C11), 118.5 (C1), 114.5 (C2), 90.7 (C5), 66.4 (C6), 60.8 (ester CH2), 59.1 (CH2 bridge), 58.6 (C9), 55.9 (OCH3), 45.0 (C16), 42.3 (C13), 40.9 (glycine CH2), 38.3 (C14), 37.3 (C15), 26.1 (C7), 22.9 (C10), 19.8 (C8), 14.5 (ester CH3)
(35) N-acetylglycine-noroxymorphone ethyl ester (ethyl 2-(2-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 95%, m.p.: 83 °C, HR-MS [M + H+]: calculated: 431.1813, measured: 431.1799; 1H-NMR (600 MHz, DMSO-d6) δ 6.56 (d, J = 8.1 Hz, H-2), 6.53 (d, J = 8.1 Hz, H-1), 4.77 (s, H-5), 4.09 (qd, J = 7.1, 1.2 Hz, ester CH2), 3.95 (d, J = 6.7 Hz, glycine CH2), 3.80 (d, J = 5.7 Hz, glycine CH2), 3.23 (d, J = 16.0 Hz, CH2 bridge), 3.02–2.89 (m, CH2 bridge, H-9, H-7), 2.59–2.53 (m, H-16), 2.53–2.51 (m, H-10), 2.43 (td, J = 12.6, 5.0 Hz, H-15), 2.08 (dt, J = 14.3, 3.2 Hz, H-7), 1.99 (td, J = 12.0, 3.7 Hz, H-16), 1.73 (ddd, J = 13.3, 5.2, 3.0 Hz, H-8), 1.29 (dt, J = 11.9, 2.7 Hz, H-15), 1.18 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 209.3 (C6), 170.5 (amide C=O), 170.4 (ester C=O), 143.8 (C4), 139.8 (C3), 130.1 (C12), 123.9 (C11), 119.5 (C1), 117.6 (C2), 89.7 (C5), 70.7 (C14), 64.0 (C9), 60.9 (ester CH2), 59.5 (CH2 bridge), 50.1 (C13), 44.2 (C16), 40.9 (glycine CH2), 36.2 (C7), 31.6 (C8), 29.8 (C15), 24.5 (C10), 14.5 (ester CH3)
(36) N-acetylglycine-noroxycodone ethyl ester (ethyl 2-(2-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetate) yield: 96% m.p.: 81 °C, HR-MS [M + H+]: calculated: 445.1969, measured: 445.1964; 1H-NMR (600 MHz, DMSO-d6) δ 6.75 (d, J = 8.2 Hz, H-2), 6.67 (d, J = 8.2 Hz, H-1), 4.85 (s, H-5), 4.09 (qd, J = 7.1, 1.4 Hz, ester CH2), 3.94 (dd, J = 17.3, 6.7 Hz, glycine CH2), 3.79 (s, glycine CH2), 3.78 (s, OCH3), 3.24 (d, J = 16.0 Hz, CH2 bridge), 3.08–2.92 (m, CH2 bridge, H-7, H-10), 2.65–2.54 (m, H-10, H-16), 2.40–2.37 (m, H-15), 2.08 (dt, J = 14.1, 3.1 Hz, H-7), 1.97 (td, J = 12.0, 3.7 Hz, H-16), 1.75 (ddd, J = 13.3, 5.1, 2.9 Hz, H-8), 1.51–1.37 (m, H-8), 1.32–1.27 (m, H-15), 1.19 (t, J = 7.1 Hz, ester CH3); 13C-NMR (150 MHz, DMSO-d6) δ 209.0 170.4 (amide C=O), 170.3 (ester C=O), (C6), 142.5 (C3), 144.8 (C4), 130.3 (C12), 126.1 (C11), 119.8 (C1), 115.3 (C2), 90.1 (C5), 70.6 (C14), 60.9 (ester CH2), 59.4 (CH2 bridge), 56.8 (OCH3), 50.2 (C13), 44.1 (C16), 40.7 (glycine CH2), 36.0 (C7), 31.8 (C8), 63.9 (C9), 29.4 (C15), 24.6 (C10), 14.5 (ester CH3)
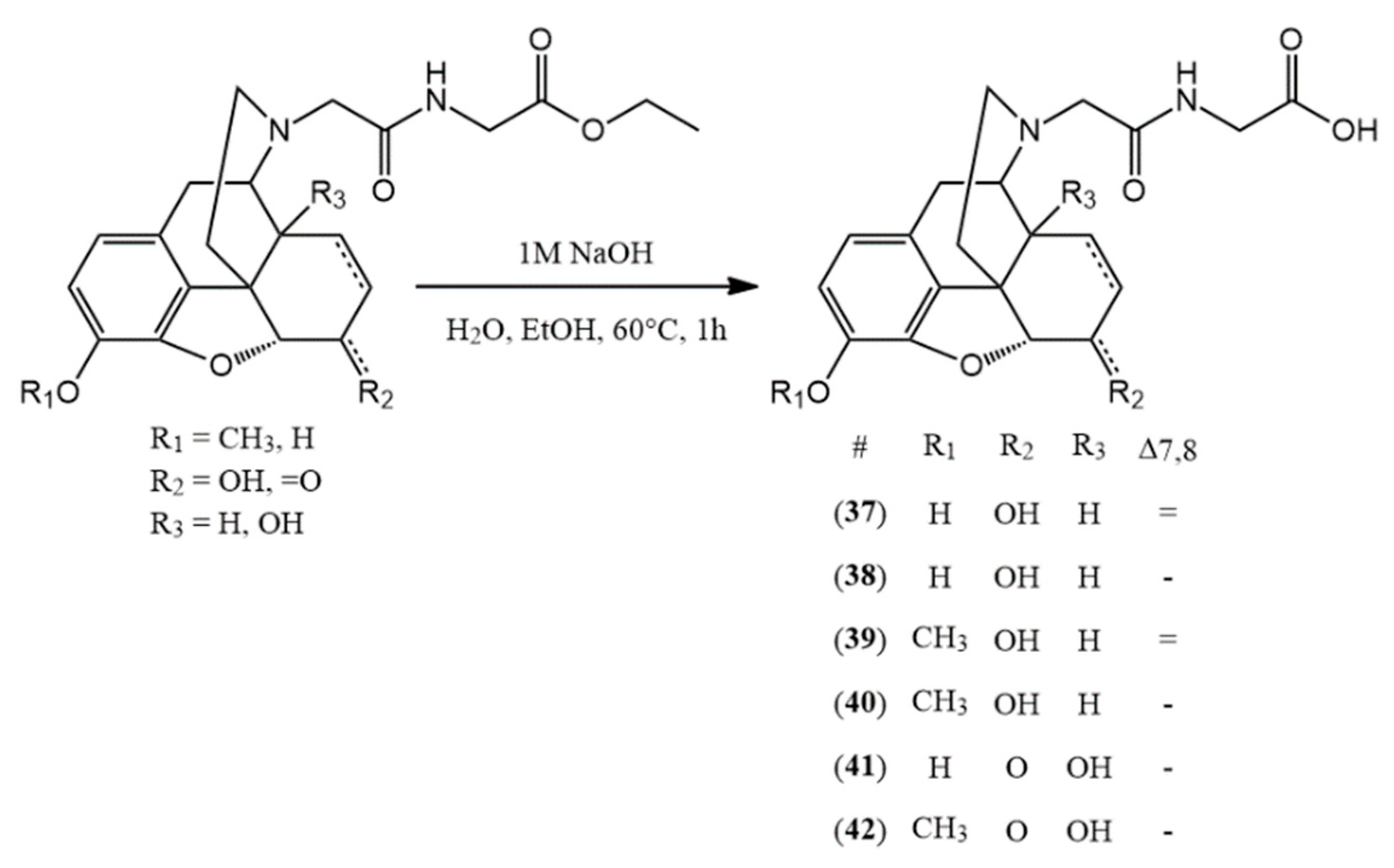
3.2.7. General Synthesis of N-Acetylglycine-Nor-Compounds
The N-acetylglycine-nor-compound ethyl ester (0.5 mmol) was dissolved in the mixture of ethanol (1 g) and water (2,5 mL). The mixture was hydrolyzed with sodium hydroxide (1 M, 0.5 mmol) for 1 h at 60 °C. The hydrolysis reaction was monitored by TLC and upon complete disapparance of the starting material, the pH was adjusted to 3–4 with 10% HCl solution. Then the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
(37) N-acetylglycine-normorphine HCl (2-(2-((7aR)-7,9-dihydroxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 98%, m.p.: > 205 °C decomp., HR-MS [M + H+]: calculated: 387.1551, measured: 387.1547; 1H-NMR (600 MHz, D2O) δ 6.65 (d, J = 8.1 Hz, H-2), 6.57 (d, J = 8.2 Hz, H-1), 5.63 (dd, J = 9.8, 2.8 Hz, H-7), 5.25 (d, J = 10.0 Hz, H-8), 4.96 (dd, J = 6.4, 1.2 Hz, H-5), 4.33–4.00 (m, H-6, H-9), 3.94 (s, glycine CH2), 3.51 (d, J = 7.1 Hz, CH2 bridge), 3.38 (s, H-16), 3.15 (d, J = 20.1 Hz, H-10), 3.02 (s, H-14), 2.42–2.23 (m, H-15), 2.05 (d, J = 14.4 Hz, H-15); 13C-NMR (150 MHz, D2O) δ 173.1 (carboxylic C=O), 165.3 (amide C=O), 145.5 (C4), 138.0 (C3), 133.1 (C7), 129.2 (C12), 125.5 (C8), 123.0 (C11), 120.3 (C1), 117.7 (C2), 90.3 (C5), 65.2 (C6), 60.4 (C9), 57.6 (CH2 bridge), 47.1 (C16), 41.7 (C13), 41.2 (glycine CH2), 38.0 (C14), 32.2 (C15), 22.0 (C10)
(38) N-acetylglycine-dihydronormorphine HCl (2-(2-((7aR)-7,9-dihydroxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 94%, m.p.: > 225 °C decomp., HR-MS [M + H+]: calculated: 389.1707, measured: 389.1765; 1H-NMR (600 MHz, D2O) δ 6.83 (d, J = 8.3 Hz, H-2), 6.65 (d, J = 8.3 Hz, H-1), 4.74 (d, J = 5.6 Hz, H-5), 4.41–4.17 (m, H-6, H-9), 4.16–4.00 (m, H-6, H-9), 3.96 (d, J = 3.6 Hz, glycine CH2) 3.31 (d, J = 14.0 Hz, H-16), 3.11 (t, J = 19.5 Hz, H-10), 2.89 (d, J = 13.5 Hz, CH2 bridge), 2.53 (m, H-14), 2.11 (s, H-15), 1.81 (d, J = 13.0 Hz, H-15), 1.47 (m, H-8), 0.95 (m, H-8); 13C-NMR (150 MHz, D2O) δ 172.9 (carboxylic C=O), 165.3 (amide C=O), 145.6 (C4), 138.5 (C3), 128.7 (C12), 123.2 (C11), 120.0 (C1), 117.1 (C2), 89.7 (C5), 66.1 (C6), 61.1 (C9), 57.0 (CH2 bridge), 47.8 (C16), 41.4 (glycine CH2), 41.2 (C13), 38.2 (C14), 32.9 (C15), 25.7 (C7), 21.6 (C10), 17.9 (C8)
(39) N-acetylglycine-norcodeine HCl (2-(2-((7aR)-7-hydroxy-9-methoxy-4,4a,7,7a-tetrahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 99%, m.p.: > 200 °C decomp., HR-MS [M + H+]: calculated: 401.1707, measured: 401.1704; 1H-NMR (600 MHz, D2O) δ 6.80 (d, J = 8.3 Hz, H-2), 6.66 (d, J = 8.3 Hz, H-1), 5.62 (d, J = 9.9 Hz, H-7), 5.25 (d, J = 10.0 Hz, H-8), 4.97 (d, J = 6.3 Hz, H-5), 4.29–4.23 (m, H-6, H-9), 4.16–4.06 (m, CH2 bridge, glycine CH2), 3.72 (s, OCH3), 3.39 (m, H-16), 3.20–3.17 (m, H-10, H-16), 3.03 (m, H-14), 2.33 (t, J = 13.0 Hz, H-15), 2.04 (d, J = 14.5 Hz, H-15); 13C-NMR (150 MHz, D2O) δ 173.2 (carboxylic C=O), 165.4 (amide C=O), 146.3 (C4), 142.2 (C3), 133.0 (C7), 128.8 (C12), 125.2 (C8), 123.8 (C11), 120.2 (C1), 114.7 (C2), 90.7 (C5), 65.7 (C6), 60.4 (C9), 56.8 (OCH3), 54.5 (CH2 bridge), 47.1 (C16), 41.3 (glycine CH2), 41.9 (C13), 37.9 (C14), 32.2 (C15), 22.1 (C10)
(40) N-acetylglycine-dihydronorcodeine HCl (2-(2-((7aR)-7-hydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 95%, m.p.: > 205 °C decomp., HR-MS [M + H+]: calculated: 403.1864, measured: 403.1852; 1H-NMR (600 MHz, D2O) δ 6.87 (d, J = 8.3 Hz, H-2), 6.74 (d, J = 8.4 Hz, H-1), 4.70 (d, J = 5.4 Hz, H-5), 4.03 (d, J = 4.7 Hz, H-6), 3.94 (s, H-9, glycine CH2), 3.76 (s, OCH3), 3.30 (d, J = 13.0 Hz, H-16), 3.12 (d, J = 19.4 Hz, H-10), 2.80–2.74 (m, CH2 bridge), 2.49 (s, H-14), 2.11 (s, H-15), 1.79 (d, J = 14.0 Hz, H-15), 1.48–1.43 (m, H-8), 0.96–0.91 (m, H-8); 13C-NMR (150 MHz, D2O) δ 172.8 (carboxylic C=O), 165.6 (amide C=O), 145.8 (C4), 141.7 (C3), 127.9 (C12), 123.3 (C11), 120.0 (C1), 115.1 (C2), 89.2 (C5), 66.0 (C6), 61.5 (C9), 57.0 (OCH3), 54.4 (CH2 bridge), 47.9 (C16), 41.4 (glycine CH2), 40.6 (C13), 38.2 (C14), 33.9 (C15), 26.0 (C7), 21.4 (C10), 17.6 (C8)
(41) N-acetylglycine-noroxymorphone HCl (2-(2-((7aR)-4a,9-dihydroxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 98%, m.p.: > 190 °C decomp., HR-MS [M + H+]: calculated: 403.1500, measured: 403.1495; 1H-NMR (600 MHz, D2O) δ 6.73 (d, J = 8.2 Hz, H-2), 6.70 (d, J = 8.3 Hz, H-1), 4.95 (s, H-5), 4.19 (d, J = 16.1 Hz, CH2 bridge), 3.99–3.88 (m, CH2 bridge, glycine CH2), 3.83 (d, J = 6.1 Hz, H-9), 3.33 (s, H-10), 3.20 (dd, J = 13.2, 4.9 Hz, H-16), 3.08 (dd, J = 20.1, 6.3 Hz, H-10), 2.90 (td, J = 14.8, 4.8 Hz, H-7), 2.68 (td, J = 13.5, 5.0 Hz, H-15), 2.22 (dt, J = 14.8, 3.2 Hz, H-7), 1.99–1.92 (m, H-8), 1.72–1.59 (m, H-8, H-15); 13C-NMR (150 MHz, D2O) δ 211.5 (C6), 172.4 (carboxylic C=O), 165.2 (amide C=O), 143.2 (C4), 138.8 (C3), 127.1 (C12), 121.5 (C11), 121.1 (C1), 118.7 (C2), 89.3 (C5), 70.7 (C14), 65.0 (C9), 53.6 (CH2 bridge), 48.7 (C13), 47.0 (C16), 41.6 (glycine CH2), 34.5 (C7), 30.4 (C8), 27.4 (C15), 23.8 (C10)
(42) N-acetylglycine-noroxycodone HCl (2-(2-((7aR)-4a-hydroxy-9-methoxy-7-oxo-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-3(2H)-yl)acetamido)acetic acid hydrochloride) yield: 93%, m.p.: > 180 °C decomp., HR-MS [M + H+]: calculated: 417.1656, measured: 417.1642; 1H-NMR (600 MHz, D2O) δ 6.89 (d, J = 8.3 Hz, H-2), 6.81 (d, J = 8.4 Hz, H-1), 4.98 (s, H-5), 4.18 (d, J = 16.1 Hz, CH2 bridge), 3.95 (d, J = 16.2 Hz, CH2 bridge), 3.86 (d, J = 6.2 Hz, H-9), 3.79 (d, J = 3.6 Hz, glycine CH2), 3.77 (s, OCH3), 3.35 (d, J = 20.1 Hz, H-10), 3.22 (dd, J = 13.0, 4.8 Hz, H-16), 3.11 (dd, J = 20.1, 6.3 Hz, H-10), 2.97–2.83 (m, H-7), 2.69 (td, J = 13.5, 5.0 Hz, H-15), 2.22 (dt, J = 14.9, 3.3 Hz, H-7), 1.98 (ddd, J = 14.5, 5.3, 3.1 Hz, H-8), 1.73–1.66 (m, H-15), 1.63 (td, J = 14.6, 3.4 Hz, H-8); 13C-NMR (150 MHz, D2O) δ 211.3, 175.1 (carboxylic C=O), 165.2 (amide C=O), (C6), 144.0 (C4), 142.8 (C3), 127.1 (C12), 122.5 (C11), 121.0 (C1), 115.6 (C2), 89.4 (C5), 70.8 (C14), 65.0 (C9), 56.5 (OCH3), 53.8 (CH2 bridge), 48.8 (C13), 47.0 (C16), 42.8 (glycine CH2), 34.4 (C7), 30.4 (C8), 27.3 (C15), 23.8 (C10)





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}