
An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia




Abstract
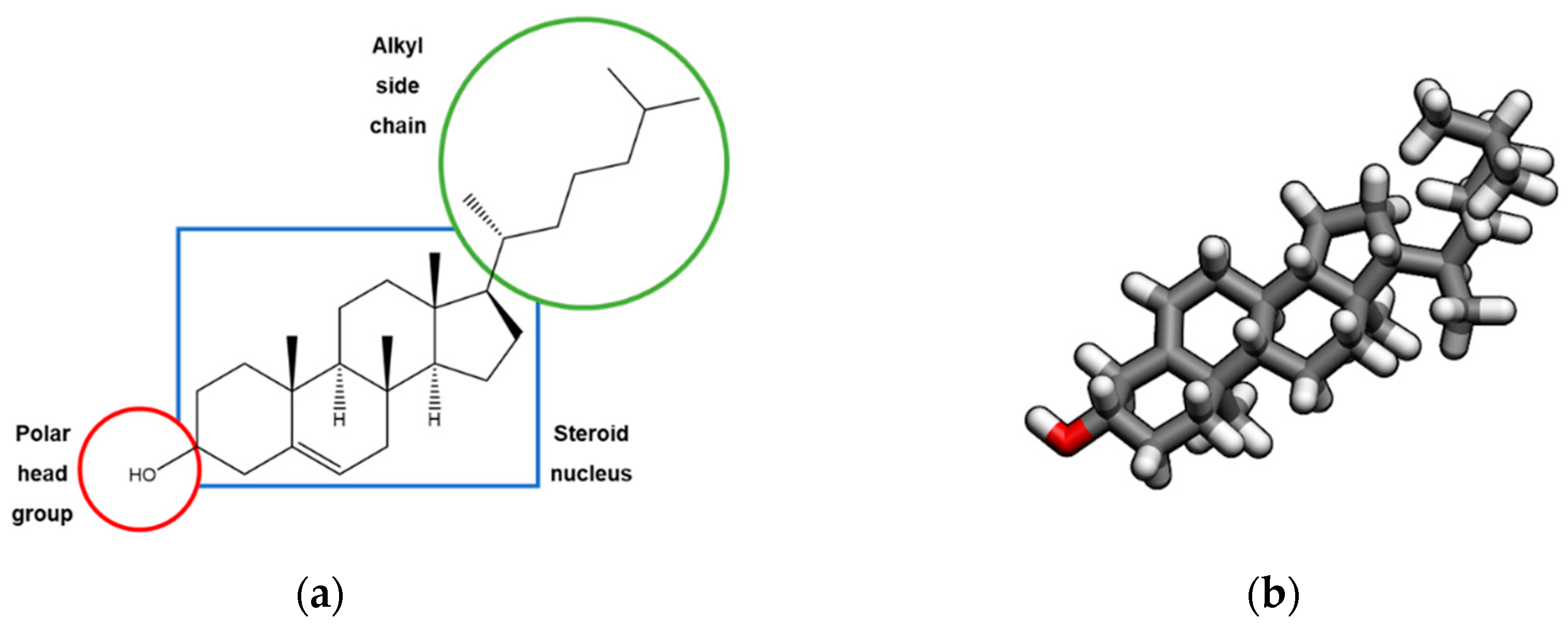
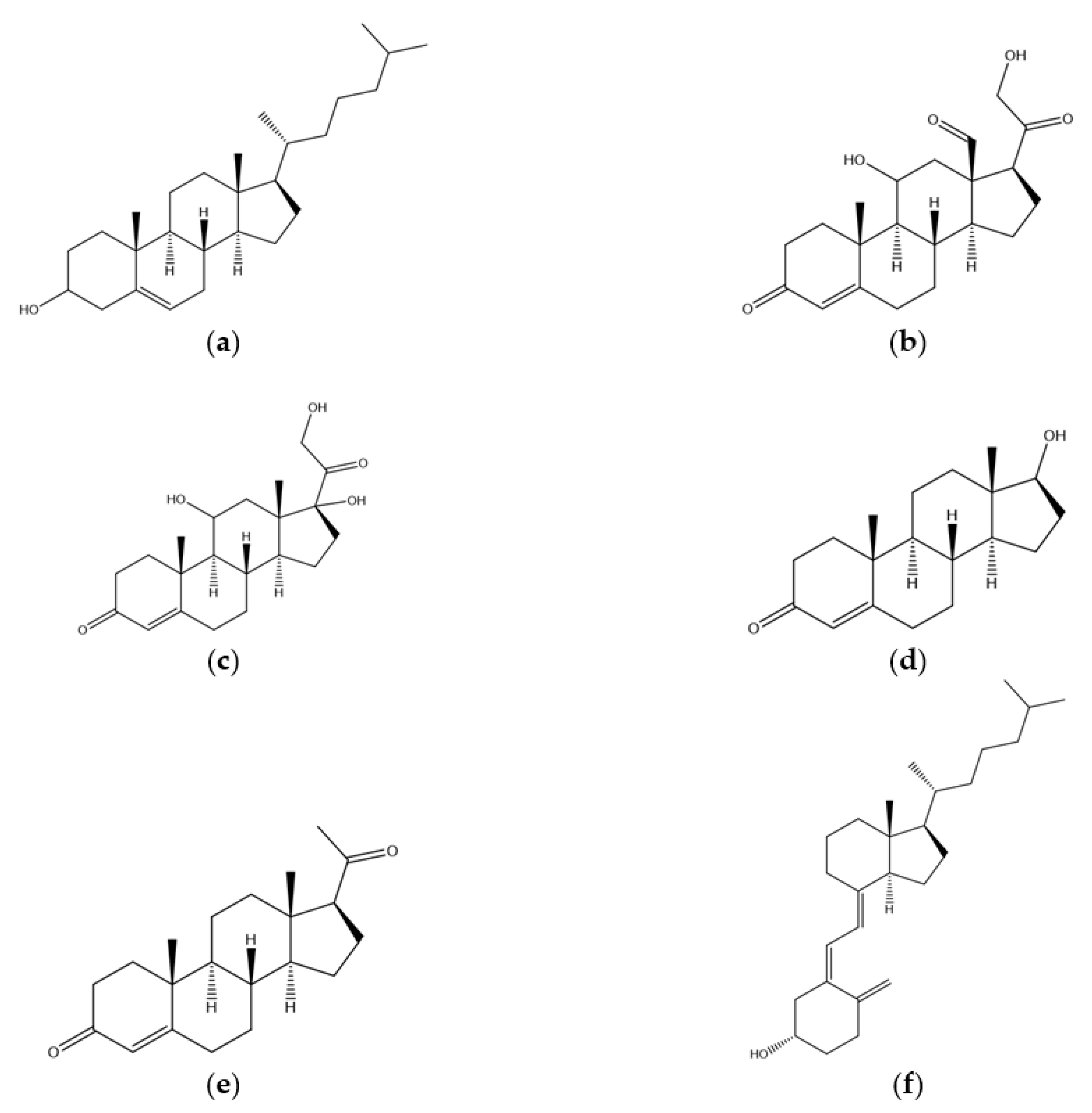
1. Introduction
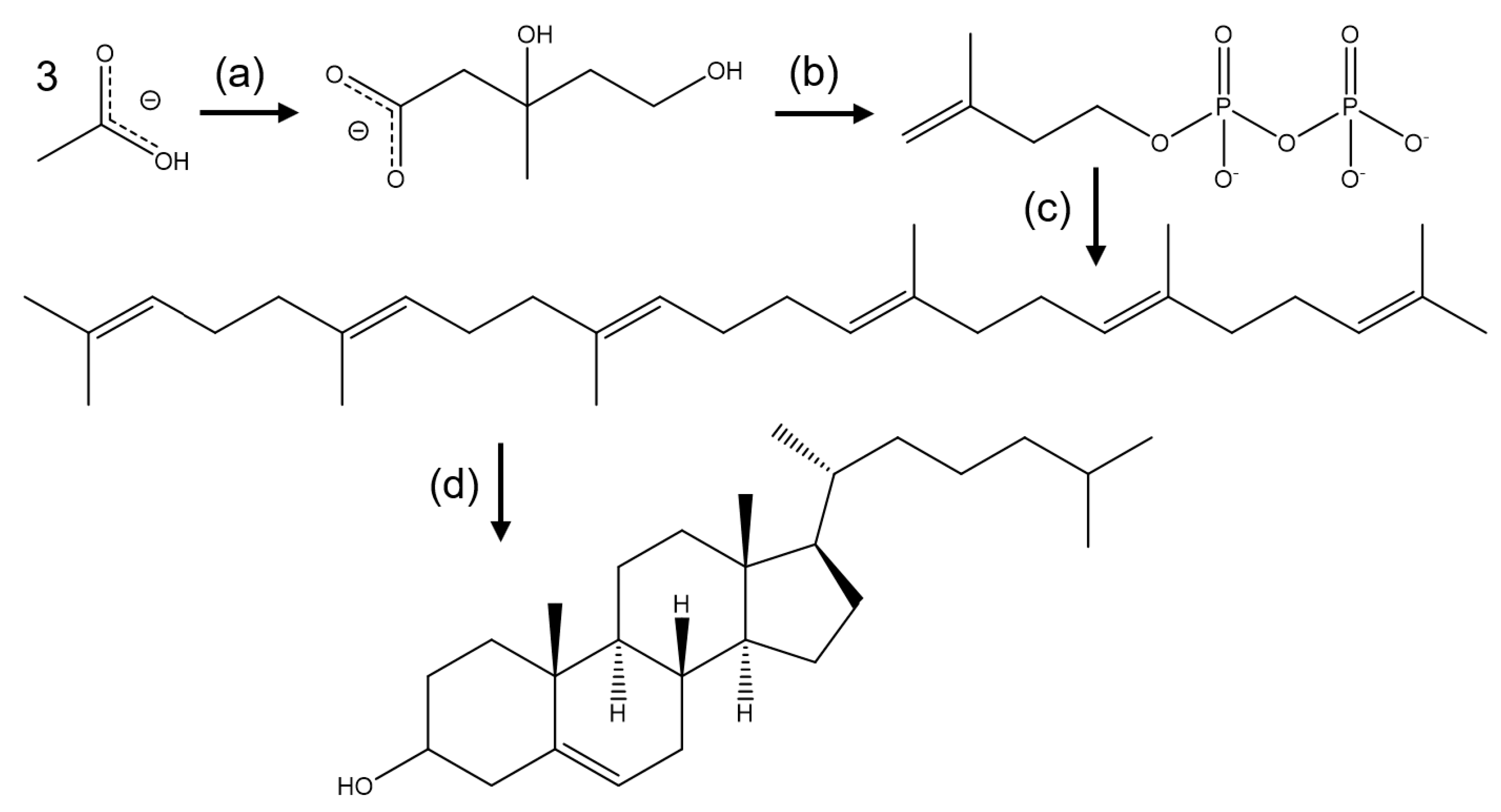
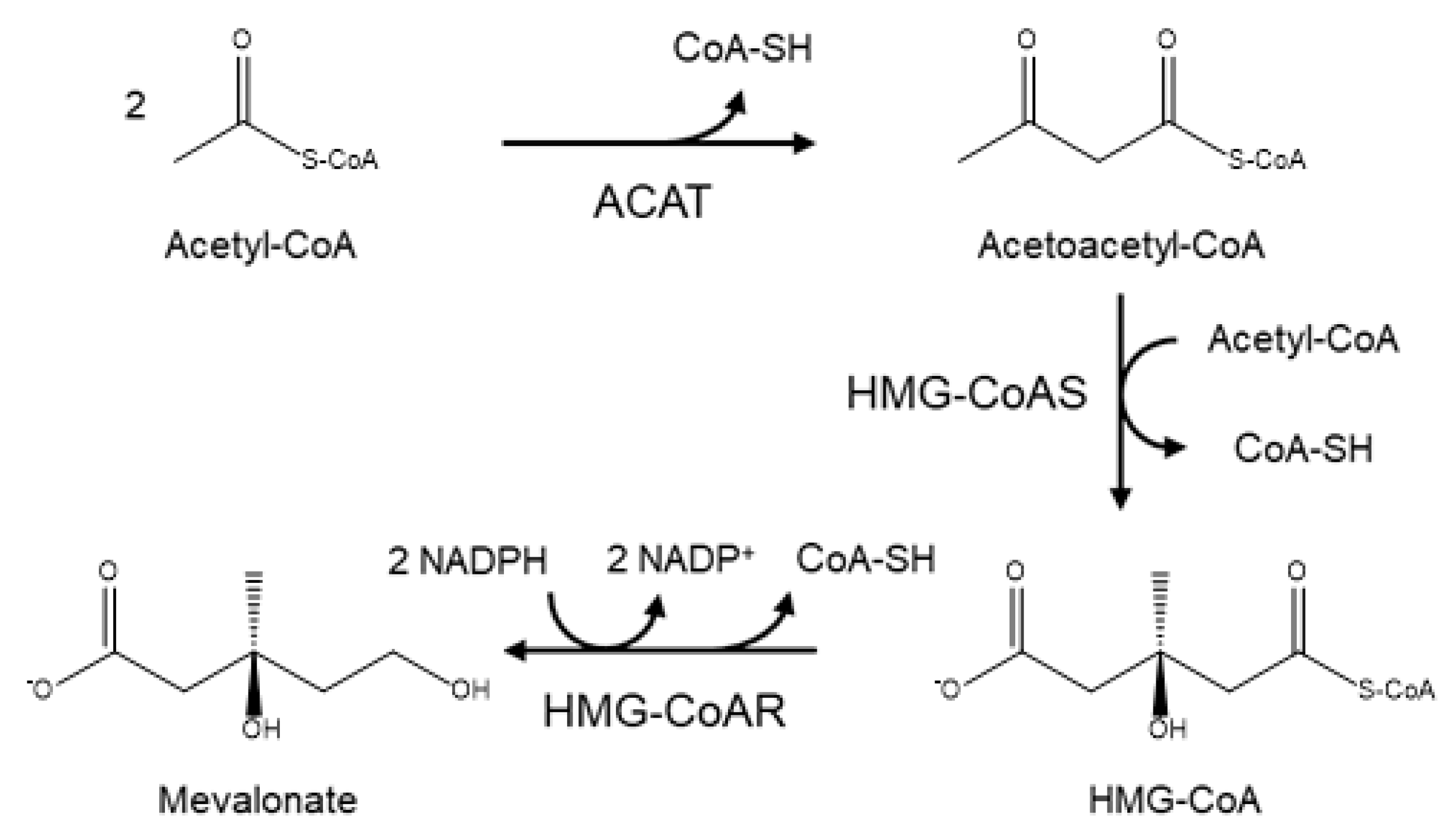
2. Biosynthesis of Cholesterol and the Role of HMG-CoA
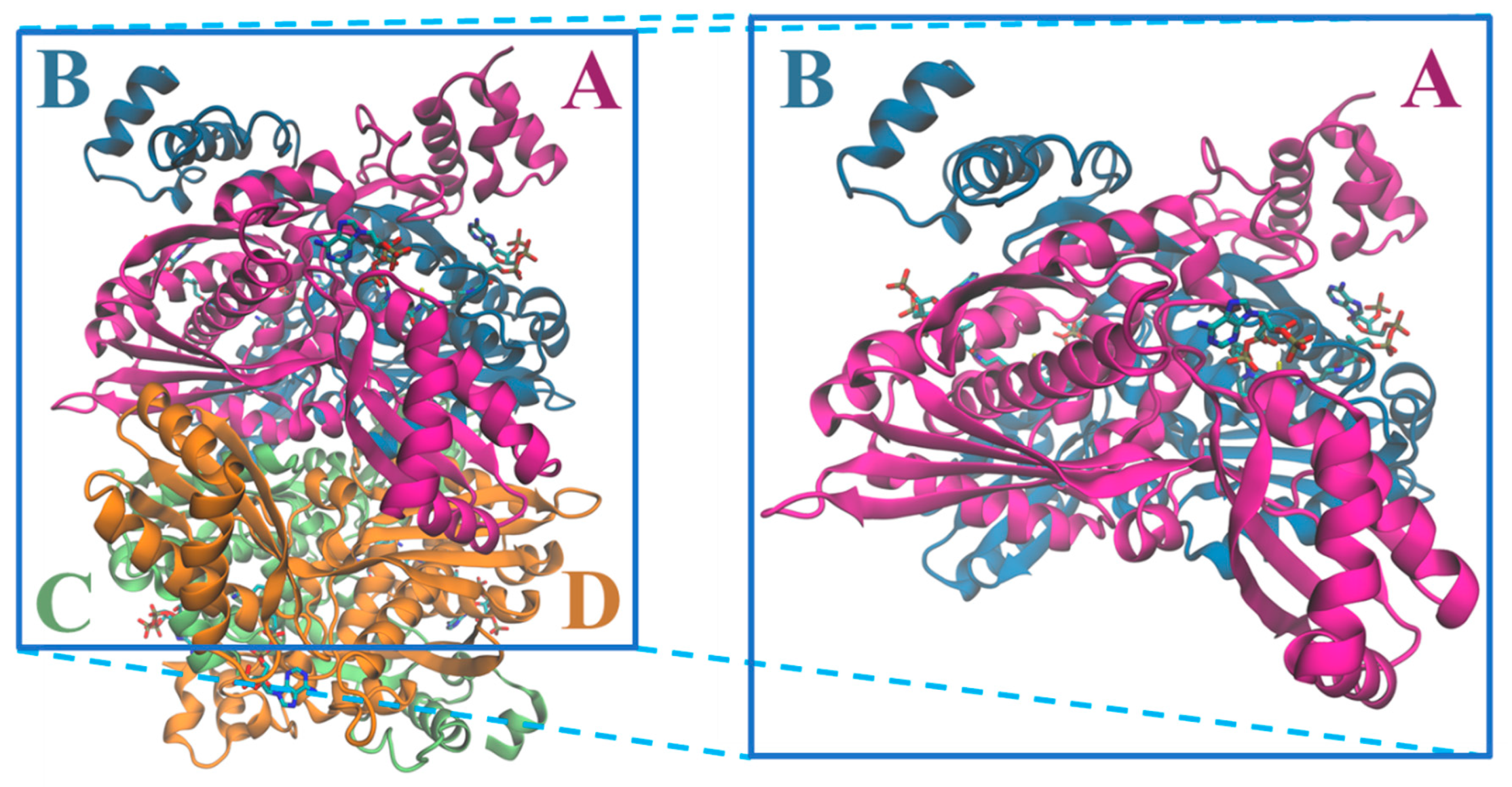
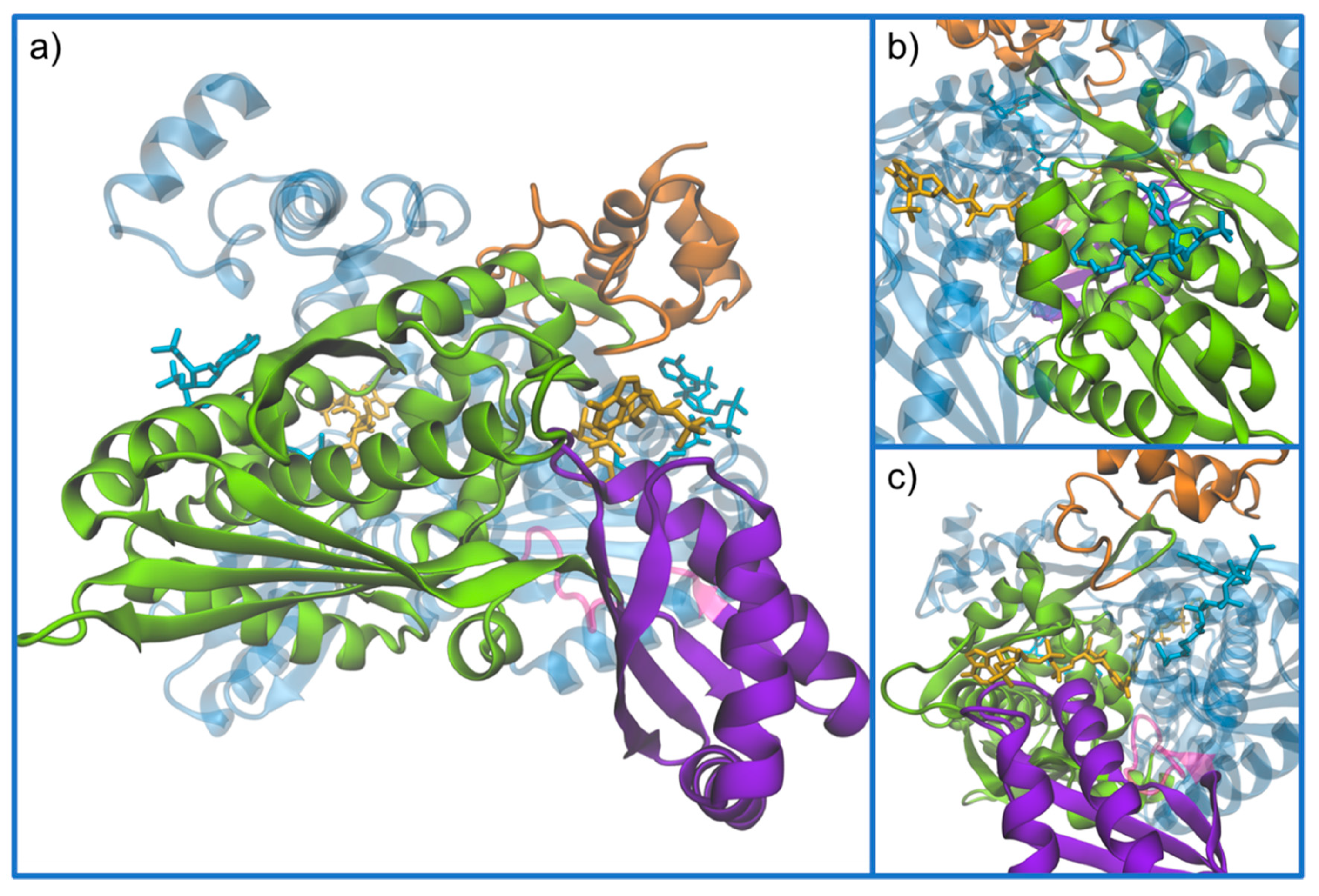
3. Structure of HMG-CoA Reductase
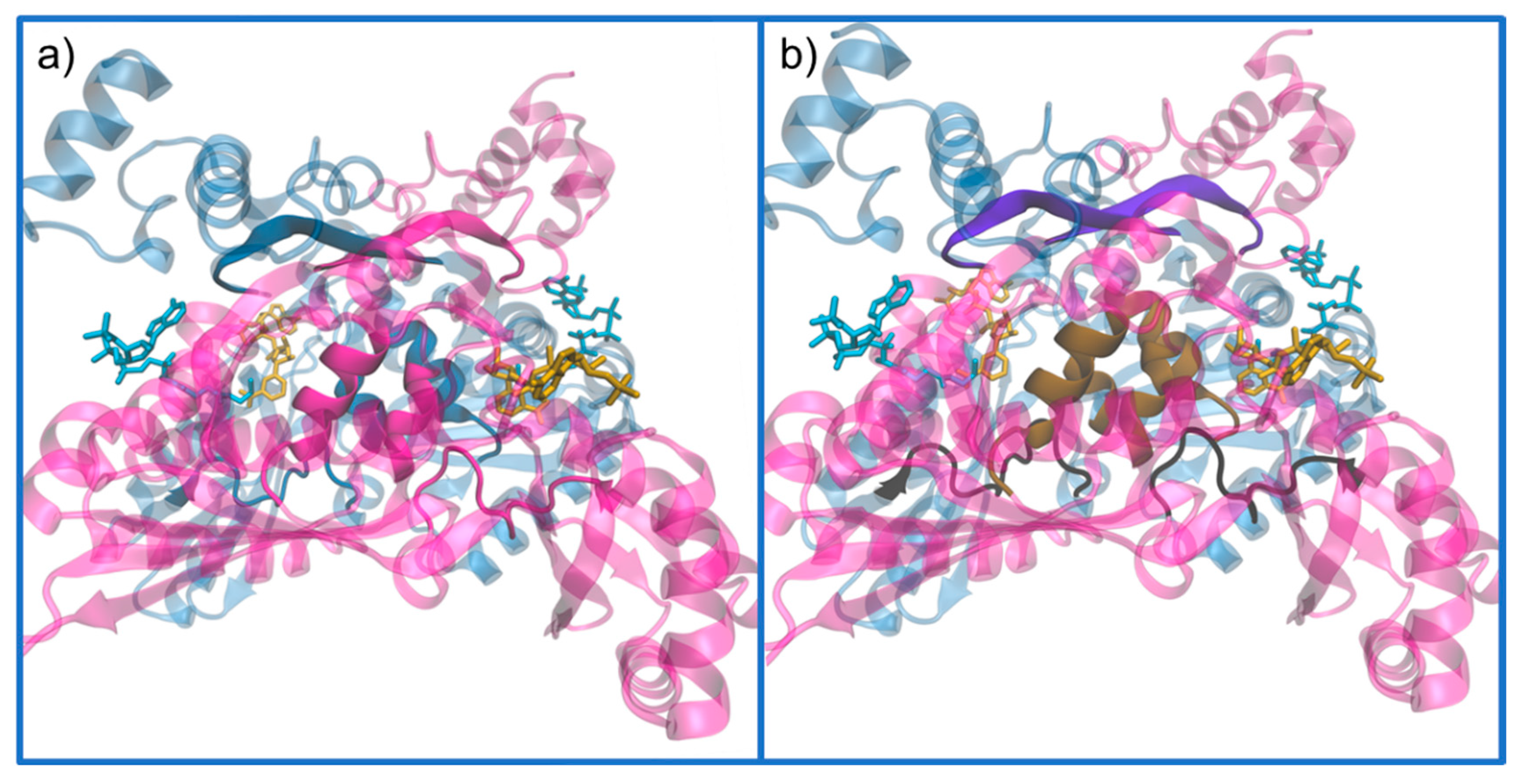
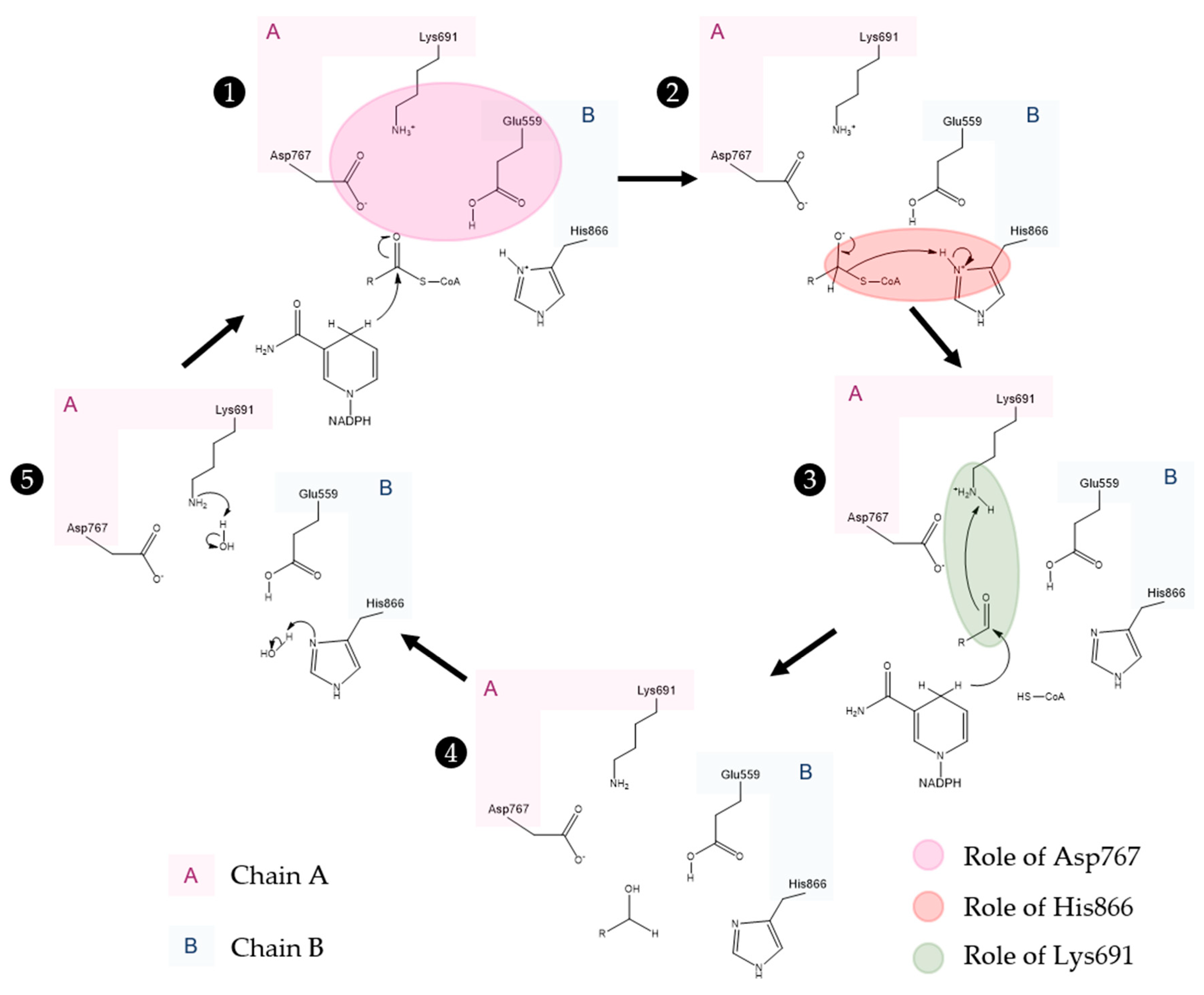
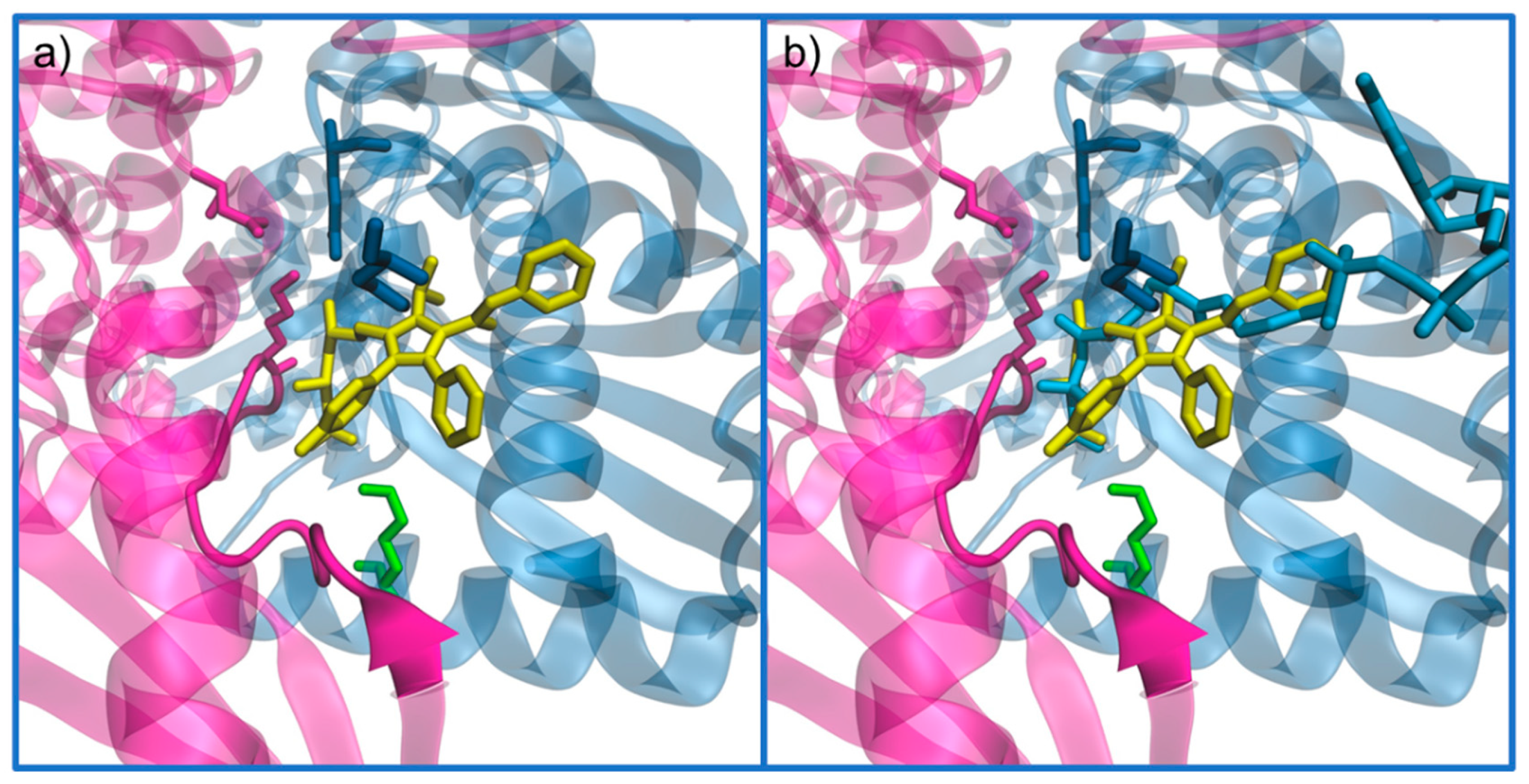
Active Site Architecture and Catalytic Mechanism of HMG-CoA Reductase
4. Regulation of HMG-CoAR
5. HMG-CoAR Inhibitors
5.1. Statins: The Most Common Inhibitor
5.2. Alternative Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Windaus, A. Über die konstitution des cholesterins und der gallensäuren. Biol. Chem. 1932, 213, 147–187. [Google Scholar] [CrossRef]
- Sheppard, A.J.; O’Dell, R.G.; Pennington, J.A.T. CHOLESTEROL | Properties and determination. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Oxford, UK, 2003; pp. 1220–1226. ISBN 978-0-12-227055-0. [Google Scholar]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2008; ISBN 9781429208925. [Google Scholar]
- Arnold, D.R.; Kwiterovich, P.O. CHOLESTEROL | Absorption, Function, and Metabolism. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Oxford, UK, 2003; pp. 1226–1237. ISBN 978-0-12-227055-0. [Google Scholar]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Lecerf, J.M.; de Lorgeril, M. Dietary cholesterol: From physiology to cardiovascular risk. Br. J. Nutr. 2011, 106, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, P.A.S.; Jones, P.J.H. Revisiting human cholesterol synthesis and absorption: The reciprocity paradigm and its key regulators. Lipids 2016, 51, 519–536. [Google Scholar] [CrossRef]
- McAuley, M.T.; Wilkinson, D.J.; Jones, J.J.; Kirkwood, T.B. A whole-body mathematical model of cholesterol metabolism and its age-associated dysregulation. BMC Syst. Biol. 2012, 6, 130. [Google Scholar] [CrossRef]
- WHO. Noncommunicable Diseases Country Profiles 2018; WHO: Geneva, Switzerland, 2018; ISBN 978-92-4-151462-0. [Google Scholar]
- Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region. Available online: https://www.who.int/healthinfo/global_burden_disease/estimates/en/ (accessed on 12 January 2020).
- Ravnskov, U.; de Lorgeril, M.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjold, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Mascitelli, L.; et al. LDL-C does not cause cardiovascular disease: A comprehensive review of the current literature. Expert Rev. Clin. Pharmacol 2018, 11, 959–970. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Ravnskov, U.; McCully, K.S. Review and hypothesis: Vulnerable plaque formation from obstruction of Vasa vasorum by homocysteinylated and oxidized lipoprotein aggregates complexed with microbial remnants and LDL autoantibodies. Ann. Clin. Lab. Sci 2009, 39, 3–16. [Google Scholar]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Hussain, M.M.; Strickland, D.K.; Bakillah, A. The mammalian low-density lipoprotein receptor family. Annu. Rev. Nutr. 1999, 19, 141–172. [Google Scholar] [CrossRef]
- Willnow, T.E. The low-density lipoprotein receptor gene family: Multiple roles in lipid metabolism. J. Mol. Med. 1999, 77, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R.; Kletzien, R.F. Current and emerging strategies for treating dyslipidemia and macrovascular disease. In Advances in Pharmacology; Enna, S.J., Williams, M., Eds.; Academic Press: Oxford, UK, 2009; Volume 57, pp. 237–251. ISSN 1054-3589. [Google Scholar]
- Parks, L.W. Metabolism of sterols in yeast. CRC Crit Rev. Microbiol. 1978, 6, 301–341. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol biosynthesis: A mechanistic overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- Steussy, C.N.; Critchelow, C.J.; Schmidt, T.; Min, J.K.; Wrensford, L.V.; Burgner, J.W., 2nd; Rodwell, V.W.; Stauffacher, C.V. A novel role for coenzyme A during hydride transfer in 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Biochemistry 2013, 52, 5195–5205. [Google Scholar] [CrossRef]
- Holstein, S.A.; Hohl, R.J. Isoprenoids: Remarkable diversity of form and function. Lipids 2004, 39, 293–309. [Google Scholar] [CrossRef]
- Johnson, E.A.; Schroeder, W.A. Microbial carotenoids. Adv. Biochem Eng. Biotechnol 1996, 53, 119–178. [Google Scholar] [CrossRef]
- Reusch, V.M., Jr. Lipopolymers, isoprenoids, and the assembly of the gram-positive cell wall. Crit. Rev. Microbiol. 1984, 11, 129–155. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yasukawa, J.; Ishii, M.; Hayashi, Y.; Miyazaki, S.; Sekimizu, K. A critical role of mevalonate for peptidoglycan synthesis in Staphylococcus aureus. Sci. Rep. 2016, 6, 22894. [Google Scholar] [CrossRef]
- Chappell, J.; Wolf, F.; Proulx, J.; Cuellar, R.; Saunders, C. Is the reaction catalyzed by 3-Hydroxy-3-methylglutaryl coenzyme a reductase a rate-limiting step for isoprenoid biosynthesis in plants? Plant Physiol 1995, 109, 1337–1343. [Google Scholar] [CrossRef]
- Haines, B.E.; Wiest, O.; Stauffacher, C.V. The increasingly complex mechanism of HMG-CoA reductase. Acc. Chem. Res. 2013, 46, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Koning, A.J.; Roberts, C.J.; Wright, R.L. Different subcellular localization of Saccharomyces cerevisiae HMG-CoA reductase isozymes at elevated levels corresponds to distinct endoplasmic reticulum membrane proliferations. Mol. Biol. Cell 1996, 7, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Hedl, M.; Tabernero, L.; Stauffacher, C.V.; Rodwell, V.W. Class II 3-hydroxy-3-methylglutaryl coenzyme A reductases. J. Bacteriol. 2004, 186, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Beach, M.J.; Rodwell, V.W. Cloning, sequencing, and overexpression of mvaA, which encodes Pseudomonas mevalonii 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Bacteriol. 1989, 171, 2994–3001. [Google Scholar] [CrossRef] [PubMed]
- Bochar, D.A.; Stauffacher, C.V.; Rodwell, V.W. Sequence comparisons reveal two classes of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mol. Genet. Metab. 1999, 66, 122–127. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef]
- Jordan-Starck, T.C.; Rodwell, V.W. Role of cysteine residues in Pseudomonas mevalonii 3-hydroxy-3-methylglutaryl-CoA reductase. Site-directed mutagenesis and characterization of the mutant enzymes. J. Biol. Chem. 1989, 264, 17919–17923. [Google Scholar]
- Jordan-Starck, T.C.; Rodwell, V.W. Pseudomonas mevalonii 3-hydroxy-3-methylglutaryl-CoA reductase. Characterization and chemical modification. J. Biol. Chem. 1989, 264, 17913–17918. [Google Scholar]
- Darnay, B.G.; Wang, Y.; Rodwell, V.W. Identification of the catalytically important histidine of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 1992, 267, 15064–15070. [Google Scholar]
- Omkumar, R.V.; Darnay, B.G.; Rodwell, V.W. Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J. Biol. Chem. 1994, 269, 6810–6814. [Google Scholar] [PubMed]
- Istvan, E.S.; Palnitkar, M.; Buchanan, S.K.; Deisenhofer, J. Crystal structure of the catalytic portion of human HMG-CoA reductase: Insights into regulation of activity and catalysis. EMBO J. 2000, 19, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Vogeli, B.; Shima, S.; Erb, T.J.; Wagner, T. Crystal structure of archaeal HMG-CoA reductase: Insights into structural changes of the C-terminal helix of the class-I enzyme. FEBS Lett. 2019, 593, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ragwan, E.R.; Arai, E.; Kung, Y. New crystallographic snapshots of large domain movements in bacterial 3-hydroxy-3-methylglutaryl coenzyme a reductase. Biochemistry 2018, 57, 5715–5725. [Google Scholar] [CrossRef]
- Miller, B.R.; Kung, Y. Structural features and domain movements controlling substrate binding and cofactor specificity in class II HMG-CoA reductase. Biochemistry 2018, 57, 654–662. [Google Scholar] [CrossRef]
- Sarver, R.W.; Bills, E.; Bolton, G.; Bratton, L.D.; Caspers, N.L.; Dunbar, J.B.; Harris, M.S.; Hutchings, R.H.; Kennedy, R.M.; Larsen, S.D.; et al. Thermodynamic and structure guided design of statin based inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med. Chem. 2008, 51, 3804–3813. [Google Scholar] [CrossRef]
- Pfefferkorn, J.A.; Choi, C.; Larsen, S.D.; Auerbach, B.; Hutchings, R.; Park, W.; Askew, V.; Dillon, L.; Hanselman, J.C.; Lin, Z.; et al. Substituted pyrazoles as hepatoselective HMG-CoA reductase inhibitors: Discovery of (3R,5R)-7-[2-(4-Fluoro-phenyl)-4-isopropyl-5-(4-methyl-benzylcarbamoyl)-2H-pyrazol-3-yl]-3,5-dihydroxyheptanoic Acid (PF-3052334) as a candidate for the treatment of hyper. J. Med. Chem. 2008, 51, 31–45. [Google Scholar] [CrossRef]
- Park, W.K.C.; Kennedy, R.M.; Larsen, S.D.; Miller, S.; Roth, B.D.; Song, Y.; Steinbaugh, B.A.; Sun, K.; Tait, B.D.; Kowala, M.C.; et al. Hepatoselectivity of statins: Design and synthesis of 4-sulfamoyl pyrroles as HMG-CoA reductase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1151–1156. [Google Scholar] [CrossRef]
- Pfefferkorn, J.A.; Song, Y.; Sun, K.L.; Miller, S.R.; Trivedi, B.K.; Choi, C.; Sorenson, R.J.; Bratton, L.D.; Unangst, P.C.; Larsen, S.D.; et al. Design and synthesis of hepatoselective, pyrrole-based HMG-CoA reductase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4538–4544. [Google Scholar] [CrossRef]
- Tabernero, L.; Rodwell, V.W.; Stauffacher, C.V. Crystal structure of a statin bound to a class II hydroxymethylglutaryl-CoA reductase. J. Biol. Chem. 2003, 278, 19933–19938. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, L.; Bochar, D.A.; Rodwell, V.W.; Stauffacher, C.V. Substrate-induced closure of the flap domain in the ternary complex structures provides insights into the mechanism of catalysis by 3-hydroxy-3-methylglutaryl–CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7167. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. The structure of the catalytic portion of human HMG-CoA reductase. Biochim. Biophys. Acta 2000, 1529, 9–18. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.; Stryer, L. Biochemistry, 8th ed.; W.H. Freeman: New York, NY, USA, 2015; ISBN 1464126100. ISBN 9781464126109. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, P.E.; Labouesse, M. The sterol-sensing domain: Multiple families, a unique role? Trends Genet. 2002, 18, 193–201. [Google Scholar] [CrossRef]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural basis for cholesterol transport-like activity of the hedgehog receptor patched. Cell 2018, 175, 1352–1364.e14. [Google Scholar] [CrossRef]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Oliveira, E.F.; Cerqueira, N.M.F.S.A.; Ramos, M.J.; Fernandes, P.A. QM/MM study of the mechanism of reduction of 3-hydroxy-3-methylglutaryl coenzyme A catalyzed by human HMG-CoA reductase. Catal. Sci. Technol. 2016, 6, 7172–7185. [Google Scholar] [CrossRef]
- Gill, J.F., Jr.; Beach, M.J.; Rodwell, V.W. Mevolonate utilization in Pseudomonas sp. M. Purification and characterization of an inducible 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Biol. Chem. 1985, 260, 9393–9398. [Google Scholar]
- Frimpong, K.; Rodwell, V.W. Catalysis by Syrian hamster 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proposed roles of histidine 865, glutamate 558, and aspartate 766. J. Biol. Chem. 1994, 269, 11478–11483. [Google Scholar] [PubMed]
- Osborne, T.F.; Gil, G.; Goldstein, J.L.; Brown, M.S. Operator constitutive mutation of 3-hydroxy-3-methylglutaryl coenzyme A reductase promoter abolishes protein binding to sterol regulatory element. J. Biol. Chem. 1988, 263, 3380–3387. [Google Scholar] [PubMed]
- Rajavashisth, T.B.; Taylor, A.K.; Andalibi, A.; Svenson, K.L.; Lusis, A.J. Identification of a zinc finger protein that binds to the sterol regulatory element. Science 1989, 245, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Rawson, R.B.; DeBose-Boyd, R.; Goldstein, J.L.; Brown, M.S. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 1999, 274, 28549–28556. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Rawson, R.B.; Brown, M.S. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch. Biochem. Biophys. 2002, 397, 139–148. [Google Scholar] [CrossRef]
- Brown, M.S.; Ye, J.; Rawson, R.B.; Goldstein, J.L. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 2000, 100, 391–398. [Google Scholar] [CrossRef]
- Sever, N.; Yang, T.; Brown, M.S.; Goldstein, J.L.; DeBose-Boyd, R.A. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 2003, 11, 25–33. [Google Scholar] [CrossRef]
- Sever, N.; Song, B.L.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 2003, 278, 52479–52490. [Google Scholar] [CrossRef]
- Faust, J.R.; Luskey, K.L.; Chin, D.J.; Goldstein, J.L.; Brown, M.S. Regulation of synthesis and degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase by low density lipoprotein and 25-hydroxycholesterol in UT-1 cells. Proc. Natl. Acad. Sci. USA 1982, 79, 5205–5209. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, T.A.; Simon, H.M.; Patel, D.D.; Bentzen, C.; Niesor, E.; Jackson, B.; Suckling, K.E. The novel cholesterol-lowering drug SR-12813 inhibits cholesterol synthesis via an increased degradation of 3-hydroxy-3-methylglutaryl-coenzyme a reductase. J. Biol. Chem. 1996, 271, 14376–14382. [Google Scholar] [CrossRef] [PubMed]
- Roitelman, J.; Masson, D.; Avner, R.; Ammon-Zufferey, C.; Perez, A.; Guyon-Gellin, Y.; Bentzen, C.L.; Niesor, E.J. Apomine, a novel hypocholesterolemic agent, accelerates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and stimulates low density lipoprotein receptor activity. J. Biol. Chem. 2004, 279, 6465–6473. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-Y.; Li, H.; Tang, J.-J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.-K.; Shi, X.-J.; Cui, H.-W.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar] [CrossRef] [PubMed]
- Toyota, Y.; Yoshioka, H.; Sagimori, I.; Hashimoto, Y.; Ohgane, K. Bisphosphonate esters interact with HMG-CoA reductase membrane domain to induce its degradation. Bioorg. Med. Chem. 2020, 28, 115576. [Google Scholar] [CrossRef]
- Li, M.-X.; Yang, Y.; Zhao, Q.; Wu, Y.; Song, L.; Yang, H.; He, M.; Gao, H.; Song, B.-L.; Luo, J.; et al. Degradation versus Inhibition: Development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme a reductase. J. Med. Chem. 2020, 63, 4908–4928. [Google Scholar] [CrossRef]
- Beg, Z.H.; Stonik, J.A.; Brewer, H.B., Jr. In vivo modulation of rat liver 3-hydroxy-3-methylglutaryl-coenzyme A reductase, reductase kinase, and reductase kinase kinase by mevalonolactone. Proc. Natl. Acad. Sci. USA 1984, 81, 7293–7297. [Google Scholar] [CrossRef]
- Panda, T.; Devi, V.A. Regulation and degradation of HMGCo-A reductase. Appl. Microbiol. Biotechnol. 2004, 66, 143–152. [Google Scholar] [CrossRef]
- Omkumar, R.V.; Rodwell, V.W. Phosphorylation of Ser871 impairs the function of His865 of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 1994, 269, 16862–16866. [Google Scholar]
- Chen, L.; Ma, M.-Y.; Sun, M.; Jiang, L.-Y.; Zhao, X.-T.; Fang, X.-X.; Man Lam, S.; Shui, G.-H.; Luo, J.; Shi, X.-J.; et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J. Lipid Res. 2019, 60, 1765–1775. [Google Scholar] [CrossRef]
- Song, B.-L.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of 3-Hydroxy-3-methylglutaryl coenzyme a reductase stimulated by δ- and γ-Tocotrienols. J. Biol. Chem. 2006, 281, 25054–25061. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.; Hardie, D.G. Regulation of HMG-CoA reductase: Identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990, 9, 2439–2446. [Google Scholar] [CrossRef]
- Sato, R.; Goldstein, J.L.; Brown, M.S. Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc. Natl. Acad. Sci. USA 1993, 90, 9261–9265. [Google Scholar] [CrossRef] [PubMed]
- Hegsted, D.M. Serum-cholesterol response to dietary cholesterol: A re-evaluation. Am. J. Clin. Nutr. 1986, 44, 299–305. [Google Scholar] [CrossRef]
- Brown, M.S.; Faust, J.R.; Goldstein, J.L.; Kaneko, I.; Endo, A. Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J. Biol. Chem. 1978, 253, 1121–1128. [Google Scholar] [PubMed]
- Endo, A.; Kuroda, M.; Tanzawa, K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976, 72, 323–326. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef]
- Moorthy, N.S.H.N.; Cerqueira, N.M.F.S.A.; Ramos, M.J.; Fernandes, P.A. Ligand based analysis on HMG-CoA reductase inhibitors. Chemom. Intell. Lab. Syst. 2015, 140, 102–116. [Google Scholar] [CrossRef]
- Gotto, A.M., Jr. Results of recent large cholesterol-lowering trials and implications for clinical management. Am. J. Cardiol. 1997, 79, 1663–1666. [Google Scholar] [CrossRef]
- Hua, X.; Nohturfft, A.; Goldstein, J.L.; Brown, M.S. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage–activating protein. Cell 1996, 87, 415–426. [Google Scholar] [CrossRef]
- Istvan, E.S. Structural mechanism for statin inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Am. Heart J. 2002, 144, S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [PubMed]
- Tobert, J.A. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tamariz, J.; Chamorro, G.; Medina-Franco, J.L. Inhibitors of HMG-CoA reductase: Current and future prospects. Mini Rev. Med. Chem. 2009, 9, 1272–1283. [Google Scholar] [CrossRef]
- Wang, C.Y.; Liu, P.Y.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G. Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 2002, 23, 482–487. [Google Scholar] [CrossRef]
- Hassanabad, A.F. Current perspectives on statins as potential anti-cancer therapeutics: Clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699. [Google Scholar] [CrossRef]
- Roca-Millan, E.; González-Navarro, B.; Izquierdo-Gómez, K.; Marí-Roig, A.; Jané-Salas, E.; López-López, J.; Velasco-Ortega, E. The application of statins in the regeneration of bone defects. Systematic review and meta-analysis. Materials 2019, 12, 2992. [Google Scholar] [CrossRef]
- Ward Natalie, C.; Watts Gerald, F.; Eckel Robert, H. Statin toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef]
- Skottheim, I.B.; Gedde-Dahl, A.; Hejazifar, S.; Hoel, K.; Asberg, A. Statin induced myotoxicity: The lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 2008, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, J.C. Low-density lipoprotein cholesterol reduction: The end is more important than the means. Am. J. Cardiol. 2007, 100, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J.; Staffa, J.A.; Shatin, D.; Andrade, S.E.; Schech, S.D.; La Grenade, L.; Gurwitz, J.H.; Chan, K.A.; Goodman, M.J.; Platt, R. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004, 292, 2585–2590. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, A.V.; Pineda, M.D.; Venkata, K.C.N. Comprehension of top 200 prescribed drugs in the US as a resource for pharmacy teaching, training and practice. Pharmacy 2018, 6, 43. [Google Scholar] [CrossRef]
- Oliveira, E.F.; Santos-Martins, D.; Ribeiro, A.M.; Brás, N.F.; Cerqueira, N.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. HMG-CoA Reductase inhibitors: An updated review of patents of novel compounds and formulations (2011–2015). Expert Opin. Ther. Pat. 2016, 26, 1257–1272. [Google Scholar] [CrossRef]
- Mo, H.; Jeter, R.; Bachmann, A.; Yount, S.T.; Shen, C.L.; Yeganehjoo, H. The potential of isoprenoids in adjuvant cancer therapy to reduce adverse effects of statins. Front. Pharmacol. 2018, 9, 1515. [Google Scholar] [CrossRef]
- Demierre, M.F.; Higgins, P.D.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and cancer prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef]
- Hindler, K.; Cleeland, C.S.; Rivera, E.; Collard, C.D. The role of statins in cancer therapy. Oncologist 2006, 11, 306–315. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; Ciaglia, E.; D’Alessandro, A.; Bifulco, M. Novel prospects of statins as therapeutic agents in cancer. Pharmacol Res. 2014, 88, 84–98. [Google Scholar] [CrossRef]
- Clendening, J.W.; Penn, L.Z. Targeting tumor cell metabolism with statins. Oncogene 2012, 31, 4967–4978. [Google Scholar] [CrossRef]
- Bjarnadottir, O.; Romero, Q.; Bendahl, P.O.; Jirstrom, K.; Ryden, L.; Loman, N.; Uhlen, M.; Johannesson, H.; Rose, C.; Grabau, D.; et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res. Treat. 2013, 138, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Hiyoshi, H.; Yanagimachi, M.; Ito, M.; Yasuda, N.; Okada, T.; Ikuta, H.; Shinmyo, D.; Tanaka, K.; Kurusu, N.; Yoshida, I.; et al. Squalene synthase inhibitors suppress triglyceride biosynthesis through the farnesol pathway in rat hepatocytes. J. Lipid Res. 2003, 44, 128–135. [Google Scholar] [CrossRef]
- Hiyoshi, H.; Yanagimachi, M.; Ito, M.; Ohtsuka, I.; Yoshida, I.; Saeki, T.; Tanaka, H. Effect of ER-27856, a novel squalene synthase inhibitor, on plasma cholesterol in rhesus monkeys: Comparison with 3-hydroxy-3-methylglutaryl-coa reductase inhibitors. J. Lipid Res. 2000, 41, 1136–1144. [Google Scholar]
- van Heek, M.; Davis, H. Pharmacology of ezetimibe. Eur. Heart J. Suppl. 2002, 4, J5–J8. [Google Scholar] [CrossRef][Green Version]
- Rosenblum, S.B.; Huynh, T.; Afonso, A.; Davis, H.R., Jr.; Yumibe, N.; Clader, J.W.; Burnett, D.A. Discovery of 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone (SCH 58235): A designed, potent, orally active inhibitor of cholesterol absorption. J. Med. Chem. 1998, 41, 973–980. [Google Scholar] [CrossRef]
- Garcia-Calvo, M.; Lisnock, J.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef]
- Rudel Lawrence, L.; Lee Richard, G.; Parini, P. ACAT2 Is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1112–1118. [Google Scholar] [CrossRef]
- Meuwese, M.C.; de Groot, E.; Duivenvoorden, R.; Trip, M.D.; Ose, L.; Maritz, F.J.; Basart, D.C.; Kastelein, J.J.; Habib, R.; Davidson, M.H.; et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: The CAPTIVATE randomized trial. JAMA 2009, 301, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.T.; Davis, M.; Kent, C.; Chapman, J.; Tomoda, H.; Omura, S.; Rudel, L.L. Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: Individual ACAT uniqueness. J. Lipid Res. 2004, 45, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Siu, F.-M.; Lok, C.-N.; Fung, Y.M.E.; Che, C.-M. Anticancer auranofin engages 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) as a target. Metallomics 2019, 11, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-H.; Chang, D.-K.; Chou, M.-J.; Huang, K.-J.; Shiuan, D. Peptide inhibitors of human HMG-CoA reductase as potential hypocholesterolemia agents. Biochem. Biophys. Res. Commun. 2015, 456, 104–109. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, K.; Han, J.; Wang, K.; Chen, H.; Bao, L.; Liu, L.; Xiong, W.; Zhang, Y.; Huang, Y.; et al. Eight new triterpenoids with inhibitory activity against HMG-CoA reductase from the medical mushroom Ganoderma leucocontextum collected in Tibetan plateau. Fitoterapia 2018, 130, 79–88. [Google Scholar] [CrossRef]
- Wang, K.; Bao, L.; Ma, K.; Zhang, J.; Chen, B.; Han, J.; Ren, J.; Luo, H.; Liu, H. A novel class of α-glucosidase and HMG-CoA reductase inhibitors from Ganoderma leucocontextum and the anti-diabetic properties of ganomycin I in KK-Ay mice. Eur. J. Med. Chem. 2017, 127, 1035–1046. [Google Scholar] [CrossRef]
- Arantes, A.A.; Falé, P.L.; Costa, L.C.B.; Pacheco, R.; Ascensão, L.; Serralheiro, M.L. Inhibition of HMG-CoA reductase activity and cholesterol permeation through Caco-2 cells by caffeoylquinic acids from Vernonia condensata leaves. Rev. Bras. Farmacogn. 2016, 26, 738–743. [Google Scholar] [CrossRef]
- Hartanti, L.; Yonas, S.M.K.; Mustamu, J.J.; Wijaya, S.; Setiawan, H.K.; Soegianto, L. Influence of extraction methods of bay leaves (Syzygium polyanthum) on antioxidant and HMG-CoA reductase inhibitory activity. Heliyon 2019, 5, e01485. [Google Scholar] [CrossRef]
- Gesto, D.S.; Cerqueira, N.M.; Ramos, M.J.; Fernandes, P.A. Discovery of new druggable sites in the anti-cholesterol target HMG-CoA reductase by computational alanine scanning mutagenesis. J. Mol. Model. 2014, 20, 2178. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Organism | Year | Resolution (Å) | Chain Length | Ligand(s) | Ref. |
|---|---|---|---|---|---|---|
| 6HR8 | Methanothermococcus thermolithotrophicus | 2019 | 2.9 | 427 | NADPH, PEG | [39] |
| 6HR7 | 2.4 | 427 | P6G, DTT | |||
| 6EEV | Delftia acidovorans | 2018 | 1.5 | 429 | MEV | [40] |
| 6EEU | 1.9 | 429 | - | |||
| 6DIO | 2.1 | 429 | NAD | |||
| 5WPK | Streptococcus pneumoniae | 2018 | 2.3 | 426 | PE4, HMG | [41] |
| 5WPJ | 2.0 | 426 | NADPH | |||
| 4I6Y | Pseudomonas mevalonii | 2013 | 1.5 | 428 | MEV | [21] |
| 4I6W | 1.7 | 428 | 1CO | |||
| 4I6A | 1.9 | 428 | HMG | |||
| 4I64 | 1.8 | 428 | - | |||
| 4I56 | 1.5 | 428 | 1CZ | |||
| 4I4B | 1.7 | 428 | NAD, 1CO, 1CV | |||
| 3QAU | Escherichia coli | 2011 | 2.3 | 458 | - | n.a. |
| 3QAE | 2.3 | 458 | - | |||
| 3CDB | Homo sapiens | 2008 | 2.3 | 441 | 9HI | [42] |
| 3CDA | 2.1 | 441 | 8HI | |||
| 3CD7 | 2.1 | 441 | 882 | |||
| 3CD5 | 2.4 | 441 | 7HI | |||
| 3CD0 | 2.4 | 441 | 6HI | |||
| 3CCZ | Homo sapiens | 1.7 | 441 | 5HI | [42] | |
| 3CCW | 2.1 | 441 | 4HI | |||
| 3CCT | 2.1 | 441 | 3HI | |||
| 2R4F | Homo sapiens | 2008 | 1.7 | 441 | RIE | [43] |
| 3BGL | Homo sapiens | 2008 | 2.2 | 441 | RID | [44] |
| 2Q6C | Homo sapiens | 2007 | 2.0 | 441 | HR1 | [45] |
| 2Q6B | 2.0 | 441 | HR2 | |||
| 2Q1L | 2.1 | 441 | 882 | |||
| 1T02 | Pseudomonas mevalonii | 2003 | 2.6 | 428 | Lovastatin | [46] |
| 1R7I | Pseudomonas mevalonii | 2003 | 2.2 | 428 | - | n.a. |
| 1R31 | 2.1 | 428 | MEV, CoA | |||
| 1HWL | Homo sapiens | 2001 | 2.1 | 467 | ADP, Rosuvastatin | [47] |
| 1HWK | 2.2 | 467 | ADP, Atorvastatin | |||
| 1HWJ | 2.3 | 467 | ADP, Cerivastatin | |||
| 1HWI | 2.3 | 467 | ADP, Fluvastatin | |||
| 1HW9 | 2.3 | 467 | ADP, Simvastatin | |||
| 1HW8 | 2.1 | 467 | ADP, Compactin | |||
| 1DQA | Homo sapiens | 2000 | 2.0 | 467 | MAH, CoA, NADP | [38] |
| 1DQ9 | Homo sapiens | 2.8 | 467 | HMG | [38] | |
| 1DQ8 | 2.1 | 467 | CoA, DTT, MAH | |||
| 1QAY | Pseudomonas mevalonii | 1999 | 2.8 | 428 | MEV, NAD | [48] |
| 1QAX | 2.8 | 428 | HMG, NAD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesto, D.S.; Pereira, C.M.S.; Cerqueira, N.M.F.S.; Sousa, S.F. An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia. Molecules 2020, 25, 3891. https://doi.org/10.3390/molecules25173891
Gesto DS, Pereira CMS, Cerqueira NMFS, Sousa SF. An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia. Molecules. 2020; 25(17):3891. https://doi.org/10.3390/molecules25173891
Chicago/Turabian StyleGesto, Diana S., Carlos M. S. Pereira, Nuno M. F. S. Cerqueira, and Sérgio F. Sousa. 2020. "An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia" Molecules 25, no. 17: 3891. https://doi.org/10.3390/molecules25173891
APA StyleGesto, D. S., Pereira, C. M. S., Cerqueira, N. M. F. S., & Sousa, S. F. (2020). An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia. Molecules, 25(17), 3891. https://doi.org/10.3390/molecules25173891