
In-Vitro and In-Silico Evaluations of Heterocyclic-Containing Diarylpentanoids as Bcl-2 Inhibitors Against LoVo Colorectal Cancer Cells


Abstract
1. Introduction
2. Results and Discussions
2.1. Chemistry
2.2. Biological Studies
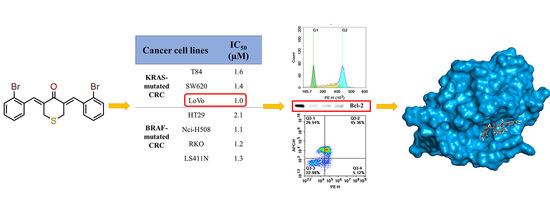
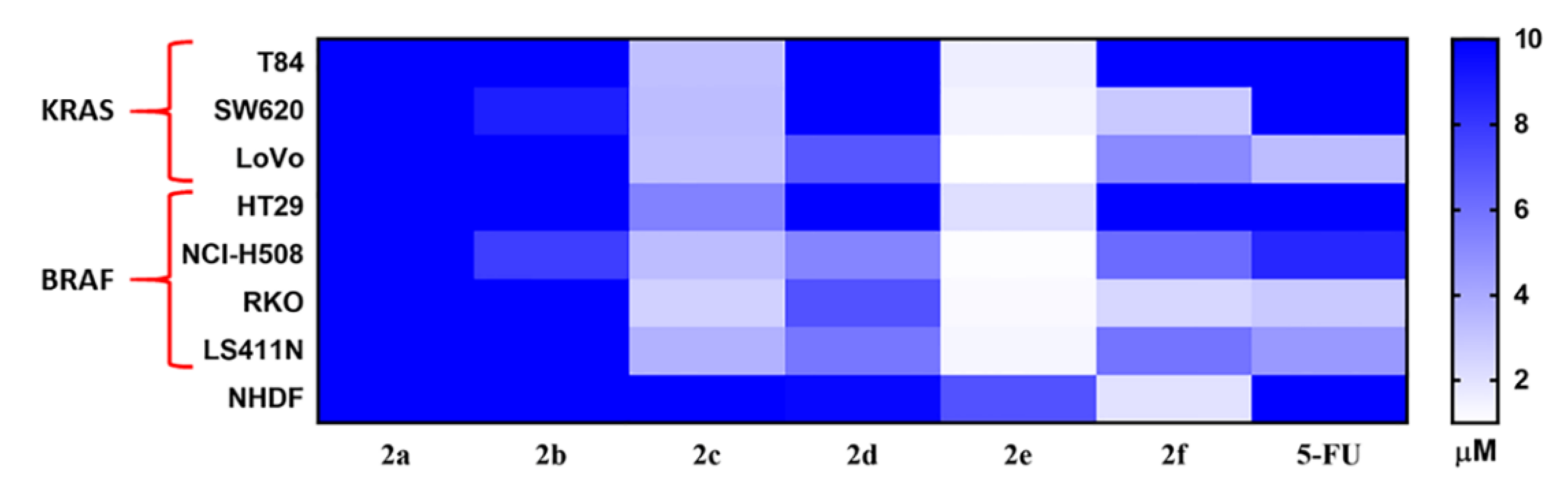
2.2.1. In-Vitro Cytotoxicity Analysis
2.2.2. In-Vitro Anti-Proliferative Analysis
2.2.3. In-Vitro Cell Apoptosis Analysis
2.2.4. In-Silico Molecular Modelling Analysis
3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of 2a–2e
3.2. Biological Evaluations
3.2.1. Cell Culture
3.2.2. MTT Assay
3.2.3. Morphological Inspection
3.2.4. Colony Formation Assay
3.2.5. Cell Cycle Analysis
3.2.6. Cell Apoptosis Analysis
3.2.7. Measurement of Intracellular Reactive Oxygen Species (ROS) Levels
3.2.8. Western Blot Analyses
3.2.9. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ren, J.-S.; Masuyer, E.; Ferlay, J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer 2012, 132, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Zheng, H.; Nebel, S.; Norris, P.S.; Aebi, S.; Lin, T.P.; Nehmé, A.; Christen, R.D.; Haas, M.; MacLeod, C.L.; et al. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997, 57, 1841–1845. [Google Scholar] [PubMed]
- Salonga, D.; Danenberg, K.D.; Johnson, M.; Metzger, R.; Groshen, S.; Tsao-Wei, D.D.; Lenz, H.J.; Leichman, C.G.; Leichman, L.; Diasio, R.; et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin. Cancer Res. 2000, 6, 1322–1327. [Google Scholar] [PubMed]
- Rosales, J.; Leong, L.A. Chemotherapy for Metastatic Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2005, 3, 525–529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sundar, S.; Symonds, R.; Decatris, M.; Kumar, D.; Osman, A.; Vasanthan, S.; O’Byrne, K. Phase II trial of Oxaliplatin and 5-Fluorouracil/Leucovorin combination in epithelial ovarian carcinoma relapsing within 2 years of platinum-based therapy. Gynecol. Oncol. 2004, 94, 502–508. [Google Scholar] [CrossRef]
- Soultati, A.; Mountzios, G.; Avgerinou, C.; Papaxoinis, G.; Pectasides, D.; Dimopoulos, M.-A.; Papadimitriou, C.A. Endothelial vascular toxicity from chemotherapeutic agents: Preclinical evidence and clinical implications. Cancer Treat. Rev. 2012, 38, 473–483. [Google Scholar] [CrossRef]
- Hatcher, H.; Planalp, R.P.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef]
- Koeberle, A.; Northoff, H.; Werz, O. Curcumin blocks prostaglandin E2 biosynthesis through direct inhibition of the microsomal prostaglandin E2 synthase-1. Mol. Cancer Ther. 2009, 8, 2348–2355. [Google Scholar] [CrossRef]
- Cho, J.-W.; Lee, K.-S.; Kim, C.-W. Curcumin attenuates the expression of IL-1β, IL-6, and TNF-α as well as cyclin E in TNF-α-treated HaCaT cells; NF-κB and MAPKs as potential upstream targets. Int. J. Mol. Med. 2007, 19, 469–474. [Google Scholar] [CrossRef]
- Shakibaei, M.; John, T.; Schulze-Tanzil, G.; Lehmann, I.; Mobasheri, A. Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem. Pharmacol. 2007, 73, 1434–1445. [Google Scholar] [CrossRef]
- Chan, M.M.-Y.; Huang, H.-I.; Fenton, M.R.; Fong, D. In Vivo Inhibition of Nitric Oxide Synthase Gene Expression by Curcumin, a Cancer Preventive Natural Product with Anti-Inflammatory Properties. Biochem. Pharmacol. 1998, 55, 1955–1962. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Anand, P.; Aggarwal, B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008, 269, 199–225. [Google Scholar] [CrossRef]
- Link, A.; Balaguer, F.; Shen, Y.; Lozano, J.-J.; Leung, H.-C.E.; Boland, C.R.; Goel, A. Curcumin Modulates DNA Methylation in Colorectal Cancer Cells. PLoS ONE 2013, 8, e57709. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, Z. The Effect of Curcumin on Breast Cancer Cells. J. Breast Cancer 2013, 16, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Maher, D.M.; Sundram, V.; Bell, M.C.; Jaggi, M.; Chauhan, S.C. Curcumin induces chemo/radio-sensitization in ovarian cancer cells and curcumin nanoparticles inhibit ovarian cancer cell growth. J. Ovarian Res. 2010, 3, 11. [Google Scholar] [CrossRef]
- Venkata, M.; Sripathy, R.; Anjana, D.; Somashekara, N.; Krishnaraju, A.; Krishanu, S.; Murali, M.; Verma, S.R.; Ramchand, C.N. In Silico, In Vitro and In Vivo Assessment of Safety and Anti-inflammatory Activity of Curcumin. Am. J. Infect. Dis. 2012, 8, 26–33. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Takada, Y.; Banerjee, S.; Newman, R.A.; Bueso-Ramos, C.E.; Price, J.E. Curcumin Suppresses the Paclitaxel-Induced Nuclear Factor-B Pathway in Breast Cancer Cells and Inhibits Lung Metastasis of Human Breast Cancer in Nude Mice. Clin. Cancer Res. 2005, 11, 7490–7498. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, Z.; Liang, G. Promising Curcumin-based Drug Design: Mono-carbonyl Analogues of Curcumin (MACs). Curr. Pharm. Des. 2013, 19, 2114–2135. [Google Scholar] [CrossRef]
- Grogan, G. Emergent mechanistic diversity of enzyme-catalysed β-diketone cleavage. Biochem. J. 2005, 388, 721–730. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.-G.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorganic Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef]
- Leong, S.W.; Faudzi, S.M.M.; Abas, F.; Aluwi, M.F.F.M.; Rullah, K.; Lam, K.W.; Bahari, M.N.A.; Ahmad, S.; Tham, C.L.; Shaari, K.; et al. Nitric oxide inhibitory activity and antioxidant evaluations of 2-benzoyl-6-benzylidenecyclohexanone analogs, a novel series of curcuminoid and diarylpentanoid derivatives. Bioorganic Med. Chem. Lett. 2015, 25, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, M.; Gao, J.; Li, J.; Fan, L.; Xiang, G.; Wang, X.; Wang, X.; Wu, X.; Sun, Y.; et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget 2015, 6, 37613–37625. [Google Scholar] [CrossRef] [PubMed]
- Nakhjiri, M.; Safavi, M.; Alipour, E.; Emami, S.; Atash, A.F.; Jafari-Zavareh, M.; Ardestani, S.K.; Khoshneviszadeh, M.; Foroumadi, A.; Shafiee, A. Asymmetrical 2,6-bis(benzylidene)cyclohexanones: Synthesis, cytotoxic activity and QSAR study. Eur. J. Med. Chem. 2012, 50, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.; Khan, S.I.; Kandi, S.K.; Raj, K.; Sun, G.; Yang, X.; Molina, A.D.C.; Ni, N.; Wang, B.-H.; Rawat, D.S. Synthesis, antimalarial activity and cytotoxic potential of new monocarbonyl analogues of curcumin. Bioorganic Med. Chem. Lett. 2013, 23, 112–116. [Google Scholar] [CrossRef]
- Subramaniam, D.; Nicholes, N.D.; Dhar, A.; Umar, S.; Awasthi, V.; Welch, D.R.; Jensen, R.A.; Anant, S. 3,5-bis(2,4-difluorobenzylidene)-4-piperidone, a novel compound that affects pancreatic cancer growth and angiogenesis. Mol. Cancer Ther. 2011, 10, 2146–2156. [Google Scholar] [CrossRef]
- Kumar, B.; Yadav, A.; Hideg, K.; Kuppusamy, P.; Teknos, T.N.; Kumar, P. A Novel Curcumin Analog (H-4073) Enhances the Therapeutic Efficacy of Cisplatin Treatment in Head and Neck Cancer. PLoS ONE 2014, 9, e93208. [Google Scholar] [CrossRef]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.-P.; Wang, S.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple Anticancer Activities of EF24, a Novel Curcumin Analog, on Human Ovarian Carcinoma Cells. Reprod. Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef]
- Pignanelli, C.; Ma, D.; Noel, M.; Ropat, J.; Mansour, F.; Curran, C.; Pupulin, S.; Larocque, K.; Wu, J.; Liang, G.; et al. Selective Targeting of Cancer Cells by Oxidative Vulnerabilities with Novel Curcumin Analogs. Sci. Rep. 2017, 7, 1105. [Google Scholar] [CrossRef]
- Sun, J.; Wang, S.; Li, H.; Jiang, W.; Hou, G.-G.; Zhao, F.; Cong, W. Synthesis, antitumor activity evaluation of some new N-aroyl-α, β-unsaturated piperidones with fluorescence. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–8. [Google Scholar] [CrossRef][Green Version]
- Pan, Y.; Wang, Y.; Cai, L.; Cai, Y.; Hu, J.; Yu, C.; Li, J.; Feng, Z.; Yang, S.; Li, X.; et al. Inhibition of high glucose-induced inflammatory response and macrophage infiltration by a novel curcumin derivative prevents renal injury in diabetic rats. Br. J. Pharmacol. 2012, 166, 1169–1182. [Google Scholar] [CrossRef]
- Katsori, A.-M.; Chatzopoulou, M.; Dimas, K.; Kontogiorgis, C.; Patsilinakos, A.; Trangas, T.; Hadjipavlou-Litina, D. Curcumin analogues as possible anti-proliferative & anti-inflammatory agents. Eur. J. Med. Chem. 2011, 46, 2722–2735. [Google Scholar] [CrossRef]
- Tan, K.-L.; Koh, S.-B.; Ee, P.-L.R.; Khan, M.; Go, M.-L. Curcumin Analogues with Potent and Selective Anti-proliferative Activity on Acute Promyelocytic Leukemia: Involvement of Accumulated Misfolded Nuclear Receptor Co-repressor (N-CoR) Protein as a Basis for Selective Activity. ChemMedChem 2012, 7, 1567–1579. [Google Scholar] [CrossRef]
- Tan, K.-L.; Ali, A.; Du, Y.; Fu, H.; Jin, H.-X.; Chin, T.-M.; Khan, M.; Go, M.-L. Synthesis and Evaluation of Bisbenzylidenedioxotetrahydrothiopranones as Activators of Endoplasmic Reticulum (ER) Stress Signaling Pathways and Apoptotic Cell Death in Acute Promyelocytic Leukemic Cells. J. Med. Chem. 2014, 57, 5904–5918. [Google Scholar] [CrossRef]
- Zamrus, S.N.H.; Akhtar, M.N.; Yeap, S.K.; Quah, C.K.; Loh, W.-S.; Alitheen, N.B.; Zareen, S.; Tajuddin, S.N.; Hussin, Y.; Shah, S.A.A. Design, synthesis and cytotoxic effects of curcuminoids on HeLa, K562, MCF-7 and MDA-MB-231 cancer cell lines. Chem. Central J. 2018, 12, 31. [Google Scholar] [CrossRef]
- Schmitt, F.; Gold, M.; Begemann, G.; Andronache, I.; Biersack, B.; Schobert, R. Fluoro and pentafluorothio analogs of the antitumoral curcuminoid EF24 with superior antiangiogenic and vascular-disruptive effects. Bioorganic Med. Chem. 2017, 25, 4894–4903. [Google Scholar] [CrossRef]
- Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P.B.; Ye, F.; Li, Y.; et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorganic Med. Chem. Lett. 2008, 18, 1525–1529. [Google Scholar] [CrossRef]
- Gindy, M.; Dwidar, I.M. 183. 3:4-Benzoxanthens. Part III. The synthesis and oxidation of 5-, 6-, 7-, and 8-methyl-3:4-benzoxanthen. J. Chem. Soc. 1953, 893–895. [Google Scholar] [CrossRef]
- Leong, S.W.; Chia, S.L.; Abas, F.; Yusoff, K. Synthesis and in-vitro anti-cancer evaluations of multi-methoxylated asymmetrical diarylpentanoids as intrinsic apoptosis inducer against colorectal cancer. Bioorganic Med. Chem. Lett. 2020, 30, 127065. [Google Scholar] [CrossRef]
- Leong, S.W.; Chia, S.L.; Abas, F.; Yusoff, K. Asymmetrical meta-methoxylated diarylpentanoids: Rational design, synthesis and anti-cancer evaluation in-vitro. Eur. J. Med. Chem. 2018, 157, 716–728. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [PubMed]
- Aharoni-Simon, M.; Shumiatcher, R.; Yeung, A.; Shih, A.Z.L.; Dolinsky, V.W.; Doucette, C.A.; Luciani, D.S. Bcl-2 Regulates Reactive Oxygen Species Signaling and a Redox-Sensitive Mitochondrial Proton Leak in Mouse Pancreatic β-Cells. Endocrinology 2016, 157, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Veis, D.J.; Sorenson, C.M.; Shutter, J.R.; Korsmeyer, S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993, 75, 229–240. [Google Scholar] [CrossRef]
- Hochman, A.; Sternin, H.; Gorodin, S.; Korsmeyer, S.; Ziv, I.; Melamed, E.; Offen, D. Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice. J. Neurochem. 1998, 71, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Nissink, J.W.M.; Murray, C.; Hartshorn, M.; Verdonk, M.L.; Cole, J.C.; Taylor, R. A new test set for validating predictions of protein-ligand interaction. Proteins Struct. Funct. Bioinform. 2002, 49, 457–471. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Z.-Y.; Li, Y.-Y.; Luo, Y.; Liu, M.-L.; Dong, H.-Y.; Wang, Y.-X.; Liu, Y.; Zhao, P.-T.; Jin, F.-G.; et al. Antiinflammatory effects of matrine in LPS-induced acute lung injury in mice. Eur. J. Pharm. Sci. 2011, 44, 573–579. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | Cmpd | Structure | Cmpd | Structure |
|---|---|---|---|---|---|
| 2a |  | 2b |  | 2c |  |
| 2d |  | 2e |  | 2f |  |

| Compound | X | R | IC50 (µM) | NHDF | Chemo- Therapeutic Index | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KRAS | BRAF | ||||||||||
| T84 | SW620 | LoVo | HT29 | NCI-H508 | RKO | LS411N | |||||
| 5-FU | - | - | >10 | >10 | 3.3 | >10 | 8.6 | 2.9 | 4.6 | >20 | - |
| 2a | C | 2-OMe | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >20 | - |
| 2b | C | 2-Br | >10 | 8.9 | >10 | >10 | 7.8 | >10 | >10 | >20 | - |
| 2c | C | 3,4-F | 3.3 | 3.2 | 3.3 | 5.4 | 3.3 | 2.6 | 3.7 | 12.9 | 2.3–5.0 |
| 2d | S | 2-OMe | >10 | >10 | 6.9 | >10 | 5.3 | 7.1 | 5.8 | 9.7 | 1.4–1.8 |
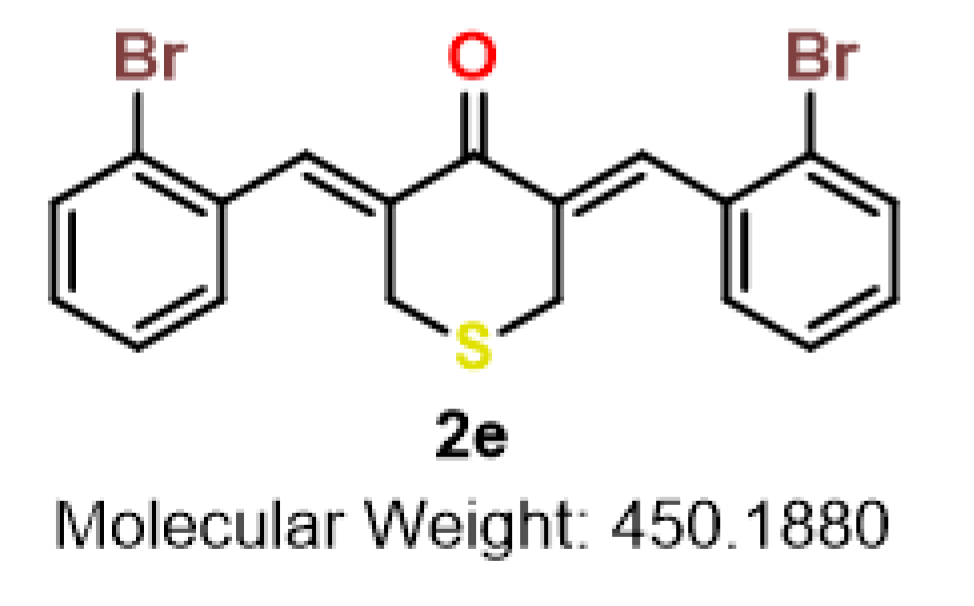
| 2e | S | 2-Br | 1.6 | 1.4 | 1.0 | 2.1 | 1.1 | 1.2 | 1.3 | 7.1 | 3.4–7.1 |
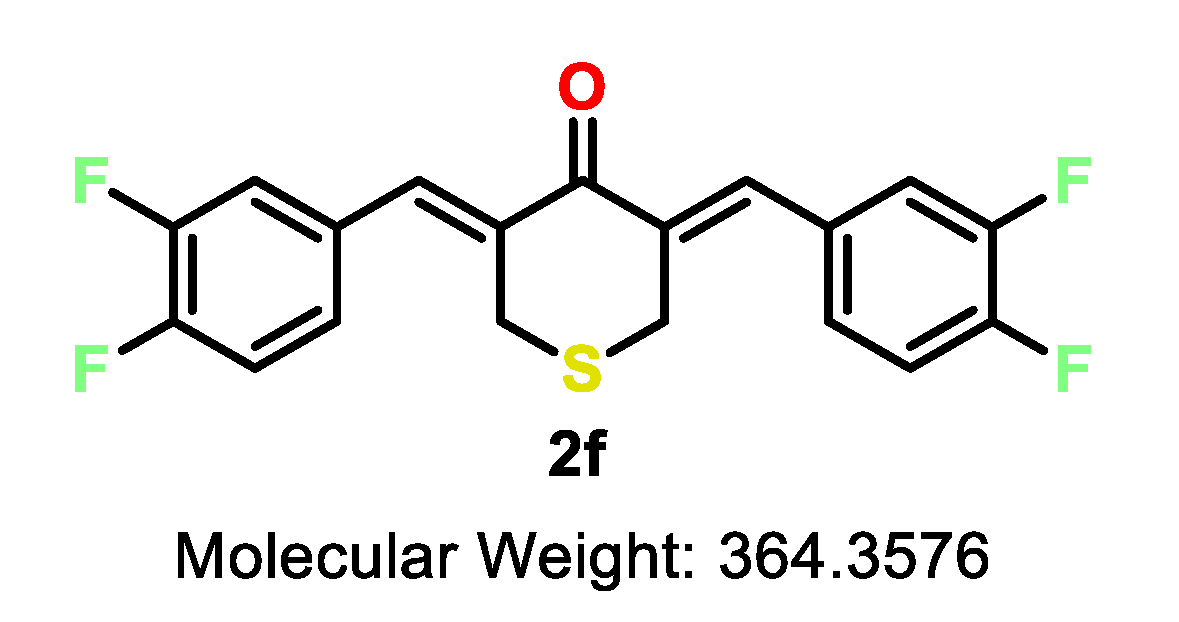
| 2f | S | 3,4-F | >10 | 2.9 | 5.1 | >10 | 6.2 | 2.4 | 5.9 | 2.0 | 0.3–0.8 |
| Compound | Structure | Interacting Amino Acid Residue | Type of Interaction | Bonding Distance (Å) |
|---|---|---|---|---|
| 2b |  | ALA-146 | Alkyl | 3.84 |
| MET-112 | Pi-Alkyl | 4.12 | ||
| ARG-143 | Pi-Cation | 2.39 | ||
| LEU-134 | Alkyl | 4.8 | ||
| Carbon | 2.49 | |||
| 2e |  | ALA-146 | Pi-Alkyl | 5.20 |
| MET-112 | Pi-Alkyl | 4.76 | ||
| Alkyl | 4.75 | |||
| ARG-143 | Alkyl | 3.93 | ||
| Pi-Cation | 3.67 | |||
| Pi-Alkyl | 5.14 | |||
| TYR-105 | Pi-Alkyl | 5.24 | ||
| PHE-101 | Pi-Sulfur | 4.78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leong, S.W.; Chia, S.L.; Abas, F.; Yusoff, K. In-Vitro and In-Silico Evaluations of Heterocyclic-Containing Diarylpentanoids as Bcl-2 Inhibitors Against LoVo Colorectal Cancer Cells. Molecules 2020, 25, 3877. https://doi.org/10.3390/molecules25173877
Leong SW, Chia SL, Abas F, Yusoff K. In-Vitro and In-Silico Evaluations of Heterocyclic-Containing Diarylpentanoids as Bcl-2 Inhibitors Against LoVo Colorectal Cancer Cells. Molecules. 2020; 25(17):3877. https://doi.org/10.3390/molecules25173877
Chicago/Turabian StyleLeong, Sze Wei, Suet Lin Chia, Faridah Abas, and Khatijah Yusoff. 2020. "In-Vitro and In-Silico Evaluations of Heterocyclic-Containing Diarylpentanoids as Bcl-2 Inhibitors Against LoVo Colorectal Cancer Cells" Molecules 25, no. 17: 3877. https://doi.org/10.3390/molecules25173877
APA StyleLeong, S. W., Chia, S. L., Abas, F., & Yusoff, K. (2020). In-Vitro and In-Silico Evaluations of Heterocyclic-Containing Diarylpentanoids as Bcl-2 Inhibitors Against LoVo Colorectal Cancer Cells. Molecules, 25(17), 3877. https://doi.org/10.3390/molecules25173877