
Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method

 , ,
, ,
Abstract
1. Introduction
2. Results
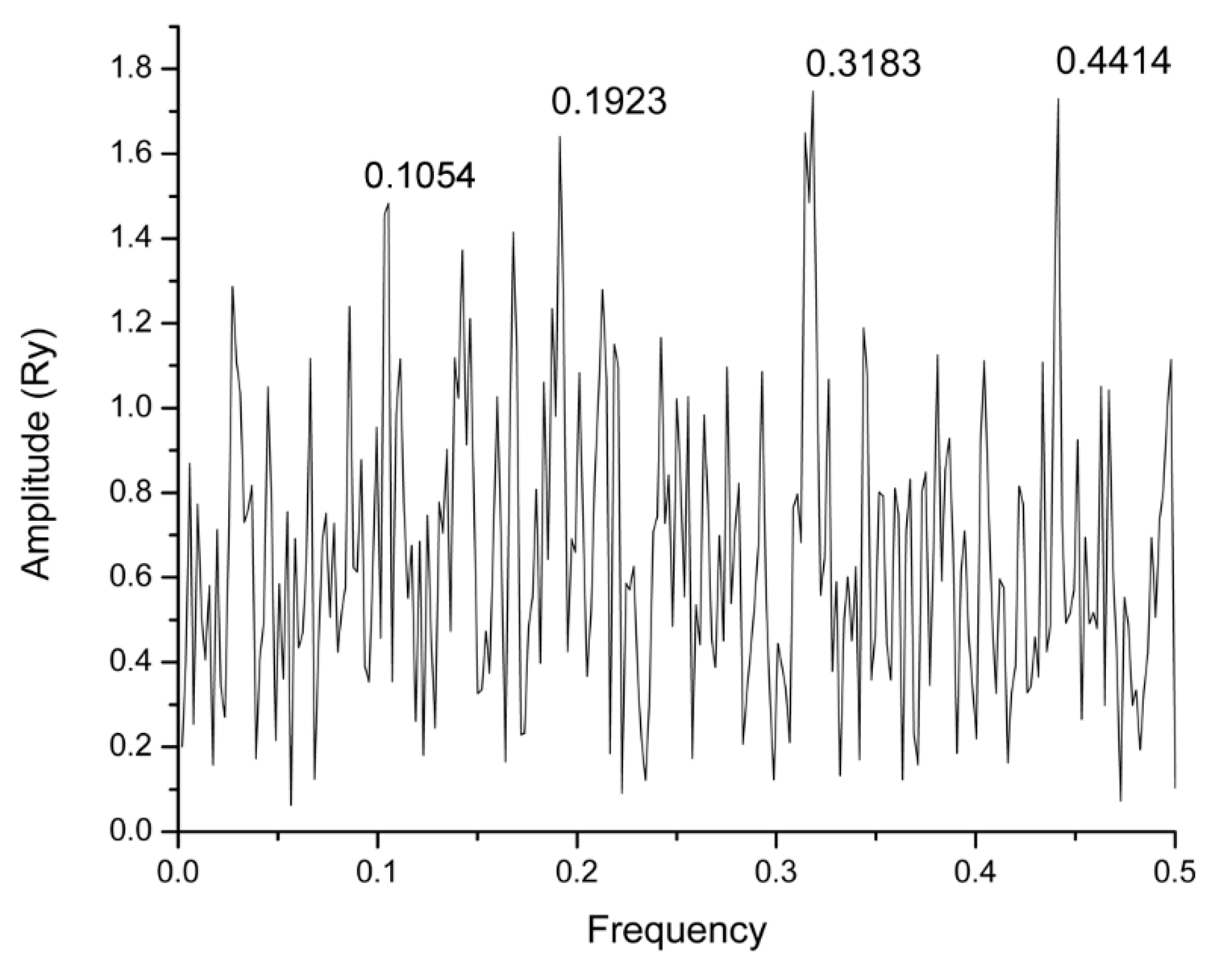
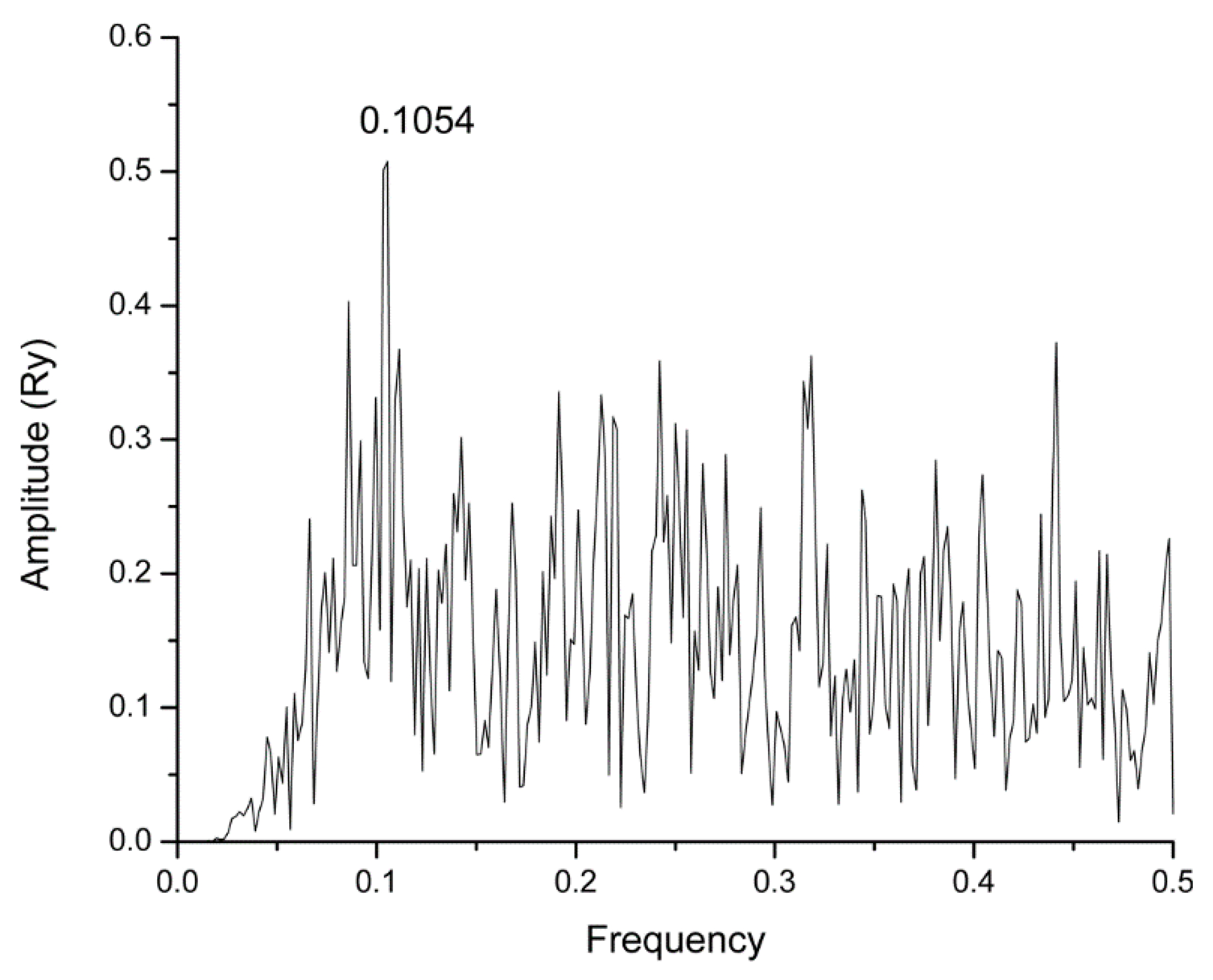
2.1. Informational Spectrum Method Analysis
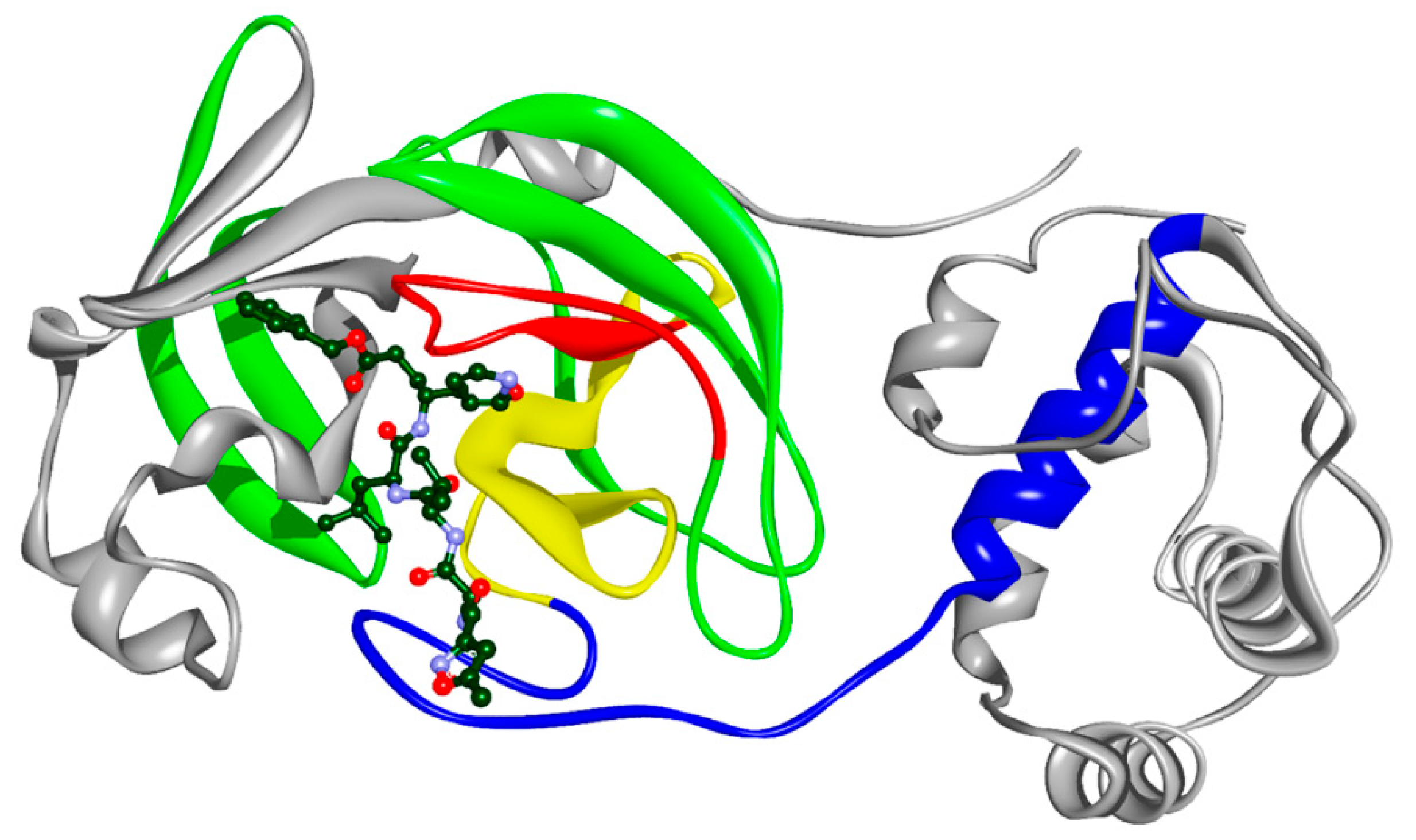
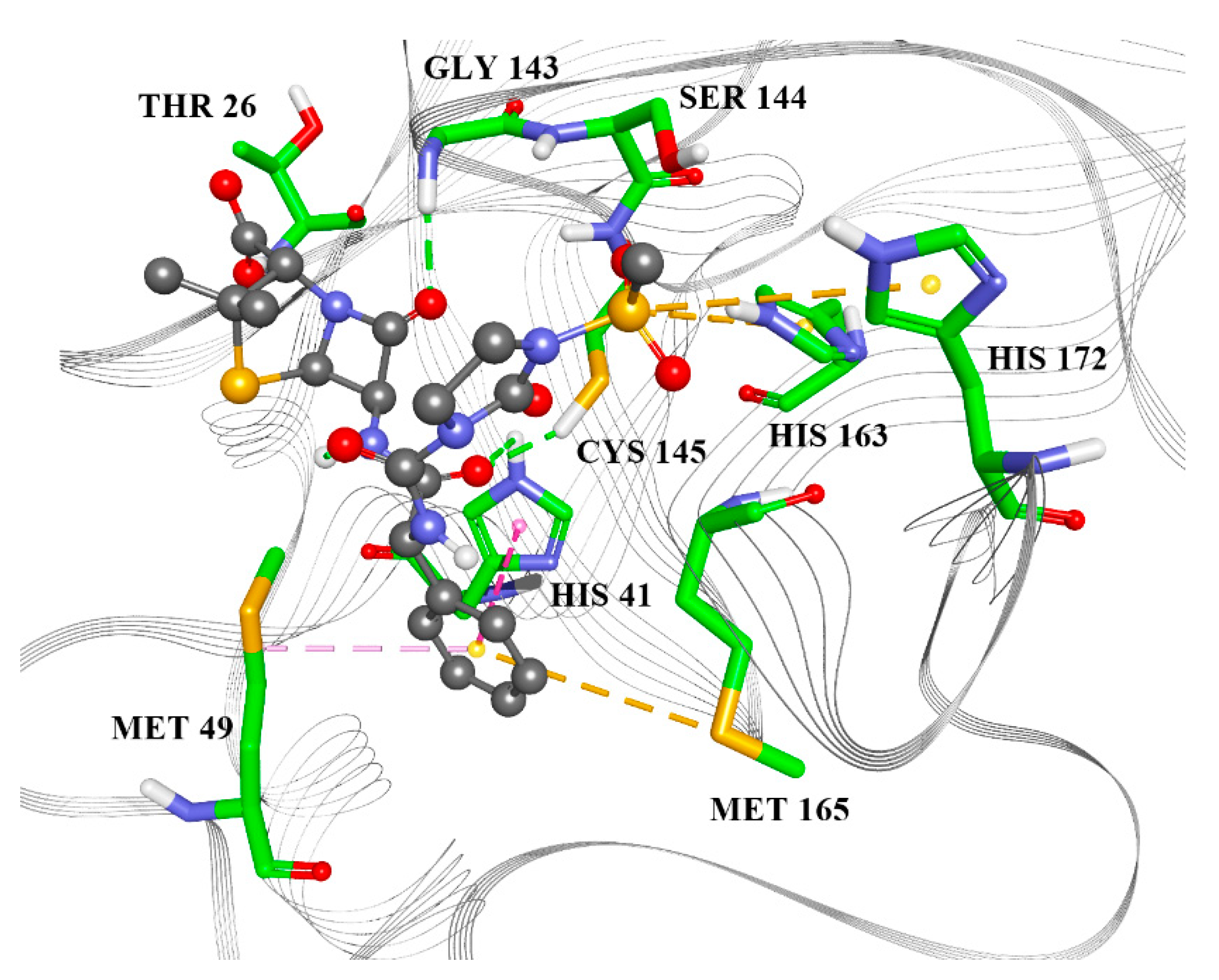
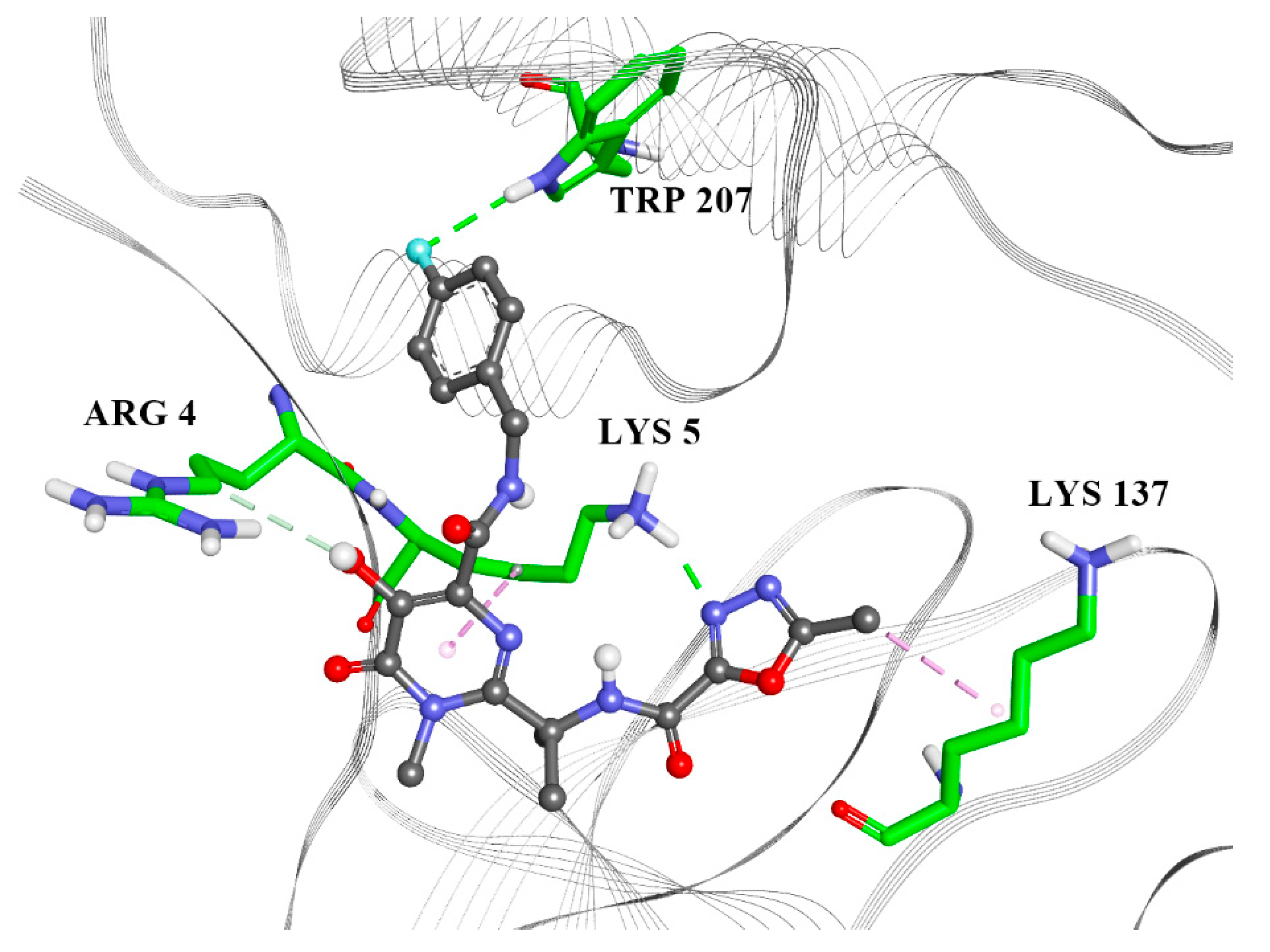
2.2. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Informational Spectrum Method
4.2. Data Preparation
4.3. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Coronavirus Disease 2019. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 15 April 2020).
- Yamey, G.; Schäferhoff, M.; Pate, M.; Chawla, M.; Ranson, K.; Hatchett, R.; Wilder, R. Funding the Development and Manufacturing of COVID-19. Vaccines 2020. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Tu, Y.F.; Chien, C.S.; Yarmishyn, A.A.; Lin, Y.Y.; Luo, Y.H.; Lin, Y.T.; Lai, W.T.; Yang, D.M.; Chou, S.J.; Yang, Y.P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef]
- Mitjà, O.; Clotet, B. Use of antiviral drugs to reduce COVID-19 transmission. Lancet Glob. Health 2020. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Strodel, B.; Olubiyi, O.; Olagunju, M.; Keutmann, M.; Loschwitz, J. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Preprints 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeldl, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of iMproved a-ketoamide inhibitors. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Thenin-Houssier, S.; De Vera, I.M.; Pedro-Rosa, L.; Brady, A.; Richard, A.; Konnick, B.; Opp, S.; Buffone, C.; Fuhrmann, J.; Kota, S.; et al. Ebselen, a small-molecule capsid inhibitor of HIV-1 replication. Antimicrob. Agents Chemother. 2016, 60, 2195–2208. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Vergara-Alert, J.; Segalés, J.; Paessler, S. Use of the informational spectrum methodology for rapid biological analysis of the novel coronavirus 2019-nCoV: Prediction of potential receptor, natural reservoir, tropism and therapeutic/vaccine target. F1000Research 2020, 9, 52. [Google Scholar] [CrossRef]
- Veljkovic, V.; Paessler, S. COVID-19 Orf3b protein: The putative biological function and the therapeutic target. (Version 1) Res. Sq. 2020. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Sencanski, M.; Sumonja, N.; Perovic, V.; Glisic, S.; Veljkovic, N.; Veljkovic, V. The Application of Information Spectrum Method on Small Molecules and Target Recognition. arXiv 2019, arXiv:1907.02713. [Google Scholar]
- Hosseini, M.; Chen, W.; Wang, C. Computational Molecular Docking and Virtual Screening Revealed Promising SARS-CoV-2 Drugs (Preprint). ChemRxiv 2020. [Google Scholar] [CrossRef]
- Matsuyama, S.; Kawase, M.; Nao, N.; Shirato, K.; Ujike, M.; Kamitani, W.; Shimojima, M.; Fukushi, S. The inhaled corticosteroid ciclesonide blocks coronavirus RNA replication by targeting viral NSP15. BioRxiv 2020. [Google Scholar] [CrossRef]
- Nakajima, K.; Ogawa, F.; Sakai, K.; Uchiyama, M.; Oyama, Y.; Kato, H.; Takeuchi, I. A Case of Coronavirus Disease 2019 Treated With Ciclesonide. In Mayo Clinic Proceedings; Elsevier: Rochester, MN, USA, 2020. [Google Scholar] [CrossRef]
- Iwabuchi, K.; Yoshie, K.; Kurakami, Y.; Takahashi, K.; Kato, Y.; Morishima, T. Therapeutic potential of ciclesonide inahalation for COVID-19 pneumonia: Report of three cases. J. Infect. Chemother. 2020. [Google Scholar] [CrossRef]
- A Trial of Ciclesonide Alone or in Combination with Hydroxychloroquine for Adults with Mild COVID-19. ClinicalTrials. gov Identifier: NCT0433058. Available online: https://clinicaltrials.gov/ct2/show/NCT04330586 (accessed on 14 July 2020).
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Aly, M.O. Molecular Docking Reveals the Potential of Aliskiren, Dipyridamole, Mopidamol, Rosuvastatin, Rolitetracycline and Metamizole to Inhibit COVID-19 Virus Main Protease. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Adler, S.; Schrier, R.W.; Berl, T.; Zhao, Q.; Czerwiec, F.S. Efficacy and safety of oral tolvaptan therapy in patients with the syndrome of inappropriate antidiuretic hormone secretion. Eur. J. Endocrinol. 2011, 164, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, Z.; Al-Shokri, S.D.; Al-soub, H.; Mohamed, M.F. COVID-19-associated SIADH: A clue in the times of pandemic! Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E882–E885. [Google Scholar] [CrossRef] [PubMed]
- Gul, S.; Ozcan, O.; Asar, S.; Okyar, A.; Baris, I.; Kavakli, I.H. In Silico Identification of Widely Used and Well Tolerated Drugs That May Inhibit SARSCov-2 3C-like Protease and Viral RNA-Dependent RNA Polymerase Activities, and May Have Potential to Be Directly Used in Clinical Trials. ChemRxiv 2020. PPR: PPR151426. [Google Scholar] [CrossRef]
- De Lucca, G.V.; Otto, M.J. Synthesis and anti-HIV activity of pyrrolo-[1, 2-d]-(1, 4)-benzodiazepin-6-ones. Bioorg. Med. Chem. Lett. 1992, 2, 1639–1644. [Google Scholar] [CrossRef]
- Smyth, L.J.; Cañadas-Garre, M.; Cappa, R.C.; Maxwell, A.P.; McKnight, A.J. Genetic associations between genes in the renin-angiotensin-aldosterone system and renal disease: A systematic review and meta-analysis. BMJ Open 2019, 9, e026777. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Bean, D.; Kraljevic, Z.; Searle, T.; Bendayan, R.; Pickles, A.; Folarin, A.; Roguski, L.; Noor, K.; Shek, A.; O’Gallagher, K.; et al. Treatment with ACE-inhibitors is associated with less severe disease with SARS-Covid-19 infection in a multi-site UK acute Hospital Trust. Medrxiv 2020. [Google Scholar] [CrossRef]
- Mahanta, S.; Chowdhury, P.; Gogoi, N.; Goswami, N.; Borah, D.; Kumar, R.; Chetia, D.; Borah, P.; Buragohain, A.K.; Gogoi, B. Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach [published online ahead of print, 25 May 2020]. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Vatansever, E.C.; Yang, K.; Kratch, K.C.; Drelich, A.; Cho, C.C.; Mellot, D.M.; Xu, S.; Tseng, C.T.K.; Liu, W.R. Targeting the SARS-CoV-2 Main Protease to Repurpose Drugs for COVID-19. BioRxiv 2020. [Google Scholar] [CrossRef]
- Tariq, A.; Mateen, R.; Sohail, A.S.; Saleem, M. Paromomycin: A Potential Dual Targeted Drug Effectively Inhibits both Spike (S1) and Main Protease of COVID-19. Int. J. Infect. Dis. 2020, 98, 166–175. [Google Scholar] [CrossRef]
- Duarte, R.R.; Copertino, D.C., Jr.; Iñiguez, L.P.; Marston, J.L.; Nixon, D.F.; Powell, T.R. Repurposing FDA-Approved Drugs for COVID-19 Using a Data-Driven Approach. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Kumar, V.; Jena, M. In silico virtual screening-based study of nutraceuticals predicts the therapeutic potentials of folic acid and its derivatives against COVID-19. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Aksoydan, B.; Dogan, B.; Sahin, K.; Shahraki, A.; Birgül-İyison, N. Screening of Clinically Approved and Investigation Drugs as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike Receptor-Binding Domain Bound with ACE2 COVID19 Target Proteins: A Virtual Drug Repurposing Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Verma, D.; Kapoor, S.; Das, S.; Thakur, K. Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) Identified from the Library of FDA Approved Drugs Using Molecular Docking Studies. Preprints.org. 2020. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Rosa, S.G.V.; Santos, W.C. Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam Salud Publica 2020, 44, e40. [Google Scholar] [CrossRef]
- Vafaei, S.; Razmi, M.; Mansoori, M.; Asadi-Lari, M.; Madjd, Z. Spotlight of Remdesivir in Comparison with Ribavirin, Favipiravir, Oseltamivir and Umifenovir in Coronavirus Disease 2019 (COVID-19) Pandemic. Favipiravir Oseltamivir Umifenovir Coronavirus Dis. 2019. [Google Scholar] [CrossRef]
- Veljkovic, V.; Cosic, I.; Dimitrijevic, B. Is it possible to analyze DNA and protein sequences by the methods of digital signal processing? IEEE Trans. Biomed. Eng. 1985, 32, 337–341. [Google Scholar] [CrossRef]
- Veljkovic, V.; Slavic, I. Simple general-model pseudopotential. Phys. Rev. Lett. 1972, 29, 105. [Google Scholar] [CrossRef]
- MOPAC2016; Stewart, J.J. Stewart Computational Chemistry; Colorado Springs, CO, USA, 2016. Available online: http://OpenMOPAC.net (accessed on 4 August 2020).
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment (Release 2017); Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Maestro, V. Schrödinger Release 2017-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2016; Available online: https://www.schrodinger.com/maestro (accessed on 4 August 2020).
- Origin; Version 9; OriginLab Corporation: Northampton, MA, USA, 2012.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Drugbank ID | F | Binding Energy (kcal/mol) |
|---|---|---|---|
| Mezlocillin | DB00948 | 0.1923 | −8.6 |
| Camazepam | DB01489 | 0.1923 | −7.5 |
| Spirapril | DB01348 | 0.1923 | −7.1 |
| Bacampicillin | DB01602 | 0.1923 | −6.8 |
| Bacitracin | DB00626 | 0.3183 | −6.6 |
| Carbinoxamine | DB00748 | 0.3183 | −6.6 |
| Paromomycin | DB01421 | 0.1923 | −6.5 |
| Nifedipine | DB01115 | 0.1923 | −6.3 |
| Gemfibrozil | DB01241 | 0.1923 | −5.9 |
| Trimethaphan | DB01116 | 0.1923 | −5.6 |
| Phensuximide | DB00832 | 0.3183 | −5.6 |
| Nitrendipine | DB01054 | 0.1923 | −5.4 |
| Paliperidone | DB01267 | 0.1923 | −5.4 |
| Levetiracetam | DB01202 | 0.1923 | −5.2 |
| Chlorambucil | DB00291 | 0.1923 | −5.2 |
| Vitamin C | DB00126 | 0.1923 | −5.1 |
| Tizanidine | DB00697 | 0.1923 | −5.0 |
| Ifosfamide | DB01181 | 0.1923 | −4.9 |
| Aminophenazone | DB01424 | 0.1923 | −4.6 |
| Mecamylamine | DB00657 | 0.1923 | −4.3 |
| Tazarotene | DB00799 | 0.1923 | −4.1 |
| Name | Drugbank ID | F | Binding Energy (kcal/mol) |
|---|---|---|---|
| Raltegravir | DB06817 | 0.1054 | −7.9 |
| Rolitetracycline | DB01301 | 0.4414 | −7.6 |
| Tolvaptan | DB06212 | 0.1054 | −7.3 |
| Ciclesonide | DB01410 | 0.1054 | −7.2 |
| Rescinnamine | DB01180 | 0.4414 | −7.2 |
| Spectinomycin | DB00919 | 0.1054 | −6.8 |
| Cefotiam | DB00229 | 0.1054 | −6.6 |
| Azatadine | DB00719 | 0.1054 | −6.5 |
| Flecainide | DB01195 | 0.1054 | −6.2 |
| Pivmecillinam | DB01605 | 0.1054 | −6.2 |
| Voriconazole | DB00582 | 0.1054 | −6.1 |
| Ambenonium | DB01122 | 0.1054 | −5.9 |
| Amitriptyline | DB00321 | 0.1054 | −5.9 |
| Azapropazone | DB07402 | 0.1054 | −5.8 |
| Miconazole | DB01110 | 0.1054 | −5.8 |
| Clofedanol | DB04837 | 0.1054 | −5.7 |
| Flutamide | DB00499 | 0.1054 | −5.7 |
| Leflunomide | DB01097 | 0.1054 | −5.7 |
| Tobramycin | DB00684 | 0.1054 | −5.7 |
| Clevidipine | DB04920 | 0.1054 | −5.6 |
| Imipramine | DB00458 | 0.1054 | −5.6 |
| Kanamycin | DB01172 | 0.1054 | −5.6 |
| Ciclopirox | DB01188 | 0.1054 | −5.5 |
| Oseltamivir | DB00198 | 0.1054 | −5.5 |
| Ospemifene | DB04938 | 0.1054 | −5.5 |
| Trimipramine | DB00726 | 0.1054 | −5.5 |
| Ebselen | DB12610 | 0.1054 | −5.3 |
| Bepridil | DB01244 | 0.1054 | −5.1 |
| Ethinamate | DB01031 | 0.1054 | −4.9 |
| Propylthiouracil | DB00550 | 0.1054 | −4.5 |
| l−Arginine | DB00125 | 0.1054 | −4.4 |
| Isoflurophate | DB00677 | 0.1054 | −4.2 |
| Ethanolamine Oleate | DB06689 | 0.1054 | −4.1 |
| l−Carnitine | DB00583 | 0.1054 | −4.1 |
| l−Lysine | DB00123 | 0.1054 | −4.0 |
| Methoxyflurane | DB01028 | 0.1054 | −3.6 |
| Residue | Mezlocillin | Camazepam | Spirapril |
|---|---|---|---|
| THR27 | X | ||
| HIS41 | X | X | X |
| GLY143 | X | ||
| SER144 | X | ||
| CYS145 | X | X | X |
| HIS163 | X | X | |
| GLU166 | X | X | |
| HIS172 | X | ||
| MET49 | X | X | X |
| MET165 | X | X |
| Raltegravir | Rolitetracycline | Tolvaptan | Ciclesonide | Rescinnamine | |
|---|---|---|---|---|---|
| ARG4 | X | X | |||
| LYS5 | X | X | X | X | X |
| ARG131 | X | X | X | ||
| LYS137 | X | X | X | ||
| TRP207 | X | ||||
| ASP289 | X | X | |||
| LEU287 | X | ||||
| GLU288 | X | ||||
| GLN127 | X | ||||
| ASP197 | X | ||||
| GLY138 | X | ||||
| GLU290 | X | ||||
| TYR126 | X | X | |||
| PHE291 | X | ||||
| LEU286 | X |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sencanski, M.; Perovic, V.; Pajovic, S.B.; Adzic, M.; Paessler, S.; Glisic, S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules 2020, 25, 3830. https://doi.org/10.3390/molecules25173830
Sencanski M, Perovic V, Pajovic SB, Adzic M, Paessler S, Glisic S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules. 2020; 25(17):3830. https://doi.org/10.3390/molecules25173830
Chicago/Turabian StyleSencanski, Milan, Vladimir Perovic, Snezana B. Pajovic, Miroslav Adzic, Slobodan Paessler, and Sanja Glisic. 2020. "Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method" Molecules 25, no. 17: 3830. https://doi.org/10.3390/molecules25173830
APA StyleSencanski, M., Perovic, V., Pajovic, S. B., Adzic, M., Paessler, S., & Glisic, S. (2020). Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules, 25(17), 3830. https://doi.org/10.3390/molecules25173830