
Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Neurodegenerative Diseases
3. Parkinson’s Disease
3.1. Symptoms
3.2. Epidemiology
3.3. Molecular Mechanisms and Causes
3.4. Treatments
4. Neuroprotective Properties of Green Tea against Parkinson’s Disease
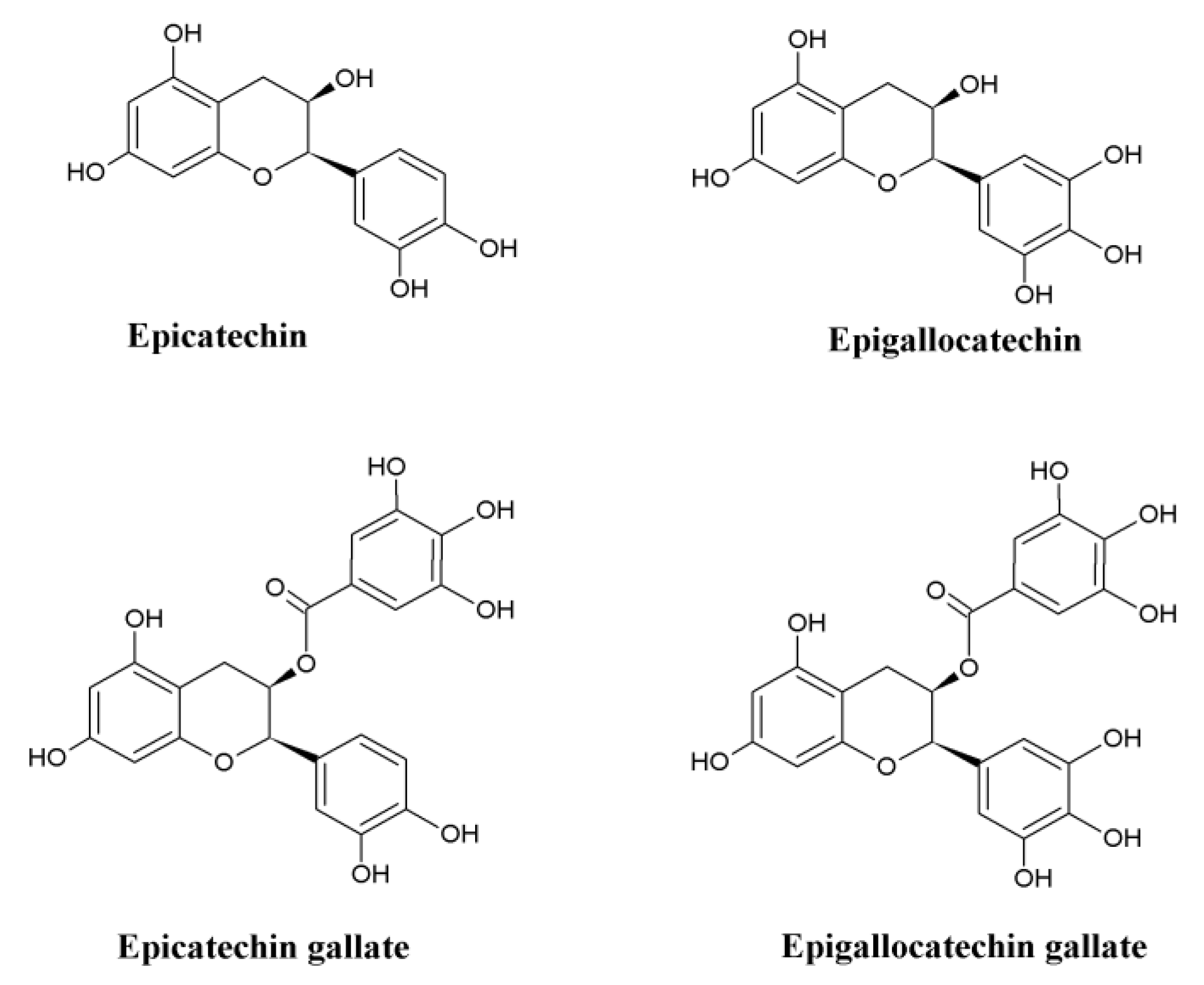
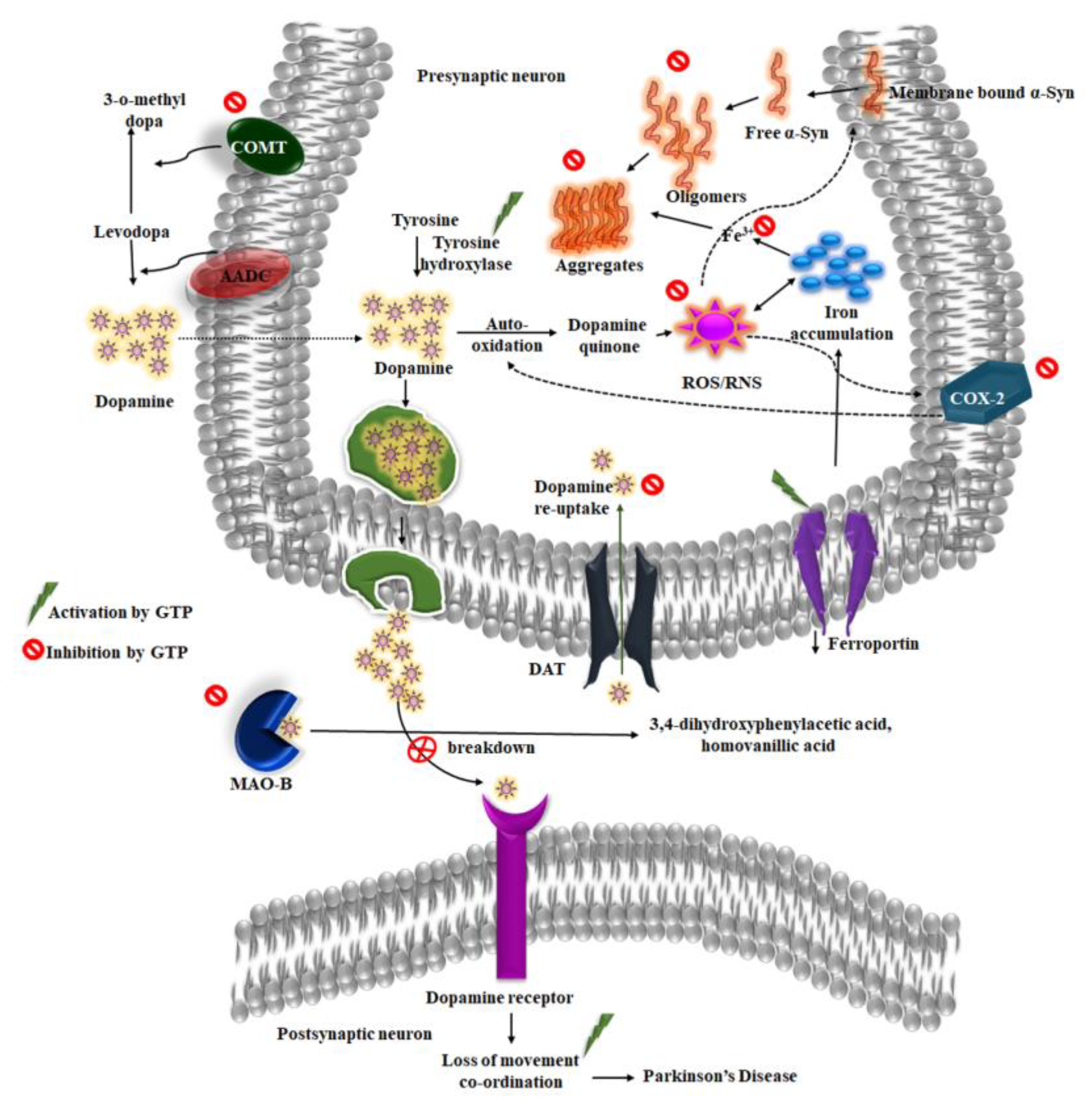
4.1. Green Tea and α-Synuclein
4.2. Dopamine and Green Tea Extract
4.3. Inhibition of MAO-B
4.4. Iron and Green Tea
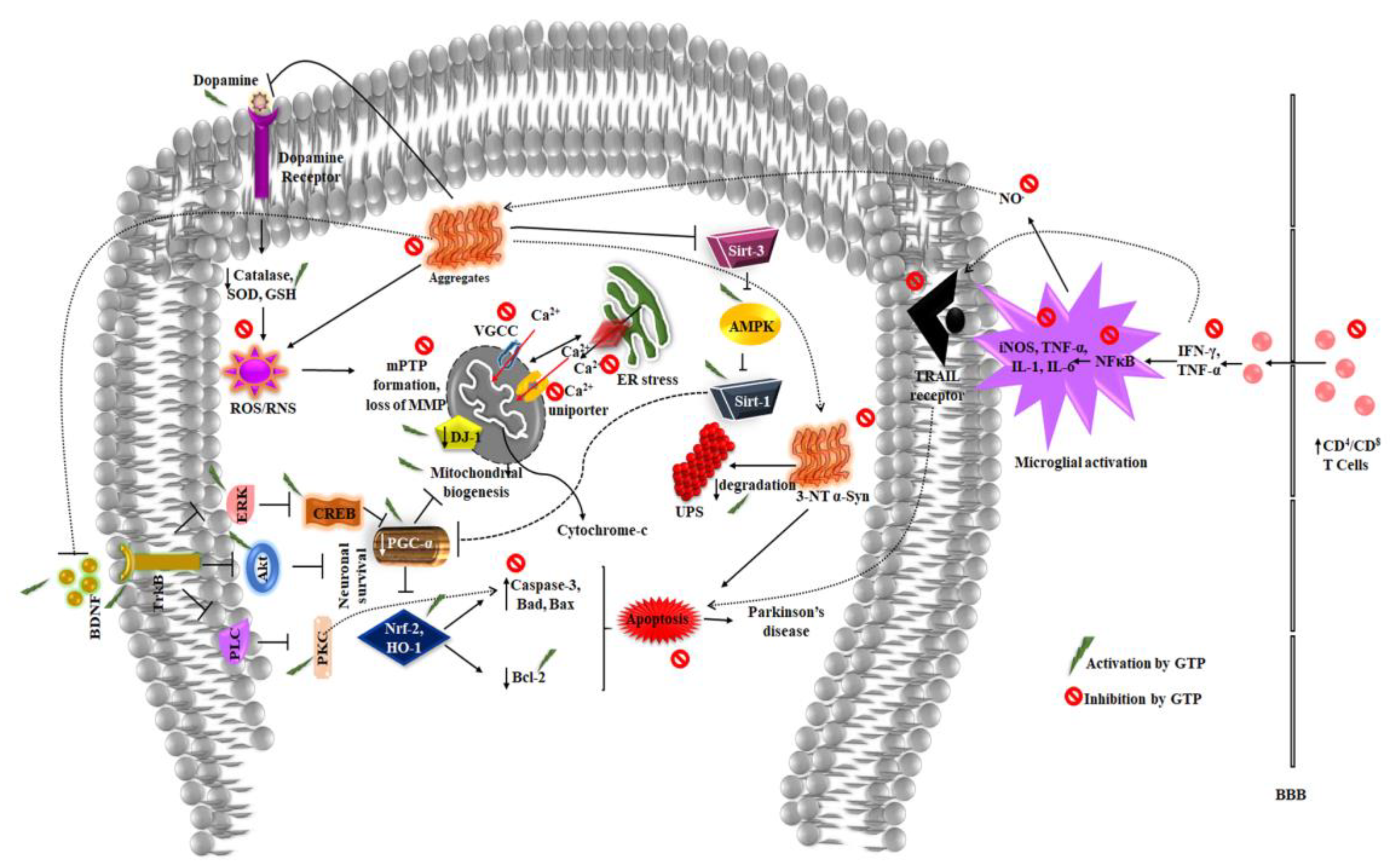
4.5. Antioxidant Potential of Green Tea in Combating Oxidative Stress
4.6. Green Tea in Alleviating Mitochondrial Dysfunction
4.7. Activation of Neurotrophic Factors and Signaling Pathways
4.8. Neuroinflammation
4.9. Alteration of the Gut Microbiome
5. Future Perspective and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinsons disease |
| AD | Alzheimer’s disease |
| HD | Huntington’s disease |
| ROS | Reactive oxygen species |
| MAO-B | Monoamine oxidase B |
| BBB | Blood brain barrier |
| EGCG | Epigallocatechin-3-gallate |
| SNpc | Substantia Nigra pars compacta |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| 6-OHDA | 6-hydroxydopamine |
| TH | Tyrosine hydroxylase |
| SOD | Superoxide dismutase |
| L-DOPA | L-dihydroxyphenylalanine |
| PKC-α | Protein kinase C-α |
| DAT | Dopamine transporters |
| COMT | Catecholamine-O-methyltransferase |
| 4-HNE | 4-hydroxyl-2-nonenal |
| MDA | Malondialdehyde |
| GSH | Glutathione |
| 3-NT | 3-Nitrotyrosine |
| mPTP | Mitochondrial membrane permeability transition pore |
| MMP | Mitochondrial membrane potential |
| PGC-1α | Peroxisome proliferator-activated receptor γ coactivator-1 α |
| TrkB | Tyrosine receptor kinase B |
| CNS | Central nervous system |
| TRAIL | TNF-related apoptosis-inducing ligand |
References
- Weisburger, J.H. Tea and health: A historical perspective. Cancer Lett. 1997, 114, 315–317. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Gimenez, R. Beneficial effects of green tea—A review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.C. Green Tea—Its cultivation, processing of the leaves for drinking materials and kinds of green tea. In Chemistry and Applications of Green Tea; Yamamoto, T., Juneja, L.R., Chu, D.C., Kim, M., Eds.; CRC Press: New York, NY, USA, 1997; pp. 1–11. [Google Scholar]
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A Review of the Role of Green Tea (Camellia sinensis) in Antiphotoaging, Stress Resistance, Neuroprotection, and Autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Dumitrascu, D.I.; Capitanescu, B.; Petcu, E.B.; Surugiu, R.; Fang, W.H.; Dumbrava, D.A. Dietary habits, lifestyle factors and neurodegenerative diseases. Neural. Regen. Res. 2020, 15, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Biswas, A. Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci. 2015, 2, 54. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Molloy, S.; Bain, P.G.; Chaudhuri, K.R.; Piccini, P. Parkinson’s disease symptoms: The patient’s perspective. Mov. Disord. 2010, 25, 1646–1651. [Google Scholar] [CrossRef]
- Lees, A.J.; Smith, E. Cognitive deficits in the early stages of Parkinson’s disease. Brain 1983, 106, 257–270. [Google Scholar] [CrossRef]
- Levin, B.E.; Llabre, M.M.; Weiner, W.J. Cognitive impairments associated with early Parkinson’s disease. Neuropsychology 1989, 39, 557–561. [Google Scholar] [CrossRef]
- Dubois, B.; Pillon, B. Cognitive deficits in Parkinson’s disease. J. Neurol. 1997, 244, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.G.; Marsden, C.D. Cognitive function in Parkinson’s disease: From description to theory. Trends Neurosci. 1990, 13, 21–29. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Auriacombe, S.; Grossman, M.; Carvell, S.; Stephen, G.; Matthew, S.B.; Howard, H.I. Verbal fluency deficits in Parkinson’s disease. Neuropsychology 1993, 7, 182–192. [Google Scholar] [CrossRef]
- Peran, P.; Rascol, O.; Demonet, J.F.; Celsis, P.; Nespoulous, J.L.; Dubois, B.; Cardebat, D. Deficit of verb generation in nondemented patients with Parkinson’s disease. Mov. Disord. 2003, 18, 150–156. [Google Scholar] [CrossRef]
- Cooper, J.A.; Sagar, H.J.; Jordan, N.; Harvey, N.S.; Sullivan, E.V. Cognitive impairment in early, untreated Parkinson’s disease and its relationship to motor disability. Brain 1991, 114, 2095–2122. [Google Scholar] [CrossRef]
- Levin, B.E.; Llabre, M.M.; Reisman, S.; Weiner, W.J.; Sanchez-Ramos, J.; Singer, C.; Brown, M.C. Visuospatial impairment in Parkinson’s disease. Neuropsychology 1991, 41, 365–369. [Google Scholar] [CrossRef]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Nielsen, H.; Kragh-Sørensen, P. Risk of dementia in Parkinson’s disease. A community-based prospective study. Neuropsychology 2001, 56, 730–736. [Google Scholar] [CrossRef]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef]
- De-Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- De-Rijk, M.C.; Breteler, M.M.; Graveland, G.A.; Ott, A.; Grobbee, D.E.; van der Meché, F.G.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neuropsychology 1995, 45, 2143–2146. [Google Scholar] [CrossRef] [PubMed]
- Quinn, N.; Critchley, P.; Marsden, C.D. Young onset Parkinson’s disease. J. Mov. Disord. 1987, 2, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Wooten, G.; Currie, L.; Bovbjerg, V.; Lee, J.; Patrie, J. Are men at greater risk for Parkinson’s disease than women? J. Neurol. Neurosurg. Psychiatry 2004, 75, 637–639. [Google Scholar] [CrossRef]
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head injury and risk of Parkinson disease: A systematic review and meta-analysis. Mov. Disord. 2013, 28, 1222–1229. [Google Scholar] [CrossRef]
- Hubble, J.P.; Cao, T.; Hassanein, R.; Neuberger, J.; Roller, W. Risk factors for Parkinson’s disease. Neuropsychology 1993, 43, 1693. [Google Scholar]
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Maguire-Zeiss, K.A.; Short, D.W.; Federoff, H.J. Synuclein, dopamine and oxidative stress: Co-conspirators in Parkinson’s disease? Brain Res. Mol. Brain Res. 2005, 134, 18–23. [Google Scholar] [CrossRef]
- Lew, M. Overview of Parkinson’s disease. Pharmacotherapy 2007, 27, 155S–160S. [Google Scholar] [CrossRef]
- Duvoisin, R.C. Overview of Parkinson’s disease. Ann. N.Y. Acad. Sci. 1992, 648, 187–193. [Google Scholar] [CrossRef]
- Schapira, A.H. Etiology and pathogenesis of Parkinson disease. Neurol. Clin. 2009, 27, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Goers, J.; Uversky, V.N.; Fink, A.L. Polycation-induced oligomerization and accelerated fibrillation of human α-synuclein in vitro. Protein Sci. 2003, 12, 702–707. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Schapira, A.; Cooper, J.; Dexter, D.; Clark, J.; Jenner, P.; Marsden, C. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Przedborski, S.; Kostic, V.; Jackson-Lewis, V.; Naini, A.B.; Simonetti, S.; Fahn, S.; Carlson, E.; Epstein, C.J.; Cadet, J.L. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced neurotoxicity. J. Neurosci. 1992, 12, 1658–1667. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Rogers, G.; Davies, D.; Pink, J.; Cooper, P. Parkinson’s disease: Summary of updated NICE guidance. BMJ 2017, 358, j1951. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.; Rogers, J.; Sibley, J. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990, 335, 1037. [Google Scholar] [CrossRef]
- Morgan, J.C.; Sethi, K.D. Emerging drugs for Parkinson’s disease. Expert Opin. Emerg. Drugs 2006, 11, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Quan, Z.; Wong, W.; Guo, J.; Zhang, R.; Yang, Q.; Dai, R.; McGeer, P.L.; Qing, H. Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2788–2796. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Sun, Q.; Chen, C.; Xu, L.; Ding, Y.; Zhou, P. (-)-Epigallocatechin-3-gallate (EGCG) inhibits fibrillation, disaggregates amyloid fibrils of α-synuclein, and protects PC12 cells against α-synuclein-induced toxicity. RSC Adv. 2017, 7, 32508–32517. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Nielsen, S.B.; Yoshimura, Y.; Vad, B.S.; Andersen, C.B.; Betzer, C.; Kaspersen, J.D.; Christiansen, G.; Pedersen, J.S.; Jensen, P.H.; et al. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J. Biol. Chem. 2014, 289, 21299–21310. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.R.; Klein, J.C.; Mackay, C.E.; Hu, M.T.M. Neuroimaging in pre-motor Parkinson’s disease. Neuroimage Clin. 2017, 15, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of Motor Dysfunction in Parkinson’s Disease as the Rationale for Drug Treatment and Rehabilitation. Parkinsons Dis. 2016, 2016, 9832839. [Google Scholar] [CrossRef] [PubMed]
- Salari, S.; Bagheri, M. In vivo, in vitro and pharmacologic models of Parkinson’s disease. Physiol. Res. 2019, 68, 17–24. [Google Scholar] [CrossRef]
- Chen, M.; Wang, T.; Yue, F.; Li, X.; Wang, P.; Li, Y.; Chan, P.; Yu, S. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral α-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 2015, 286, 383–392. [Google Scholar] [CrossRef]
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.; Mandel, S. Green tea polyphenol (-)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem. 2001, 78, 1073–1082. [Google Scholar] [CrossRef]
- Mandel, S.; Maor, G.; Youdim, M.B. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: Effect of neuroprotective drugs R-apomorphine and green tea polyphenol (-)-epigallocatechin-3-gallate. J. Mol. Neurosci. 2004, 24, 401–416. [Google Scholar] [CrossRef]
- Kalfon, L.; Youdim, M.B.; Mandel, S.A. Green tea polyphenol (-)-epigallocatechin-3-gallate promotes the rapid protein kinase C- and proteasome-mediated degradation of Bad: Implications for neuroprotection. J. Neurochem. 2007, 100, 992–1002. [Google Scholar] [CrossRef]
- Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001, 21, 2929–2938. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Fei, J.; Zhou, X.; Jankovic, J.; Le, W. Effects of green tea polyphenols on dopamine uptake and on MPP+ -induced dopamine neuron injury. Life Sci. 2003, 72, 1073–1083. [Google Scholar] [CrossRef]
- Huot, P.; Fox, S.H.; Brotchie, J.M. Dopamine Reuptake Inhibitors in Parkinson’s Disease: A Review of Nonhuman Primate Studies and Clinical Trials. J. Pharmacol. Exp. Ther. 2016, 357, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Peng, N.; Li, X.P.; Le, W.D. (-)-Epigallocatechin gallate regulates dopamine transporter internalization via protein kinase C-dependent pathway. Brain Res. 2006, 1097, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Wen, Y.; Yamabe, N.; Fukui, M.; Bishop, S.C.; Zhu, B.T. Dual beneficial effects of (-)-epigallocatechin-3-gallate on levodopa methylation and hippocampal neurodegeneration: In vitro and in vivo studies. PLoS ONE 2010, 5, e11951. [Google Scholar] [CrossRef]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef]
- Lin, S.M.; Wang, S.W.; Ho, S.C.; Tang, Y.L. Protective effect of green tea (-)-epigallocatechin-3-gallate against the monoamine oxidase B enzyme activity increase in adult rat brains. Nutrition 2010, 26, 1195–1200. [Google Scholar] [CrossRef]
- Batista-Nascimento, L.; Pimentel, C.; Menezes, R.A.; Rodrigues-Pousada, C. Iron and neurodegeneration: From cellular homeostasis to disease. Oxid. Med. Cell. Longev. 2012, 2012, 128647. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, L.; Liang, Q.; Sun, Q.; Chen, C.; Zhang, Y.; Ding, Y.; Zhou, P. Metal chelator EGCG attenuates Fe(III)-induced conformational transition of α-synuclein and protects AS-PC12 cells against Fe(III)-induced death. J. Neurochem. 2017, 143, 136–146. [Google Scholar] [CrossRef]
- Bao, G.H.; Xu, J.; Hu, F.L.; Wan, X.C.; Deng, S.X.; Barasch, J. EGCG inhibit chemical reactivity of iron through forming anNgal-EGCG-iron complex. Biometals 2013, 26, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Langley, M.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin Gallate Has a Neurorescue Effect in a Mouse Model of Parkinson Disease. J. Nutr. 2017, 147, 1926–1931. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, L.; Kalfon, L.; Amit, T.; Youdim, M.B.; Mandel, S.A. Low dosage of rasagiline and epigallocatechin gallate synergistically restored the nigrostriatal axis in MPTP-induced parkinsonism. Neurodegener Dis. 2010, 7, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Danaraddi, S.; Koneru, A.; Hunasgi, S.; Ramalu, S.; Vanishree, M. Natural ways to prevent and treat oral cancer. J. Oral Res. Rev. 2014, 6, 34–39. [Google Scholar] [CrossRef]
- Chen, D.; Zhou, Y.; Lyons, K.E.; Pahwa, R.; Reddy, M.B. Green Tea Consumption Reduces Oxidative Stress in Parkinson’s Disease Patients. J. Behav. Brain Sci. 2015, 5, 194–202. [Google Scholar] [CrossRef]
- Siddique, Y.H.; Jyoti, S.; Naz, F. Effect of epicatechingallate dietary supplementation on transgenic Drosophila model of Parkinson’s disease. J. Diet. Suppl. 2014, 11, 121–130. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef]
- Ishiakawa, N.; Nakashima, I. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J. Cell Sci. 2000, 113, 635–641. [Google Scholar]
- Qin, Z.; Hu, D.; Han, S.; Reaney, S.H.; Di Monte, D.A.; Fink, A.L. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J. Biol. Chem. 2007, 282, 5862–5870. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, S.; Castro, E.; Rocha-Pereira, P.; Rebelo, I.; Rocha, S.; Santos-Silva, A. The effect of green tea in oxidative stress. Clin. Nutr. 2006, 25, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, M.; Miyazaki, I.; Murakami, S.; Kita, T.; Asanuma, M. L-Theanine protects against excess dopamine-induced neurotoxicity in the presence of astrocytes. J. Clin. Biochem. Nutr. 2016, 59, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3-18-deficient mice. Neurobiol. Dis. 2012, 45, 973–982. [Google Scholar] [CrossRef]
- Iwata-Ichikawa, E.; Kondo, Y.; Miyazaki, I.; Asanuma, M.; Ogawa, N. Glial cells protect neurons against oxidative stress via transcriptional up-regulation of the glutathione synthesis. J. Neurochem. 1999, 72, 2334–2344. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef]
- Guo, S.; Yan, J.; Yang, T.; Yang, X.; Bezard, E.; Zhao, B. Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson’s disease through inhibition of ROS-NO pathway. Biol. Psychiatry 2007, 62, 1353–1362. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Duda, J.E.; Giasson, B.I.; Chen, Q.; Gur, T.L.; Hurtig, H.I.; Stern, M.B.; Gollomp, S.M.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am. J. Pathol. 2000, 157, 1439–1445. [Google Scholar] [CrossRef]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Perez, D.A.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Epigallocatechin-3-Gallate Protects and Prevents Paraquat-Induced Oxidative Stress and Neurodegeneration in Knockdown dj-1-β Drosophila melanogaster. Neurotox Res. 2018, 34, 417. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Grady, J.P.; Cosgrave, E.M.; Bennison, E.; Chen, C.; Hepplewhite, P.D.; Morris, C.M. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Guo, S.; Bezard, E.; Zhao, B. Protective effect of green tea polyphenols on the SH-SY5Y cells against 6-OHDA induced apoptosis through ROS-NO pathway. Free Radic. Biol. Med. 2005, 39, 682–695. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Zhang, H.; Diao, X.; Zhao, S.; Zhou, W. Reduction in Autophagy by (-)-Epigallocatechin-3-Gallate (EGCG): A Potential Mechanism of Prevention of Mitochondrial Dysfunction After Subarachnoid Hemorrhage. Mol. Neurobiol. 2017, 54, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1α Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front. Mol. Neurosci. 2017, 10, 443. [Google Scholar] [CrossRef]
- Ye, Q.; Ye, L.; Xu, X.; Huang, B.; Zhang, X.; Zhu, Y.; Chen, X. Epigallocatechin-3-gallate suppresses 1-methyl-4-phenyl-pyridine-induced oxidative stress in PC12 cells via the SIRT1/PGC-1α signaling pathway. BMC Complement. Altern. Med. 2012, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. Global PD Gene Expression (GPEX) Consortium. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, H.F.; Zhang, Z.F.; Liu, Z.G.; Pei, X.R.; Wang, J.B.; Cai, M.Y.; Li, Y. Long-term administration of green tea catechins prevents age-related spatial learning and memory decline in C57BL/6 J mice by regulating hippocampal cyclic amp-response element binding protein signaling cascade. Neuroscience 2009, 159, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Kim, S.; Lee, S.Y.; Park, J.A.; Kim, S.J.; Chun, H.S. Protective effect of the green tea component, L-theanine on environmental toxins-induced neuronal cell death. Neurotoxicology 2008, 29, 656–662. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, H.; Boyd, C.; Spencer, J.P.; Williams, R.J.; Cadenas, E.; Rice-Evans, C. MAPK signaling in neurodegeneration: Influences of flavonoids and of nitric oxide. Neurobiol. Aging 2002, 23, 861–880. [Google Scholar] [CrossRef]
- Cavanaugh, J.E.; Jaumotte, J.D.; Lakoski, J.M.; Zigmond, M.J. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J. Neurosci. Res. 2006, 84, 1367–1375. [Google Scholar] [CrossRef]
- Di Cristo, G.; Berardi, N.; Cancedda, L.; Pizzorusso, T.; Putignano, E.; Ratto, G.M.; Maffei, L. Requirement of ERK activation for visual cortical plasticity. Science 2001, 292, 2337–2340. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Ding, M.L.; Ma, H.; Man, Y.G.; Lv, H.Y. Protective effects of a green tea polyphenol, epigallocatechin-3-gallate, against sevoflurane-induced neuronal apoptosis involve regulation of CREB/BDNF/TrkB and PI3K/Akt/mTORsignalling pathways in neonatal mice. Can. J. Physiol. Pharmacol. 2017, 95, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Kida, S.; Serita, T. Functional roles of CREB as a positive regulator in the formation and enhancement of memory. Brain Res. Bull. 2014, 105, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Timmons, S.; Coakley, M.F.; Moloney, A.M.; O’Neill, C. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci. Lett. 2009, 467, 30–35. [Google Scholar] [CrossRef]
- Chao, J.; Lau, W.K.; Huie, M.J.; Ho, Y.S.; Yu, M.S.; Lai, C.S.; Wang, M.; Yuen, W.H.; Lam, W.H.; Chan, T.H.; et al. A pro-drug of the green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG) prevents differentiated SH-SY5Y cells from toxicity induced by 6-hydroxydopamine. Neurosci. Lett. 2010, 469, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S.; Buendia, E. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152. [Google Scholar] [CrossRef]
- Zhou, T.; Zhu, M.; Liang, Z. (-)-Epigallocatechin-3-gallate modulates peripheral immunity in the MPTP-induced mouse model of Parkinson’s disease. Mol. Med. Rep. 2018, 17, 4883–4888. [Google Scholar] [CrossRef]
- Tseng, H.C.; Wang, M.H.; Chang, K.C.; Soung, H.S.; Fang, C.H.; Lin, Y.W.; Li, K.Y.; Yang, C.C.; Tsai, C.C. Protective Effect of (-)-Epigallocatechin-3-gallate on Rotenone-Induced Parkinsonism-like Symptoms in Rats. Neurotox Res. 2020, 37, 669–682. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Youdim, M.B. Gene and protein expression profiles of anti- and pro-apoptotic actions of dopamine, R-apomorphine, green tea polyphenol (-)-epigallocatechine-3-gallate, and melatonin. Ann N.Y. Acad. Sci. 2003, 993, 351–393. [Google Scholar] [CrossRef]
- Genc, S.; Kizildag, S.; Genc, K.; Ates, H.; Atabey, N. Interferon gamma and lipopolysaccharide upregulate TNF-related apoptosis-inducing ligand expression in murine microglia. Immunol. Lett. 2003, 85, 271–274. [Google Scholar] [CrossRef]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.B.; Zhou, L.; Wang, Y.Z.; Wang, X.; Zhou, Y.; Ho, W.Z.; Li, J.L. Neuroprotective Activity of (-)-Epigallocatechin Gallate against Lipopolysaccharide-Mediated Cytotoxicity. J. Immunol. Res. 2016, 2016, 4962351. [Google Scholar] [CrossRef] [PubMed]
- Bitu-Pinto, N.; da Silva-Alexandre, B.; Neves, K.R.; Silva, A.H.; Leal, L.K.; Viana, G.S. Neuroprotective Properties of the Standardized Extract from Camellia sinensis (Green Tea) and Its Main Bioactive Components, Epicatechin and Epigallocatechin Gallate, in the 6-OHDA Model of Parkinson’s Disease. Evid. Based Complement. Altern. Med. 2015, 2015, 161092. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.M.; O., J.J.; Jeon, B.S. Inhibition of inducible nitric oxide synthase expression and cell death by (-)-epigallocatechin-3-gallate, a green tea catechin, in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Clin. Neurosci. 2010, 17, 1165–1168. [Google Scholar] [CrossRef]
- Singh, S.; Das, T.; Ravindran, A.; Chaturvedi, R.K.; Shukla, Y.; Agarwal, A.K.; Dikshit, M. Involvement of nitric oxide in neurodegeneration: A study on the experimental models of Parkinson’s disease. Redox Rep. 2005, 10, 103–109. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, C.S.; Kim, D.J.; Cho, M.H.; Jin, B.K.; Pie, J.E.; Chung, W.G. Prevention of nitric oxide-mediated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease in mice by tea phenolic epigallocatechin 3-gallate. Neurotoxicology 2002, 23, 367–374. [Google Scholar] [CrossRef]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; McAuliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [CrossRef]
- Bortolanza, M.; Padovan-Neto, F.E.; Cavalcanti-Kiwiatkoski, R.; Santos-Pereira, M.D.; Mitkovski, M.; Raisman-Vozari, R.; Del-Bel, E. Are cyclooxygenase-2 and nitric oxide involved in the dyskinesia of Parkinson’s disease induced by L-DOPA? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140190. [Google Scholar] [CrossRef]
- Mollace, V.; Muscoli, C.; Masini, E.; Cuzzocrea, S.; Salvemini, D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 2005, 57, 217–252. [Google Scholar] [CrossRef]
- Sánchez-Pernaute, R.; Ferree, A.; Cooper, O.; Yu, M.; Brownell, A.L.; Isacson, O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. J. Neuroinflamm. 2004, 1, 6. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.C.; Pal, R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin. Neurosci. 2017, 19, 71–80. [Google Scholar] [PubMed]
- Levites, Y.; Youdim, M.B.; Maor, G.; Mandel, S. Attenuation of 6-hydroxydopamine (6-OHDA)-induced nuclear factor-kappaB (NF-kappaB) activation and cell death by tea extracts in neuronal cultures. Biochem. Pharmacol. 2002, 63, 21–29. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Pereira, P.A.B.; Aho, V.T.E.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Oral and nasal microbiota in Parkinson’s disease. Parkinsonism. Relat. Disord. 2017, 38, 61–67. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, M.; Xue, J.; Xiang, L.; Li, Y.; Xiao, J.; Xiao, G.; Wang, H.L. EGCG ameliorates neuronal and behavioral defects by remodeling gut microbiota and TotM expression in Drosophila models of Parkinson’s disease. FASEB J. 2020, 34, 5931–5950. [Google Scholar] [CrossRef]
- Stefani, M.; Rigacci, S. Beneficial properties of natural phenols: Highlight on protection against pathological conditions associated with amyloid aggregation. Biofactors 2014, 40, 482–493. [Google Scholar] [CrossRef]
- Bu-Abbas, A.; Nunez, X.; Clifford, M.N.; Walker, R.; Ioannides, C. A comparison of the antimutagenic potential of green, black and decaffeinated teas: Contribution of flavanols to the antimutagenic effect. Mutagenesis 1996, 11, 597–603. [Google Scholar] [CrossRef][Green Version]
- Ogura, R.; Ikeda, N.; Yuki, K.; Morita, O.; Saigo, K.; Blackstock, C.; Nishiyama, N.; Kasamatsu, T. Genotoxicity studies on green tea catechin. Food Chem. Toxicol. 2008, 46, 2190–2200. [Google Scholar] [CrossRef]
- Chengelis, C.P.; Kirkpatrick, J.B.; Regan, K.S.; Radovsky, A.E.; Beck, M.J.; Morita, O.; Tamaki, Y.; Suzuki, H. 28-Day oral (gavage) toxicity studies of green tea catechins prepared for beverages in rats. Food Chem. Toxicol. 2008, 46, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Ramot, Y.; Malarkey, D.E.; Blackshear, P.; Kissling, G.E.; Travlos, G.; Nyska, A. Fourteen-week toxicity study of green tea extract in rats and mice. Toxicol. Pathol. 2010, 38, 1070–1084. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults—Results of a systematic review. Regul. Toxicol. Pharmacol. 2018, 95, 412–433. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (-)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef]
- Carvalho, A.N.; Firuzi, O.; Gama, M.J.; Horssen, J.V.; Saso, L. Oxidative Stress and Antioxidants in Neurological Diseases: Is There Still Hope? Curr. Drug Targets 2017, 18, 705–718. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Dai, W.; Ruan, C.; Zhang, Y.; Wang, J.; Han, J.; Shao, Z.; Sun, Y.; Liang, J. Bioavailability enhancement of EGCG by structural modification and nano-delivery: A review. J. Funct. Foods 2020, 103732. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sl. NO. | Model Used | Mode of Action | Reference |
|---|---|---|---|
| 1 | Cell free in vitro system | EGCG inhibits the aggregation of and induces disaggregation of α-synuclein | [50,51,52,53,54] |
| 2 | PC12 cells | EGCG protected PC12 cells from α-synuclein induced toxicity | [50] |
| 3 | AS-PC12 cells | EGCG protected AS-PC12 cells from Fe(III) induced toxicity by reducing the formation of ROS | [52] |
| 4 | NB SH-SY5Y cells | EGCG offered neuroprotection by PKC mediated degradation of pro-apoptotic protein Bad | [61] |
| 5 | CHO cells expressing DAT | EGCG prevented neurodegeneration by inhibiting the ability of dopamine transporters (DAT) to actively uptake MPP+ and being transported to presynaptic dopaminergic neurons | [63] |
| 6 | DAT-PC12 cells | EGCG induces the internalization of DAT through the activation of PKC thereby preventing the reuptake of dopamine from the synaptic cleft | [65] |
| 7 | Cell free in vitro system | EGCG treatment chelates Fe3+ and prevents the fibrillization and toxic oligomer formation | [71] |
| 8 | SN4741 cells | EGCG downregulates cell cycle proteins cyclin D1, E and helps in neurite outgrowth and neuronal differentiation | [74] |
| 9 | Primary neuronal and neuron-astrocyte cells | L-theanine, protected cells from dopamine-induced toxicity by inducing glutathione production | [84] |
| 10 | CGN culture | EGCG inhibited apoptosis of neuronal cultures from mitochondrial oxidative stressors | [88] |
| 11 | SH-SY5Y cells | EGCG protected cells from 6-OHDA induced neurotoxicity; suppressing the buildup of ROS, restoring MMP, and maintaining calcium homeostasis | [97] |
| 12 | PC12 cells | EGCG protects cells from Oxy-Hb induced stress; inhibits Ca2+-influx through voltage-gated calcium channels | [98] |
| 13 | PC12 cells | EGCG protected cells from MPTP toxicity by the activation of PGC-1α via SIRT-1 signaling | [102] |
| 14 | SH-SY5Y cells | L-theanine exhibited neuroprotective effect against rotenone and dieldrin toxicity by downregulating HO-1, caspase-3, inducing neurotrophic factors BDNF and GDNF and activating ERK1/2 pathway | [105] |
| 15 | SH-SY5Y cells | EGCG ameliorates 6-OHDA toxicity via Akt signaling pathway and prevents apoptosis by downregulating caspase-3 activity | [116] |
| 16 | SH-SY5Y cells | EGCG inhibits TRAIL ligand expression as well as TRAIL receptor DR5 | [120] |
| 17 | Macrophage cells | EGCG protects macrophage cells from LPS induced toxicity by inducing the expression of IFN-γ | [123] |
| 18 | Neuronal cultures | Green tea extract attenuated 6-OHDA induced NF-κB activation and cell death | [134] |
| Sl. No. | Model Used | Activity Observed | Reference |
|---|---|---|---|
| 1 | Cynomolgus monkeys | Catechin-rich tea polyphenol extract improved motor impairments and restored TH and dopamine levels in MPTP PD model. | [58] |
| 2 | C57/BL mice | Green tea extract and EGCG reduced the loss of dopamine by modulating the antioxidant enzymes in MPTP PD model. | [59] |
| 3 | C57/BL mice | In MPTP PD model EGCG reduced the expression of α-synuclein and prevented apoptosis by downregulating the expression of Bax and increasing the expression of PKC-α | [60] |
| 4 | Long-Evans Rats | EGCG inhibited MAO-B in aged rat brain | [69] |
| 5 | C57 mice | EGCG induced ferroportin expression and offered neuroprotection | [73] |
| 6 | PD affected individuals | Green tea consumption showed a marked increase in the antioxidant enzymes catalase, SOD, and reduced the oxidation of proteins and lipids | [77] |
| 7 | Drosophila | Epicatechin gallate restored locomotor activity and reduced lipid peroxidation, oxidative stress | [78] |
| 8 | Human | Green tea exerts beneficial effect, by reducing oxidative stress and protects the individual against oxidative stress diseases | [83] |
| 9 | Sprague-Dawley Rats | Green tea polyphenol exhibits neuroprotective effect against 6-OHDA by reducing lipid peroxidation, 3-NT level. | [89] |
| 10 | Knockdown dj-1-β Drosophila | EGCG prevented oxidative stress and neurodegeneration induced by paraquat. | [93] |
| 11 | C57BL/6J mice | Long-term administration of EGCG prevented age-related cognitive decline and improved locomotor activity by increasing the expression of CREB and post-synaptic proteins PSD95, CAMKII. | [104] |
| 12 | C57/BL6 mice | A combination of Rasagiline and EGCG restored mice from MPTP induced parkinsonism by increasing the expression of BDNF, phosphorylated PKC-α as well as Ras and its downstream effector Akt | [74] |
| 13 | C57/BL6 mice | EGCG protects from sevoflurane by regulating the expression of BDNF-TrkB and activating Akt signaling | [113] |
| 14 | C57BL/6J mice | EGCG reduced CD4+ to CD8+ ratio downregulating the expression of TNF-α, IL-6 in MPTP treated mice | [118] |
| 15 | Male Wistar Rats | EGCG reduced rotenone induced parkinsonism like symptoms in rats by downregulating the expression of TNF-α, IL-1, IL-6 | [119] |
| 16 | Male Wistar rats | Standardized green tea extract and its active constituents downregulated the expression of inflammatory mediators COX-2 and iNOS by 6-OHDA | [124] |
| 17 | C57BL/6 mice | EGCG inhibited iNOS expression and cell death induced by MPTP | [125] |
| 18 | PINK1 null mutant Drosophila | EGCG rescued flies from motor, neuronal deficits and significantly remodeled gut microbiota | [138] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malar, D.S.; Prasanth, M.I.; Brimson, J.M.; Sharika, R.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review. Molecules 2020, 25, 3926. https://doi.org/10.3390/molecules25173926
Malar DS, Prasanth MI, Brimson JM, Sharika R, Sivamaruthi BS, Chaiyasut C, Tencomnao T. Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review. Molecules. 2020; 25(17):3926. https://doi.org/10.3390/molecules25173926
Chicago/Turabian StyleMalar, Dicson Sheeja, Mani Iyer Prasanth, James Michael Brimson, Rajasekharan Sharika, Bhagavathi Sundaram Sivamaruthi, Chaiyavat Chaiyasut, and Tewin Tencomnao. 2020. "Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review" Molecules 25, no. 17: 3926. https://doi.org/10.3390/molecules25173926
APA StyleMalar, D. S., Prasanth, M. I., Brimson, J. M., Sharika, R., Sivamaruthi, B. S., Chaiyasut, C., & Tencomnao, T. (2020). Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review. Molecules, 25(17), 3926. https://doi.org/10.3390/molecules25173926